

Abstract
Clostridium difficile infection (CDI) causes nearly half a million cases of diarrhea and colitis in the United States each year. Although the importance of the gut microbiota in C. difficile pathogenesis is well recognized, components of the human gut flora critical for colonization resistance are not known. Culture-independent high-density Roche 454 pyrosequencing was used to survey the distal gut microbiota for 39 individuals with CDI, 36 subjects with C. difficile-negative nosocomial diarrhea (CDN), and 40 healthy control subjects. A total of 526,071 partial 16S rRNA sequence reads of the V1 to V3 regions were aligned with 16S databases, identifying 3,531 bacterial phylotypes from 115 fecal samples. Genomic analysis revealed significant alterations of organism lineages in both the CDI and CDN groups, which were accompanied by marked decreases in microbial diversity and species richness driven primarily by a paucity of phylotypes within the Firmicutes phylum. Normally abundant gut commensal organisms, including the Ruminococcaceae and Lachnospiraceae families and butyrate-producing C2 to C4 anaerobic fermenters, were significantly depleted in the CDI and CDN groups. These data demonstrate associations between the depletion of Ruminococcaceae, Lachnospiraceae, and butyrogenic bacteria in the gut microbiota and nosocomial diarrhea, including C. difficile infection. Mechanistic studies focusing on the functional roles of these organisms in diarrheal diseases and resistance against C. difficile colonization are warranted.
INTRODUCTION
Nosocomial diarrhea is a common complication in hospitalized patients. Clostridium difficile is the most common infectious cause of nosocomial diarrhea, representing 15 to 25% of antibiotic-associated diarrhea and most cases of colitis associated with antibiotic use. The mortality rate for Clostridium difficile infection (CDI) is estimated to be 1 to 2.5%, contributing to 14,000 deaths per year in the United States (1–3). Antibiotic-induced perturbation of the gut microbiota is widely believed to provide C. difficile an undesirable advantage, allowing the organism to proliferate and elaborate its toxins in the background of susceptible flora. Although standard antibiotic therapy (e.g., metronidazole or oral vancomycin treatment) is highly effective in suppressing C. difficile, it does not prevent relapse. Indeed, 15 to 30% of patients experience relapse within 3 months following antibiotic treatment. Multiple recurrences of CDI are not uncommon (4–6). Fecal microbiota transplantation (FMT) is known to cure >80 to 90% of recurrent C. difficile infections (7–11). Following FMT, the gut microbiota of the recipient resembles that of the donor (12). These observations indicate that the donor's bacteria are capable of restoring the structure and function of the recipient's gut microbial communities. The high efficacy rate of FMT provides proof of the principle that the gut microbiota confers resistance against C. difficile colonization. More importantly, it offers the promise that identification of key microbial factors important for colonization resistance could lead to novel probiotic therapy.
Some 100 trillion microorganisms inhabit and colonize the human gut (13, 14). Early studies indicated that randomly selected bacterial isolates from the gut flora were partially effective in suppressing C. difficile. A few species, including Lactobacillus, Enterococcus, Bifidobacterium, and Bacteroides species, have been shown to have inhibitory activities against C. difficile (15, 16). A number of recent studies have investigated the gut microbiota associated with CDI by using culture-based methods, 16S rRNA microarrays, and 16S clonal sequencing (17–19). In most studies, decreases in microbial diversity and richness and/or altered microbial compositions were observed. Interestingly, in 20 patients with asymptomatic C. difficile carriage, the gut microbial profile closely resembled that of healthy adults (19), suggesting that the normal gut microbiota may protect hosts from developing C. difficile infection. Two recent studies provided additional evidence that resistance against C. difficile colonization could be engineered in mice using a single Lachnospiraceae isolate or a defined cocktail of six murine gut isolates (20, 21). Taken together, the data on fecal transplantation in clinical settings and experimental studies in animals suggest that it may be possible to manipulate the gut microbiota using defined human gut isolates as probiotics to treat and/or to prevent CDI.
Thanks to the Human Microbiome Project (22), our knowledge of the gut microbiome in health and disease has increased rapidly in recent years. The microbial factors required for resistance against C. difficile and other enteric pathogens, however, remain elusive. The primary goal of the present study was to identify candidate commensal organisms that may be exploited for C. difficile therapy. We examined 549,643 partial prokaryotic 16S rRNA gene sequences from stool specimens from subjects with CDI, subjects with C. difficile-negative nosocomial diarrhea (CDN), and healthy control (HC) subjects, identifying 3,531 bacterial phylotypes from 115 fecal specimens. Our 16S microbiome analysis revealed significant alterations in microbial community structures associated with CDI and CDN, accompanied by markedly decreased microbial diversity and fewer bacterial phylotypes, compared with healthy controls. Several essential types of bacteria that produce butyrate and short-chain fatty acids (SCFAs), all of which are members of Clostridium clusters IV and XIVa in the Ruminococcaceae or Lachnospiraceae family, were absent or markedly depleted in CDI and CDN. The SCFA-producing bacteria were classified in silico from examinations of the genera determined from the deep-sequencing data sets. These results implicate Ruminococcaceae, Lachnospiraceae, and butyrogenic bacteria in the pathogenesis of nosocomial diarrhea and C. difficile infection.
MATERIALS AND METHODS
Subjects and sample collection.
The University of Florida institutional review board reviewed and approved the study design. Stool samples submitted to the clinical microbiology laboratory at Shands Hospital at the University of Florida for C. difficile microbiological testing were stored at 4°C and archived at −80°C, within 24 h after sample collection, for fecal microbiome analysis. Recent studies showed that storage of fecal samples at 4°C for up to 48 h had little effect on the microbiome composition (23). CDI was defined as the presence of diarrhea (a change in bowel habits with more than three unformed bowel movements in a 24-h period before sample collection) with the presence of C. difficile toxin in the stool specimen. C. difficile-negative nosocomial diarrhea (CDN) was defined as the presence of diarrhea (using the same criteria as for CDI) with the absence of C. difficile toxin in the stool specimen. Control fecal samples were collected from healthy individuals as outpatients, stored at 4°C for up to 4 h, and then archived at −80°C until microbiome analysis. Clinical microbiological testing for C. difficile was performed using the C. Diff Quik Chek Complete test (Techlab, Blacksburg, VA) or a GeneXpert multiplex PCR assay for detection of the toxin B gene (Cepheid, Sunnyvale, CA), as the platform for C. difficile testing in the clinical microbiology laboratory was converted to the more-sensitive PCR-based assay during the study. The presence of the toxin B gene (tcdB) was confirmed in all samples in the CDI group using the toxin B-specific PCR assay and in-house primers, as described in the supplemental material. Very weak amplification products also were observed in samples from seven subjects in the CDN group and one healthy control subject.
DNA extraction, bacterial 16S rRNA gene amplification, and sequence analysis.
Frozen fecal specimens were thawed, and genomic DNA was extracted using a MO-BIO PowerSoil DNA isolation kit (Carlsbad, CA). The V1 to V3 hypervariable regions of bacterial 16S rRNA gene segments were amplified in 4 replicates using broad-range rRNA PCR primers 27F and 534R, and the PCR products were pooled. The reverse primer (534R) included a barcode sequence unique to each sample, allowing PCR amplicons to be multiplexed and sequenced simultaneously. The amplicons were gel-purified, pooled, and subjected to pyrosequencing using Titanium chemistry on the Roche 454 GS-FLX platform. Pyrosequencing reads were filtered, trimmed, and aligned with the Silva nonredundant 16S reference database, release 108 (http://www.arb-silva.de) (24). Using a robust sequence analysis pipeline similar to the approach employed by Huse et al. (25), reads were assigned taxonomic classifications using USEARCH (http://www.drive5.com) and were dereplicated to unique reference sequence-based operational taxonomic units (refOTUs) using UCLUST (http://www.drive5.com). Detailed methods are described in the supplemental material.
Statistical methods.
Weighted and unweighted UniFrac analyses (26) were used to measure β-diversity between microbial communities, and results were plotted using principal coordinate analysis. Comparisons between phylogenetic taxa were performed using unpaired t tests (GraphPad, La Jolla, CA) and Minitab (version 15), at α < 0.05. Heat maps were generated using R and Matrix2png (27). Linear discriminant analysis coupled with effect size measurements (LEfSe) (28) was used to identify bacterial taxa that were differentially abundant or depleted between sample groups.
Nucleotide sequence accession number.
All sequence data sets have been deposited in the NCBI Sequence Read Archive (SRA), under accession no. SRA058490.
RESULTS
Samples and acquisition of 16S sequence data.
We sampled gut microbial communities from 39 subjects with C. difficile infection (CDI group), 36 subjects with C. difficile-negative nosocomial diarrhea (CDN group), and 40 healthy control subjects (HC group) (Table 1). Of the 39 samples in the CDI group, 9 (23.1%) were from subjects with recurrent episodes. Five subjects (13.9%) in the CDN group had a history of CDI. Most subjects in the CDI and CDN groups (69.2% and 86.1%, respectively) had mild to moderate disease. A total of 87.2% of subjects in the CDI group had received antimicrobials within 3 months prior to sampling, compared with 58.3% and 22.5% of subjects in the CDN and HC groups, respectively. Similar proportions of subjects in the CDI and CDN groups (43.6% and 41.7%, respectively) received concomitant systemic antimicrobial therapy. Five subjects (12.8%) in the CDI group received antibiotic treatment for CDI (metronidazole or oral vancomycin treatment) prior to fecal sampling. After trimming and quality control, a total of 526,071 partial high-quality 16S rRNA sequence reads of the V1 to V3 regions were available for analysis (∼4,575 ± 1,616 reads per sample, with an average amplicon length of ∼492 nucleotides) (see Table S1 in the supplemental material).
Table 1.
Demographic and baseline clinical characteristics of samples used in this study
| Characteristics | HC (n = 40)a | CDI (n = 39) | CDN (n = 36) |
|---|---|---|---|
| Age (yr [mean ± SD])b | 60.9 ± 9.1 | 54.7 ± 20.1 | 54.6 ± 20.0 |
| Female (n [%]) | 28 (70) | 16 (41) | 19 (53) |
| Caucasian (n [%])b | 27 (68) | 28 (72) | 24 (67) |
| Previous episode of CDI (n [%]) | 0 | 9 (23.1) | 5 (13.9) |
| Disease severity (n [%])c | |||
| Mild or moderate | NA | 27 (69.2) | 31 (86.1) |
| Severe | NA | 8 (20.5) | 5 (13.9) |
| Severe-complicated | NA | 4 (10.3) | 0 |
| White blood cell count (mean ± SD]) (cells/μl)d | NA | 11.9 ± 7.6 | 8.7 ± 5.4 |
| Received antibiotics within 3 mo (n [%]) | 9 (22.5) | 34 (87.2) | 21 (58.3) |
| Concomitant systemic antibiotic therapy (n [%]) | 0 | 17 (43.6) | 15 (41.7) |
| Treatment for CDI in previous 24 h (n [%]) | NA | 5 (12.8) | NA |
HC, healthy control; CDI, C. difficile infection; CDN, C. difficile negative nosocomial diarrhea; NA, not applicable or data not available.
There were no significant between-group differences.
Disease severity was defined according to reference 43; mild or moderate, white blood cell count of <15,000 cells/μl and serum creatinine level <1.5 times the premorbid level; severe, white blood cell count of ≥15,000 cells/μl or serum creatinine level ≥1.5 times the premorbid level; severe-complicated, hypotension or shock, ileus, or megacolon.
P = 0.47, CDI group versus CDN group.
Microbial diversity and species richness of the gut microbiota in CDI and CDN.
We identified a total of 3,531 refOTUs from 115 fecal samples, which belonged to 150 different genera in 15 different phyla. A total of 2,986 (84.6%) of the 3,531 refOTUs identified were found in healthy controls. In contrast, only 1,392 (39.4%) and 1,582 (44.8%) refOTUs were detected in the CDI and CDN groups, respectively. Among the most abundant refOTUs (defined as >0.1% of all pyrosequencing reads), 124 were shared in more than one-half of the healthy control samples but only 9 were detected in at least one-half of the samples in the CDI group. Species richness was significantly lower in the CDI and CDN groups than in the healthy control group, and both the microbial diversity (as determined by the Shannon diversity index) and the species evenness were markedly reduced (Fig. 1). Interestingly, microbial richness and diversity did not differ significantly between the CDI and CDN groups (P > 0.05).
Fig 1.
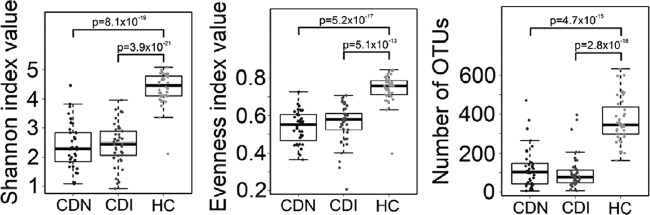
Decreased microbial diversity, evenness, and species richness in the gut microbiota associated with C. difficile infection and C. difficile-negative nosocomial diarrhea, shown distributed in box plot form. (Left) The Shannon diversity index was used to estimate microbial diversity for each group. (Middle) The species evenness index was calculated using the formula J′ = H′/H′max, where H′ is the Shannon diversity index and H′max is the maximal value of H′ (i.e., ln S, where S is the total number of species in the community). (Right) Species richness was defined as the number of refOTUs identified in each sample. All three indices were significantly lower in the CDI and CDN groups than in the healthy control group (Student's t test). No significant difference was observed between the CDI group and the CDN group. Each dot represents an individual fecal sample. P values are shown above the bars for each group comparison.
For the CDI group, when samples were stratified by disease severity, no differences in microbial diversity, species richness, or composition were observed (see Fig. S1 and S2 in the supplemental material). A comparison of initial versus recurrent disease revealed a trend toward lower species richness in recurrent disease; however, no significant difference in gut microbial diversity was observed (see Fig. S3 in the supplemental material). Furthermore, concomitant antibiotic exposure did not significantly alter microbial diversity, species richness, or the aggregate microbial composition (see Fig. S4 in the supplemental material). Thus, these data demonstrated that many normally abundant phylotypes were depleted in nosocomial diarrhea and CDI and suggest that factors other than the gut microbiota may modulate disease severity during CDI.
For the CDN group, when samples were stratified by antibiotic-associated versus non-antibiotic-associated diarrhea, no significant differences in microbial diversity or composition were observed (see Fig. S5 in the supplemental material). Furthermore, gut microbial communities were not significantly different in subjects who received concomitant antibiotic therapy versus those who did not (see Fig. S4 in the supplemental material).
Intestinal dysbiosis in CDI and CDN.
To characterize the global changes in microbial community structures, we applied the UniFrac method (26) to analyze the phylogenetic relatedness of pyrosequencing reads from all samples. In both weighted and unweighted UniFrac analyses, gut microbial communities associated with CDI and CDN clustered separately from those in healthy controls (Fig. 2A). Calculation of UniFrac distances within and between groups revealed that microbial communities within the CDI group were more similar to one another than to microbial communities in healthy subjects (Fig. 2B). The average UniFrac distance between pairs of samples in the CDI group was significantly greater than that between pairs of samples in the healthy control group (Fig. 2B), indicating greater heterogeneity in gut microbial communities associated with CDI. A similar trend was observed for the CDN group, compared with healthy controls. No appreciable differences in community structures or memberships were observed between the CDI and CDN groups. Thus, these data suggest that disease state (i.e., diarrheal state versus health) or antibiotic treatment (either prior or concomitant) accounted for the greatest amounts of variation among all samples, although interindividual differences in the gut microbiota also contributed to variability in the data.
Fig 2.
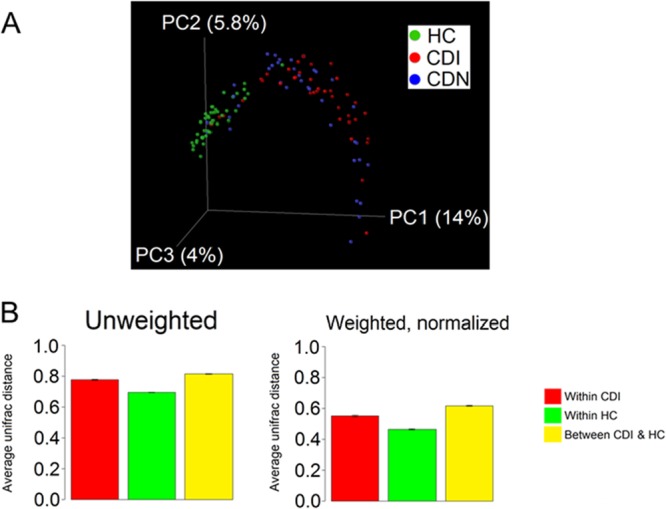
Comparison of microbial community compositions, revealing an altered gut microbiota in CDI. (A) Unweighted UniFrac analysis was used to generate distances between C. difficile-positive fecal samples (CDI), C. difficile-negative diarrheal samples (CDN), and healthy control samples (HC). Scatterplots were then generated using principal coordinate analysis. The percentage of variation explained by each principal coordinate (PC) is indicated on the axes. Each point represents a microbial community. The difference between communities in the CDI and HC groups was significant (P < 0.001, t test with permutation). (B) Average UniFrac distance between pairs of samples within each group, indicating greater heterogeneity in gut microbial communities in the CDI and CDN groups than in the healthy control group. Error bars, standard error of the mean.
Paucity of Firmicutes sequences in the aggregate gut microbiota associated with CDI and CDN.
The Firmicutes phylum includes the most numerous and diverse bacterial species in the human gut. In healthy controls, Firmicutes sequences were dominant at 74.9% of all reads (averaging ∼322 phylotypes per subject) (Fig. 3). Only 21.5% of all reads were assigned to the Bacteroidetes phylum, with ∼37 phylotypes per subject. The abundance of Firmicutes sequences was substantially lower in the CDI and CDN groups (43.2% and 32.9%, respectively). This depletion of Firmicutes sequences was accompanied by marked decreases in bacterial phylotypes (CDI, 75 phylotypes; CDN, 86 phylotypes; HC, 322 phylotypes) (Fig. 3).
Fig 3.
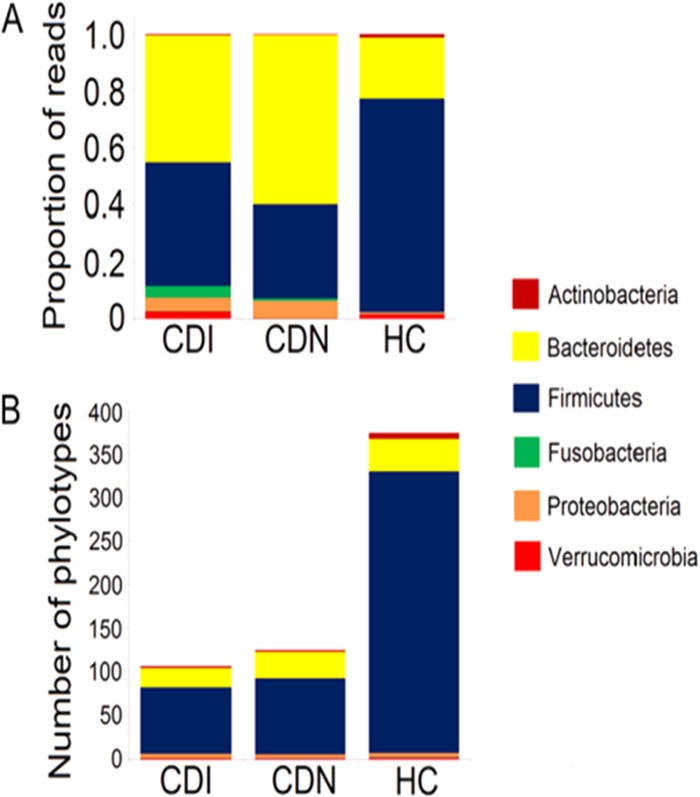
Paucity of Firmicutes sequences and phylotypes in CDI and CDN. (A) The mean proportions of Firmicutes sequences were smaller in the CDI and CDN groups (P = 5.70 × 10−7 and P = 1.08 × 10−8, respectively; Student's t test) than in the HC group. (B) The mean numbers of Firmicutes phylotypes (refOTUs) were lower in the CDI and CDN groups (P = 8.81 × 10−19 and P = 7.16 × 10−16, respectively; Student's t test) than in the HC group.
At the family level, Lachnospiraceae (45.8%), Ruminococcaceae (17.4%), and Bacteroidaceae (16.1%) sequences dominated the healthy fecal microbiota, and sequences from other families constituted <3% of all pyrosequencing reads (Fig. 4A). Lachnospiraceae and Ruminococcaceae sequences were significantly underrepresented in the CDI and CDN groups (Lachnospiraceae: CDI, 11.2%; CDN, 12.3%; HC, 45.8%; Ruminococcaceae: CDI, 3.0%; CDN, 2.8%; HC, 17.4%) (see Fig. S6 in the supplemental material). Several genera were enriched in association with CDI. For example, Veillonella (CDI, 4.5%; HC, 0.6%), Enterococcus (CDI, 7.1%; HC, 0.05%), and Lactobacillus (CD, 3.7%; HC, 0.4%) sequences were unusually abundant. Enterococcus and Lactobacillus are both lactic acid bacteria from the order Lactobacillales. Enterococcus sequences were found in 84.6% of the CDI samples, compared with only 22.5% of the samples from healthy controls. Sequences from the Gammaproteobacteria class, which includes many clinically important Gram-negative pathogens, also were enriched. Interestingly, Desulfovibrionaceae sequences from the Deltaproteobacteria class, which includes a large number of sulfate-reducing, anaerobic, Gram-negative bacteria, were depleted in CDI. Interindividual variations in the proportions of major phyla among samples were observed in all three groups, as in previous studies (29) (see Fig. S7 in the supplemental material). These results indicate that decreases in gut microbial diversity and richness in CDI and CDN are driven primarily by the loss of phylotypes within the Lachnospiraceae and Ruminococcaceae families.
Fig 4.
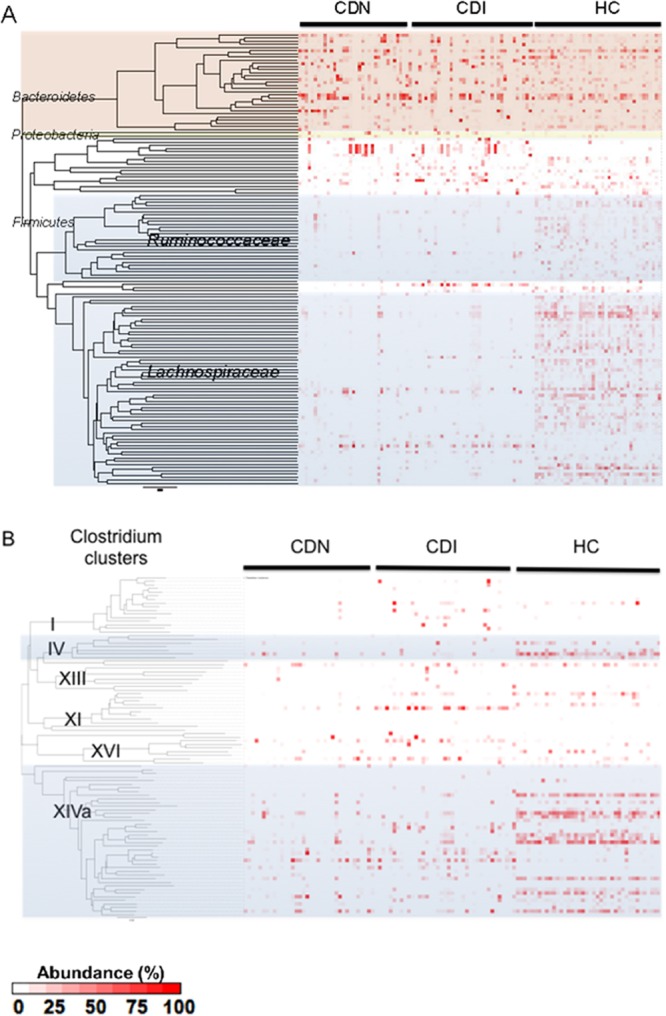
(A) Proportions of bacterial taxa in each sample, as inferred from 16S rRNA gene sequence data. Each column corresponds to an individual fecal sample. Each row corresponds to a specific bacterial phylotype or refOTU, arrayed based on phylogenetic relationships. Only the most prominent refOTUs (>0.1% abundance) are included in this heatmap. The relative abundance of each phylotype is represented by the color code. Members of the Lachnospiraceae and Ruminococcaceae families are shaded in gray. The Bacteroidetes phylum is shown in light red, and the Proteobacteria phylum in yellow. (B) Proportions of bacterial taxa within Clostridium clusters. Each column represents an individual fecal sample. Each row corresponds to a refOTU assigned to one of the 19 Clostridium clusters. Members of Clostridium clusters XIVa and IV are shaded in gray.
Depletion of butyrate-producing bacteria in CDI and CDN.
The vast majority of Firmicutes sequences (68.4%) were assigned to the Clostridia class. The Clostridia class has been divided into 19 clusters on the basis of growth, metabolic, and morphological parameters (30) and includes members of the Lachnospiraceae and Ruminococcaceae families that were significantly depleted in CDI. To identify specific Clostridium clusters that were most depleted in CDI and CDN, we used an association table of full-length sequences from species previously assigned to Clostridium clusters (29, 31) and assigned 17.8% of all reads to 19 Clostridium clusters (Fig. 4B). Smaller proportions of reads were assigned for the CDI (16.4%) and CDN (10.0%) groups than for the healthy control group (25.0%). Strikingly, members of Clostridium cluster XIVa (the Eubacterium rectale-Clostridium coccoides group) and to a lesser extent cluster IV (the Clostridium leptum group) were significantly depleted in the CDI and CDN groups, compared with the HC group (cluster XIVa: CDI, 5.5%; CDN, 6.6%; HC, 18.4%; cluster IV: CDI, 0.47%; CDN, 0.40%; HC, 2.95%). As Clostridium clusters IV and XIVa contain a large number of commensal organisms, including butyrate-producing anaerobic bacteria important for the metabolism and development of colonic epithelial cells, these data suggest a potential role for butyrate-producing organisms in nosocomial diarrhea and CDI.
Next, we used LEfSe (28) to determine specific bacteria taxa that were differentially abundant. We identified Blautia, Pseudobutyrivibrio, Roseburia, Faecalibacterium, Anaerostipes, Subdoligranulum, Ruminococcus, Streptococcus, Dorea, and Coprococcus as the most differentially depleted genera associated with CDI (Fig. 5A). The same 10 genera also were identified as the most differentially depleted genera in CDN. Many of these genera harbor butyrate producers belonging to Clostridium cluster IV or XIVa. The overall abundance of these 10 genera combined was significantly lower in the CDI and CDN groups than in the control group (CDI, 5.4%; CDN, 4.9%; HC, 37.2%), although there were interindividual variations in the relative abundance of each genus (Fig. 5B). Of the 3,531 total refOTUs identified in our data set, 119 refOTUs were significantly depleted in CDI; 74 refOTUs were members of the 10 most depleted genera, and 39 refOTUs were uncultured bacteria of the Lachnospiraceae or Ruminococcaceae family. When the CDI data set was compared with CDN data, Clostridium difficile was the most differentially enriched species associated with CDI.
Fig 5.
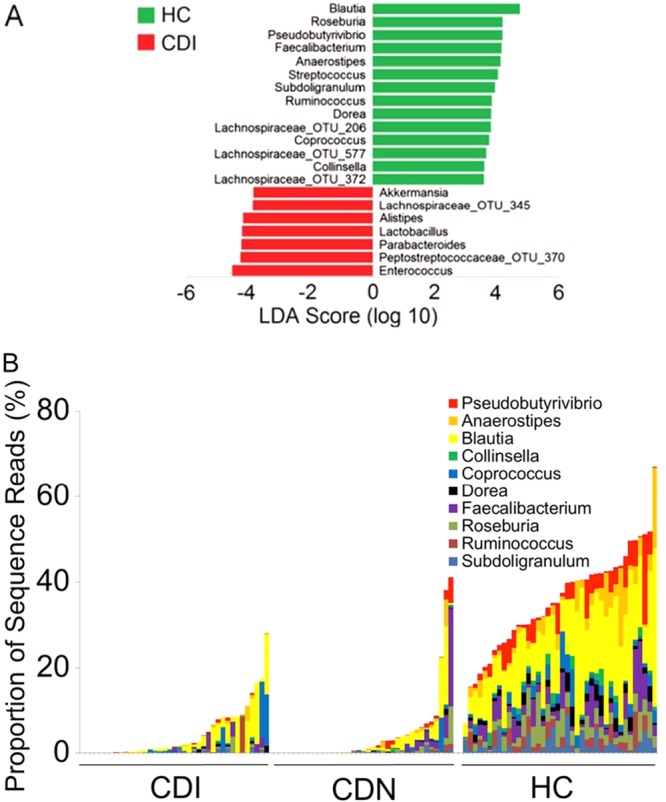
Bacteria genera most depleted or enriched in CDI and CDN. (A) Genera most differentially depleted (most of which are butyric acid-producing anaerobic bacteria) or enriched in the C. difficile-associated microbiota versus healthy control microbiota, as identified by linear discriminant analysis (LDA) coupled with effect size measurements. Bacterial taxa depleted in CDI are indicated with positive LDA scores (green), and taxa enriched in CDI are indicated with negative scores (red). Only taxa that met the significant LDA threshold of 3.6 are shown. The same genera were also depleted in CDN (data not shown). (B) Interindividual variations in the relative abundance of selected genera. The overall abundance (sequence reads, y axis) of the 10 most differentially depleted genera (indicated in the key) was significantly lower in the CDI and CDN groups than in the HC group (P = 4.0 × 10−21 and P = 9.3 × 10−20, respectively; Student's t test). Each bar corresponds to an individual sample.
Since butyrate producers supply energy to gut epithelial cells and are thought to protect against colitis, we asked whether the proportions of 16S reads assigned to genera known to produce butyrate as their main fermentation product differed among groups. The relative abundance of butyrate-producing bacteria was significantly lower in the CDI and CDN groups (CDI, 6.3%; CDN, 3.1%; HC, 17.2%) (Fig. 6A). The major butyrate producers depleted included Roseburia (CDI, 0.17%; CDN, 0.45%; HC, 3.4%), Faecalibacterium (CDI, 0.37%; CDN, 0.83%; HC, 3.2%), Subdoligranulum (CDI, 0.18%; CDN, 0.094%; HC, 2.0%), Anaerostipes (CDI, 0.2%; CDN, 0.016%; HC, 3.1%), and Pseudobutyrivibrio (CDI, 0.065%; CDN, 0.44%; HC, 3.3%) (Fig. 6B). Fusobacterium spp. were the dominant butyrate producers in the aggregate gut microbiota of subjects with CDI (CDI, 4.1%; HC, 0.03%; P = 0.099), but this was driven by the dominance of Fusobacterium in the gut microbiota of four subjects (see Fig. S8 in the supplemental material).
Fig 6.

Acetate, butyrate, and lactate fermenters in the gut microbiota. (A) Relative proportions of acetate, butyrate, and lactate fermenters in the gut microbiota in the CDI (red), CDN (yellow), and HC (green) cohorts pyrosequenced. Sequence reads were classified at the genus level according to the primary metabolic end product of carbohydrate fermentation, and results were compared using Student's t test. Genera that were ambiguously defined as producing both acetate and butyrate or other short-chain fatty acids, such as succinate, propionate, formate, or ethanol, comprising minor constituents of the gut microbiota (<1% of sequence reads), were excluded in this analysis. Error bars, standard error of the mean. ∗, P < 0.05; ∗∗, P < 0.01; ∗∗∗, P < 0.001. CDI, C. difficile infection; CDN, C. difficile-negative nosocomial diarrhea; HC, healthy control; NS, not significant. (B) Relative proportions of major butyrate-producing bacteria. (C) Relative proportions of major acetogens by genera. (D) Relative proportions of primary lactic acid-producing organisms.
Anaerobic gut bacteria produce a variety of fermentation products, including acetate, lactate, and other short-chain fatty acids (SCFAs), in addition to butyrate. A comparison of major acetogens (Fig. 6C) revealed that the genera Blautia (CDI, 2.1%; CDN, 1.6%; HC, 14.4%) and Dorea (CDI, 0.12%; CDN, 0.10%; HC, 1.5%) were significantly depleted in the CDI and CDN groups, compared with the control group. These acetogens were replaced by Bacteroides spp. (CDI, 29.5%; CDN, 45.7%; HC, 16.1%), which produce the SCFAs succinate, lactate, and formate (C2 to C4) besides acetate. Lactobacillus spp. and Enterococcus spp., both primary lactic acid producers, were more prevalent and more abundant in the CDI and CDN groups than in the HC group (Fig. 6D).
DISCUSSION
A complex community of microorganisms inhabiting the gut plays important roles in human health and disease, including digestion, metabolism, immune system development, and resistance to infection. The mammalian gut is a primary site of exposure to invading pathogens. The importance of the indigenous microbiota in resistance to pathogens is well exemplified by Clostridium difficile infection, which is one of the most common causes of gastroenteritis-associated deaths in the United States. Although the gut microbiota is believed to protect the gut against CDI, specific components that confer colonization resistance are not known. Consistent with previous studies (17, 19, 20, 32), our 16S genomic analysis demonstrated a profound alteration of the gut microbiota, or dysbiosis, which was characterized by markedly decreased biodiversity and species richness. Importantly, several types of normally abundant butyrate-producing anaerobic bacteria in the Ruminococcaceae and Lachnospiraceae families were significantly depleted in CDI. Interestingly, the microbial community structure in the aggregate gut microbiota in CDI was similar to that in C. difficile-negative nosocomial diarrhea (CDN).
Several studies have investigated the changes in the gut microbiota in C. difficile-associated disease. Using culture-based methods, Hopkins and colleagues reported a decrease in species diversity (33) and reduced numbers of Bacteroides, Prevotella, and Bifidobacteria in 4 subjects with CDI (18). The fecal samples in that study were obtained following metronidazole therapy, which might have confounded the microbial profile. Using 16S rRNA clonal sequencing, markedly decreased species richness and diversity were observed in fecal samples from 3 subjects with recurrent CDI, compared with 4 subjects with initial CDI and 3 uninfected control subjects (17), supporting a role for the gut microbiota in C. difficile-associated disease. Manges et al. compared the intestinal microbiota of 25 subjects who developed CDI during hospitalization with that of 50 matched control subjects using a 16S rRNA microarray (34). After adjustment for antibiotic use, only small numbers of organisms within the Bacteroidetes and Firmicutes phyla were associated with the subsequent development of CDI during hospitalization. Interestingly, no reduction in the diversity of the gut microbiota was observed in CDI, compared with controls. Their findings may be explained by the fact that the fecal samples were collected early in the hospital course (within 72 h after admission) and changes in the composition of the gut microbiota due to antibiotic exposure may have occurred following sample collection and contributed to subsequent C. difficile colonization and disease. Indeed, antibiotic exposure during hospitalization was strongly associated with the risk of subsequent CDI in this study. Ecological succession of gut microbial communities following antibiotic treatment and exposure to C. difficile also has been reported in animal models (35).
As proof of the principle of using selected gut microbes in microbiota transplantation, Lawley et al. demonstrated that a defined mixture of six phylogenetically diverse bacteria was capable of inducing major shifts in the gut microbial composition and resolving C. difficile disease in a mouse model of CDI (20). In another study, Reeves et al. (21) showed that a single component of the murine gut microbiota, a member of the Lachnospiraceae family, could restore colonization resistance against CDI in germfree mice. Members of the human gut microbiota and the murine gut microbiota, however, are phylogenetically distinct. Human-associated gut bacteria that can confer resistance against C. difficile colonization have not yet been identified. This study presents an in-depth analysis of the human gut microbiome associated with C. difficile infection, with a goal of identifying candidate human-associated bacteria that can be investigated in future studies or potentially exploited for novel probiotic therapy.
In this study, the microbial communities in CDI and CDN clustered separately from those of healthy individuals. This distinct clustering between healthy individuals and individuals with nosocomial diarrhea suggests that depletion of gut commensal organisms that normally live in symbiosis with the host could lead to colonic dysfunction and diarrhea. Members of the Lachnospiraceae and Ruminococcaceae families, which include butyrogenic bacteria, were significantly depleted in the CDI and CDN groups. Interestingly, few differences in microbial diversity, species richness, and community membership (with the exception of C. difficile reads) between the CDI and CDN groups were observed. The gut microbiota was disordered in nosocomial diarrhea, with low microbial diversity and a paucity of Firmicutes organisms, regardless of C. difficile status. The nonspecificity of dysbiosis in CDI and CDN may be due to the high rates of prior antibiotic exposure (CDI, 87.2%; CDN, 58.3%; HC, 22.5%) and suggests that the gut microbiota of the CDN group may be susceptible to C. difficile infection. This lack of difference between CDI and CDN also suggests that C. difficile acquisition or infection alone had little effect on the global composition or structure of the gut microbiota. Consistent with this hypothesis, a recent study showed no significant differences in the composition of the gut microbiota in elderly subjects with asymptomatic C. difficile carriage (i.e., culture-positive subjects), compared with culture-negative subjects (19).
In a cross-sectional study of 10 subjects using 16S clonal sequencing analysis, Chang et al. (17) reported lower overall microbial diversity in recurrent C. difficile disease, compared to initial infections. Although our study showed a trend toward lower species richness, no significant difference in microbial diversity in recurrent disease was found (see Fig. S3 in the supplemental material). We also observed no correlation between disease severity and ecological metrics of the gut microbiota (see Fig. S1 in the supplemental material). While it is generally believed that C. difficile infection requires prior disruption of the normal gut flora, after infection is initiated disease severity may be modulated by host and bacterial factors such as inflammatory responses, antibody levels, and/or expression of virulent toxins. In patients with more-severe disease, normal commensal bacteria may be lost as a result of intense host inflammatory responses; therefore, the overall gut microbiota does not predict or correlate with disease severity.
The present study identified several specific members of the Ruminococcaceae and Lachnospiraceae families that were markedly depleted in CDI and nosocomial diarrhea. A great majority of these species are bacterial phylotypes with the greatest similarity to butyrate-producing anaerobic bacteria. Butyric acid has well documented anti-inflammatory effects and is the preferred energy source for ATP synthesis in colonocytes. Many short-chain fatty acids (SCFAs), such as butyric acid, are known to decrease the permeability of the intestinal epithelial lining by increasing the expression of tight junction proteins. In addition, it has been proposed that butyric acid can reinforce colonic defense barriers by increasing antimicrobial peptide levels and mucin production (36, 37). Thus, for nosocomial diarrhea, depletion of butyrate-producing organisms may result in epithelial dysfunction and increased osmotic load within the intestinal lumen, leading to diarrhea. For C. difficile infection, disruption of the gut microbiota and depletion of beneficial butyrogenic bacteria in the gut may lead to increased susceptibility to infection by compromising the host defenses against C. difficile. If so, then replenishing butyrogenic bacteria to allow in situ production of butyric acid or other inhibitory compounds in the gut lumen may offer a novel therapeutic strategy for the treatment of C. difficile infection or nosocomial diarrhea.
Early studies investigating the role of SCFAs in colonization resistance have yielded mixed results. According to Rolfe (38), butyric acid was the only SCFA in the cecum of conventional hamsters that reached a concentration high enough to inhibit C. difficile growth in vitro. Using this model, the study showed a direct correlation between the inhibitory concentrations of SCFAs and the susceptibility of hamsters to C. difficile colonization, thus suggesting a role for SCFAs in resistance against C. difficile colonization. However, Su et al. subsequently introduced SCFAs directly into the colons of gnotobiotic mice (by gastric gavage, direct injection into the ceca, and use of Clostridium butyricum to allow butyric acid production in situ) and found that SCFAs alone could not inhibit the growth of C. difficile in vivo (39). Using an in vitro model of resistance to C. difficile colonization, Borriello and Barclay also failed to find a correlation between the SCFA profiles of fecal emulsions and their inhibitory activities against C. difficile (40). In a more-recent study, intestinal dysbiosis induced by the epidemic C. difficile strain 027/BI in mice was associated with an altered SCFA profile characterized by marked reductions in butyrate and acetate levels (20). Thus, it remains unclear whether butyric acid or other antimicrobial compounds that remain to be identified are the key molecules in resistance to colonization by C. difficile. However, viable bacteria are likely the most important components of colonization resistance in the gastrointestinal tract (40). While the specific bacteria responsible for colonization resistance in humans remain unknown, our study suggests that depletion of butyrate-producing bacteria in clusters IV and XIVa may render the host susceptible to C. difficile infection, and replenishment of these bacteria in the gut may be of therapeutic benefit in nosocomial diarrhea, including CDI. Consistent with this hypothesis, after antibiotic pretreatment of mice followed by C. difficile challenge, the gut microbiota of clinically well animals was dominated by Firmicutes, whereas Proteobacteria predominated in clinically ill animals (41).
This study has several limitations. First, the samples for the CDI and CDN groups were one-time fecal specimens collected at the time of diarrhea for clinical microbiological testing. Although microbiome examinations of the gut microbiota just prior to C. difficile acquisition or disease onset would be ideal, such fecal specimens are difficult to obtain from human subjects. The depletion of butyrogenic bacteria at the time of disease onset could be the result, rather than the cause, of gut inflammation. Thus, the causal relationship should be assessed mechanistically using animal models of C. difficile infection and challenge. Nonetheless, the present study identifies a list of candidate organisms at the family and genus levels that could serve as the basis for selecting specific microbes for future functional and colonization resistance studies using human microbiota-associated animal models.
Second, some subjects in the CDN group had diarrheal symptoms that could have been attributed to C. difficile-associated disease. Subjects with diarrhea were included in the CDI or CDN group according to the results of clinical C. difficile tests (either the C. Diff Quik Chek Complete test or a GeneXpert multiplex PCR assay, as the clinical platform was changed during the course of the study). To explore the possibility of false-negative clinical test results and/or C. difficile colonization, we performed additional in-house toxin B gene PCR assays for all samples. For seven subjects in the CDN group and one healthy control subject, very weak toxin B amplification products were observed. In contrast, all samples for the CDI group showed strong amplification products. Although we could not exclude the possibility of misclassification in the CDN group based on these data, the aggregate gut microbiota was not significantly altered when the seven CDN samples with weak C. difficile toxin B signals were excluded from our analysis (see Fig. S9 in the supplemental material).
Third, our findings of depletion of butyrate producers relied on predictions about short-chain fatty acid production for the inferred phylotypes (refOTUs). Our genomic analysis was based on ∼500-bp 16S partial rRNA gene sequences, which may have misclassified certain bacteria at the species or strain level. In addition, there are variations in SCFA production among different bacterial species within a given genus, and data on SCFA production are lacking for many of the gut-associated organisms identified in our study. Given these uncertainties, we have focused our classification at the genus level, which has been described in more detail in the literature. Levels of fecal SCFAs and butyrate could be directly measured in future studies, to confirm our findings.
In summary, the present study identified Lachnospiraceae, Ruminococcaceae, and butyrate-producing anaerobic bacteria as significantly depleted in C. difficile infection and nosocomial diarrhea. Recent advances in high-throughput anaerobic culture techniques (42) should facilitate culturing of representative microbes identified here in the human fecal microbiota for further mechanistic and functional studies in animal models, thereby creating a discovery pipeline for much-needed microbiota-targeted probiotic therapy and the development of novel prognostic and diagnostic strategies for the treatment of nosocomial diarrhea and CDI.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the University of Florida Department of Medicine and in part by National Institutes of Health and National Center for Research Resources CTSI grant 1UL1RR029890. G.P.W. was supported in part by NIAID grant KO8-AI077713.
We thank members of the Wang laboratory for help and suggestions and Cindy Wang and Enrique Sanchez for technical assistance with the project. We are grateful to Paul O'Toole at University College, Cork, Ireland, for providing a list of species belonging to Clostridium clusters.
No potential conflicts of interest are declared.
Footnotes
Published ahead of print 26 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.00845-13.
REFERENCES
- 1.Ananthakrishnan AN. 2011. Clostridium difficile infection: epidemiology, risk factors and management. Nat. Rev. Gastroenterol. Hepatol. 8:17–26 [DOI] [PubMed] [Google Scholar]
- 2.O'Keefe SJ. 2010. Tube feeding, the microbiota, and Clostridium difficile infection. World J. Gastroenterol. 16:139–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parkes GC, Sanderson JD, Whelan K. 2009. The mechanisms and efficacy of probiotics in the prevention of Clostridium difficile-associated diarrhoea. Lancet Infect. Dis. 9:237–244 [DOI] [PubMed] [Google Scholar]
- 4.Pepin J, Alary ME, Valiquette L, Raiche E, Ruel J, Fulop K, Godin D, Bourassa C. 2005. Increasing risk of relapse after treatment of Clostridium difficile colitis in Quebec, Canada. Clin. Infect. Dis. 40:1591–1597 [DOI] [PubMed] [Google Scholar]
- 5.Petrella LA, Sambol SP, Cheknis A, Nagaro K, Kean Y, Sears PS, Babakhani F, Johnson S, Gerding DN. 2012. Decreased cure and increased recurrence rates for Clostridium difficile infection caused by the epidemic C. difficile BI strain. Clin. Infect. Dis. 55:351–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, Speelman P, Dijkgraaf MG, Keller JJ. 2013. Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med. 368:407–415 [DOI] [PubMed] [Google Scholar]
- 7.Bakken JS. 2009. Fecal bacteriotherapy for recurrent Clostridium difficile infection. Anaerobe 15:285–289 [DOI] [PubMed] [Google Scholar]
- 8.Borody TJ. 2000. “Flora power”: fecal bacteria cure chronic C. difficile diarrhea. Am. J. Gastroenterol. 95:3028–3029 [DOI] [PubMed] [Google Scholar]
- 9.Brandt LJ, Aroniadis OC, Mellow M, Kanatzar A, Kelly C, Park T, Stollman N, Rohlke F, Surawicz C. 2012. Long-term follow-up of colonoscopic fecal microbiota transplant for recurrent Clostridium difficile infection. Am. J. Gastroenterol. 107:1079–1087 [DOI] [PubMed] [Google Scholar]
- 10.Famularo G, Trinchieri V, De Simone C. 2001. Fecal bacteriotherapy or probiotics for the treatment of intestinal diseases? Am. J. Gastroenterol. 96:2262–2264 [DOI] [PubMed] [Google Scholar]
- 11.Russell G, Kaplan J, Ferraro M, Michelow IC. 2010. Fecal bacteriotherapy for relapsing Clostridium difficile infection in a child: a proposed treatment protocol. Pediatrics 126:e239–e242 [DOI] [PubMed] [Google Scholar]
- 12.Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ. 2010. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J. Clin. Gastroenterol. 44:354–360 [DOI] [PubMed] [Google Scholar]
- 13.Berg RD. 1996. The indigenous gastrointestinal microflora. Trends Microbiol. 4:430–435 [DOI] [PubMed] [Google Scholar]
- 14.Young VB, Schmidt TM. 2008. Overview of the gastrointestinal microbiota. Adv. Exp. Med. Biol. 635:29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naaber P, Smidt I, Stsepetova J, Brilene T, Annuk H, Mikelsaar M. 2004. Inhibition of Clostridium difficile strains by intestinal Lactobacillus species. J. Med. Microbiol. 53:551–554 [DOI] [PubMed] [Google Scholar]
- 16.Rolfe RD, Helebian S, Finegold SM. 1981. Bacterial interference between Clostridium difficile and normal fecal flora. J. Infect. Dis. 143:470–475 [DOI] [PubMed] [Google Scholar]
- 17.Chang JY, Antonopoulos DA, Kalra A, Tonelli A, Khalife WT, Schmidt TM, Young VB. 2008. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J. Infect. Dis. 197:435–438 [DOI] [PubMed] [Google Scholar]
- 18.Hopkins MJ, Macfarlane GT. 2002. Changes in predominant bacterial populations in human faeces with age and with Clostridium difficile infection. J. Med. Microbiol. 51:448–454 [DOI] [PubMed] [Google Scholar]
- 19.Rea MC, O'Sullivan O, Shanahan F, O'Toole PW, Stanton C, Ross RP, Hill C. 2012. Clostridium difficile carriage in elderly subjects and associated changes in the intestinal microbiota. J. Clin. Microbiol. 50:867–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lawley TD, Clare S, Walker AW, Stares MD, Connor TR, Raisen C, Goulding D, Rad R, Schreiber F, Brandt C, Deakin LJ, Pickard DJ, Duncan SH, Flint HJ, Clark TG, Parkhill J, Dougan G. 2012. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 8:e1002995. 10.1371/journal.ppat.1002995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reeves AE, Koenigsknecht MJ, Bergin IL, Young VB. 2012. Suppression of Clostridium difficile in the gastrointestinal tracts of germfree mice inoculated with a murine isolate from the family Lachnospiraceae. Infect. Immun. 80:3786–3794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. 2007. The Human Microbiome Project. Nature 449:804–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu GD, Lewis JD, Hoffmann C, Chen YY, Knight R, Bittinger K, Hwang J, Chen J, Berkowsky R, Nessel L, Li H, Bushman FD. 2010. Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiol. 10:206. 10.1186/1471-2180-10-206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35:7188–7196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huse SM, Dethlefsen L, Huber JA, Mark Welch D, Relman DA, Sogin ML. 2008. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 4:e1000255. 10.1371/journal.pgen.1000255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. 2011. UniFrac: an effective distance metric for microbial community comparison. ISME J. 5:169–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pavlidis P, Noble WS. 2003. Matrix2png: a utility for visualizing matrix data. Bioinformatics 19:295–296 [DOI] [PubMed] [Google Scholar]
- 28.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. 2011. Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Claesson MJ, Cusack S, O'Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, Marchesi JR, Falush D, Dinan T, Fitzgerald G, Stanton C, van Sinderen D, O'Connor M, Harnedy N, O'Connor K, Henry C, O'Mahony D, Fitzgerald AP, Shanahan F, Twomey C, Hill C, Ross RP, O'Toole PW. 2011. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc. Natl. Acad. Sci. U. S. A. 108(Suppl 1):4586–4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson JL, Francis BS. 1975. Taxonomy of the Clostridia: ribosomal ribonucleic acid homologies among the species. J. Gen. Microbiol. 88:229–244 [DOI] [PubMed] [Google Scholar]
- 31.Collins MD, Lawson PA, Willems A, Cordoba JJ, Fernandez-Garayzabal J, Garcia P, Cai J, Hippe H, Farrow JA. 1994. The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. Int. J. Syst. Bacteriol. 44:812–826 [DOI] [PubMed] [Google Scholar]
- 32.Shahinas D, Silverman M, Sittler T, Chiu C, Kim P, Allen-Vercoe E, Weese S, Wong A, Low DE, Pillai DR. 2012. Toward an understanding of changes in diversity associated with fecal microbiome transplantation based on 16S rRNA gene deep sequencing. mBio 3:e00338–12. 10.1128/mBio.00338-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hopkins MJ, Sharp R, Macfarlane GT. 2001. Age and disease related changes in intestinal bacterial populations assessed by cell culture, 16S rRNA abundance, and community cellular fatty acid profiles. Gut 48:198–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manges AR, Labbe A, Loo VG, Atherton JK, Behr MA, Masson L, Tellis PA, Brousseau R. 2010. Comparative metagenomic study of alterations to the intestinal microbiota and risk of nosocomial Clostridium difficile-associated disease. J. Infect. Dis. 202:1877–1884 [DOI] [PubMed] [Google Scholar]
- 35.Peterfreund GL, Vandivier LE, Sinha R, Marozsan AJ, Olson WC, Zhu J, Bushman FD. 2012. Succession in the gut microbiome following antibiotic and antibody therapies for Clostridium difficile. PLoS One 7:e46966. 10.1371/journal.pone.0046966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cook SI, Sellin JH. 1998. Review article: short chain fatty acids in health and disease. Aliment. Pharmacol. Ther. 12:499–507 [DOI] [PubMed] [Google Scholar]
- 37.Wong JM, de Souza R, Kendall CW, Emam A, Jenkins DJ. 2006. Colonic health: fermentation and short chain fatty acids. J. Clin. Gastroenterol. 40:235–243 [DOI] [PubMed] [Google Scholar]
- 38.Rolfe RD. 1984. Role of volatile fatty acids in colonization resistance to Clostridium difficile. Infect. Immun. 45:185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su WJ, Waechter MJ, Bourlioux P, Dolegeal M, Fourniat J, Mahuzier G. 1987. Role of volatile fatty acids in colonization resistance to Clostridium difficile in gnotobiotic mice. Infect. Immun. 55:1686–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borriello SP, Barclay FE. 1986. An in-vitro model of colonisation resistance to Clostridium difficile infection. J. Med. Microbiol. 21:299–309 [DOI] [PubMed] [Google Scholar]
- 41.Reeves AE, Theriot CM, Bergin IL, Huffnagle GB, Schloss PD, Young VB. 2011. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile infection. Gut Microbes 2:145–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, Gordon JI. 2011. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc. Natl. Acad. Sci. U. S. A. 108:6252–6257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cohen SH, Gerding DN, Johnson S, Kelly CP, Loo VG, McDonald LC, Pepin J, Wilcox MH. 2010. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect. Control Hosp. Epidemiol. 31:431–455 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.