

Abstract
Acute diarrheal disease (ADD) can be caused by a range of pathogens, including bacteria, viruses, and parasites. Conventional diagnostic methods, such as culture, microscopy, biochemical assays, and enzyme-linked immunosorbent assays (ELISA), are laborious and time-consuming and lack sensitivity. Combined, the array of tests performed on a single specimen can increase the turnaround time (TAT) significantly. We validated a 19plex laboratory-developed gastrointestinal pathogen panel (GPP) using Luminex xTAG analyte-specific reagents (ASRs) to simultaneously screen directly in fecal specimens for diarrhea-causing pathogens, including bacteria (Campylobacter jejuni, Salmonella spp., Shigella spp., enterotoxigenic Escherichia coli [ETEC], Shiga toxin-producing E. coli [STEC], E. coli O157:H7, Vibrio cholerae, Yersinia enterocolitica, and toxigenic Clostridium difficile), parasites (Giardia lamblia, Cryptosporidium spp., and Entamoeba histolytica), and viruses (norovirus GI and GII, adenovirus 40/41, and rotavirus A). Performance characteristics of GPP ASRs were determined using 48 reference isolates and 254 clinical specimens. Stool specimens from individuals with diarrhea were tested for pathogens using conventional and molecular methods. Using the predictive methods as standards, the sensitivities of the GPP ASRs were 100% for adenovirus 40/41, norovirus, rotavirus A, Vibrio cholerae, Yersinia enterocolitica, Entamoeba histolytica, Cryptosporidium spp., and E. coli O157:H7; 95% for Giardia lamblia; 94% for ETEC and STEC; 93% for Shigella spp.; 92% for Salmonella spp.; 91% for C. difficile A/B toxins; and 90% for Campylobacter jejuni. The overall comparative performance of the GPP ASRs with conventional methods in clinical samples was 94.5% (range, 90% to 97%), with 99% (99.0% to 99.9%) specificity. Implementation of the GPP ASRs enables our public health laboratory to offer highly sensitive and specific screening and identification of the major ADD-causing pathogens.
INTRODUCTION
In a typical year, there are two cases of acute gastroenteritis for every three people in the United States. Those cases either cause financial loss, such as lost wages and medical expenses, or lead to serious health complications or even death (1). Despite public health efforts in food safety education and monitoring, water treatment systems, and overall sanitation, pathogen-induced acute diarrheal disease (ADD) is the leading cause of morbidity and mortality worldwide (2). The industrialization of the food supply, which helps make food inexpensive and plentiful, has also enabled pathogens to spread through the population rapidly and more broadly. In addition, new pathogens have emerged and come to the consumer in an increasing variety of food vehicles (3). Creating effective national surveillance for an emerging pathogen depends on developing new clinical and laboratory practices as well as allowing greater flexibility for reference laboratories to add new diagnostic testing strategies more efficiently.
According to the Centers for Disease Control and Prevention (CDC), 43% of laboratory-confirmed bacterial enteric infections in the United States are caused by Salmonella spp., followed by Campylobacter spp. (33%), Shigella spp. (17%), Shiga toxin-producing Escherichia coli (STEC) (4.1%), and Yersinia spp. (0.9%) (2). Most bacterial agents can be identified using conventional culture techniques, but the length of time for clinical identification varies. Enteric viruses such as norovirus, rotavirus, and adenovirus are the most common causes of nonbacterial gastroenteritis worldwide. They are most often transmitted in humans via the fecal-oral route. The infective dose can be very low and easily spread in aerosols or by contact with contaminated surfaces, resulting in large outbreaks. Current detection methods include enzyme immunoassay (EIA), latex agglutination, immunochromatography technologies, or single-target molecular testing, such as conventional or real-time PCR. Average turnaround times (TAT) for currently available methods range from 2 to 4 days (4). Parasites such as Giardia, Cryptosporidium, and Entamoeba account for a significant number of gastroenteritis cases and may cause waterborne disease in the United States. Outbreaks of giardiasis and cryptosporidiosis, which occur via fecal-oral transmission, are associated with consumption of contaminated food and drinking water (5, 6). Microscopy and immunological assays are considered the gold standard detection methods, but the prevalence of parasites in stool is so low that multiple specimens are required for a definitive identification, resulting in an increased in detection time (7).
Public health response to emerging and recurring ADD outbreaks requires efficient epidemiological investigations and sensitive, rapid, and reliable laboratory diagnosis. The diverse sources of ADD in outbreaks of any size often complicate public health response. Once specimens arrive in the laboratory, testing algorithms for each possible pathogen can be a daunting task, as they require a wide array of laboratory techniques and vast staff knowledge and experience. Current budget cuts at local and state levels coupled with shrinking laboratory staff numbers compound the task of producing a fast and sensitive diagnosis. Jones et al. determined that 71% of ADD outbreaks had no confirmed etiology and that in 45%, the suspected food vehicle could not be identified due to inadequate resources for epidemiologic investigation, collection, and fast testing of clinical samples, thus limiting successful food-borne outbreak investigations (8).
We have validated a laboratory-developed test (LDT) based on gastrointestinal (GI) pathogen-specific xTAG analyte-specific reagents (ASRs) (Luminex Corporation, Austin, TX) for the detection of ADD-causing agents that effectively shorten the time for identification of multiple pathogens in a single reaction with a TAT of less than 6 h from specimen receipt. The objective of this study was to evaluate the performance of the gastrointestinal pathogen panel (GPP) ASRs for 19 pathogens and toxins and the potential role of multiplex molecular testing during routine clinical screening and public health outbreak response. To our knowledge, this is the first report on the use of Luminex xTAG ASRs for high-throughput detection of multiple GI pathogens of clinical and public health importance.
MATERIALS AND METHODS
Culture isolates.
The study compiled 48 culture isolates from retrospectively identified clinical specimens from the City of Milwaukee Health Department Laboratory (MHDL) and from other reference laboratories. The control isolates were obtained from the American Type Culture Collection (ATCC) (Manassas, VA), ZeptoMetrix (Buffalo, NY), and the CDC (see Table S1 in the supplemental material). Bacterial isolates were stored at −80°C in 10% glycerol. Isolates were subcultured on blood agar plates and incubated for 24 to 48 h prior to the second plating. A 0.5 McFarland standard suspension was created from plates. A 1-ml volume of a 1:10 dilution of McFarland standard suspension was created in 900 μl of easyMAG lysis buffer (bioMérieux, Durham, NC) spiked with 0.1 g of solid or 100 μl of liquid stool previously tested by the GPP ASRs and confirmed to be negative for all targets. Viral isolates were obtained as quantified suspensions from ZeptoMetrix at concentrations of approximately 1 × 109 PFU/ml. A 1-ml volume of a 1:100 dilution in 900 μl of easyMAG lysis buffer was also spiked with 0.1 g of negative stool. Cryptosporidium and Giardia lamblia concentrations were 2 × 103 oocytes/ml and cysts/ml, respectively, while Entamoeba histolytica was obtained at 1 × 106 parasites/ml. In addition to isolates, we tested 10 previously reported negative stool suspensions to determine if target cross-reactivity could occur. Additional 1:10 dilutions of spiked stools were performed for each isolate to determine the GPP ASR limit of detection (LoD). Stool suspensions were stored at 2 to 8°C until nucleic acid extraction.
Clinical specimens.
A total of 254 clinical stool specimens were collected from June 2011 to June 2012. Specimens were submitted from either outbreak cases or sporadic suspected cases of gastroenteritis originating in hospitals, long-term-care facilities, child care facilities, area restaurants, and the Milwaukee refugee screening facility. Stool specimens were also obtained from several clinical and public health partners. Stool specimens were transported either in Cary-Blair transport media or as raw stool in sterile cups. All clinical specimens used in this study were obtained under Milwaukee Health Department (MHD) Institutional BioSafety Committee (IBC) guidelines and approval.
Nucleic acid extraction.
Solid stool (0.1 g), 100 μl of liquid stool, or 100 μl of a spiked pure culture suspension was added to a Bertin SK38 Soil Mix Bead tube (BioAmerica Inc., Miami, FL) containing 900 μl of easyMAG lysis buffer (bioMérieux, Durham, NC). Tubes were vortexed for 5 min and allowed to settle for 10 min at room temperature. Tubes were centrifuged at 5,000 rpm for 5 min using an Eppendorf 5417R centrifuge (Eppendorf, Hamburg, Germany). Cleared supernatant (200 μl) was removed for nucleic acid extraction and added to an easyMAG extraction cartridge containing 10 μl of MS2 bacteriophage lysate (ZeptoMetrix) at 1.0 × 109 PFU/ml. Stool specimens were extracted using easyMAG extraction protocol Specific B with an input volume of 200 μl and an elution volume of 100 μl. Purified nucleic acid was stored at −20°C until multiplex PCR and detection.
Multiplex PCR.
A multiplex PCR master mix was created using the following ASRs (Luminex Corporation, Austin, TX): xTAG Giardia-01-A053, xTAG Cryptosporidium-01-A054, xTAG E. histolytica-01-A063, xTAG Y. enterocolitica-01-A013, xTAG Salmonella-01-A014, xTAG E. coli ST-01-A018, xTAG E. coli LT-01-A020, xTAG Shigella-01-A038, xTAG C. difficile toxin A-01-A045, xTAG C. difficile toxin B-01-A062, xTAG Campylobacter-02-A048, xTAG Vibrio-01-A056, xTAG E. coli O157-01-A065, xTAG Shiga Toxin 1-01-A078, xTAG Shiga Toxin 2-01-A076, xTAG Norovirus GI-01-A057, xTAG Norovirus GII-01-A022, xTAG Rotavirus A-01-A027, xTAG Adenovirus-01-A029, and xTAG MS2-01-A055 (used for internal control). The master mix was prepared by adding 0.167 μl for each of the xTAG ASRs to 12.5 μl 2× SuperScript One-Step reaction mix (Invitrogen, Carlsbad, CA), 3.56 μl molecular analysis-grade water (Invitrogen), 0.1 μl 5 M tetramethylammonium chloride (TMAC) (Sigma, St. Louis, MO), and 0.5 μl SuperScript II Platinum TaqMix (Invitrogen). A 20-μl volume of this master mix (reaction mix) was used for each PCR. The reaction mix was dispensed in a chilled 200-μl strip cap tube or plate, and 5 μl of nucleic acid was added to each reaction mix for a total volume of 25 μl. PCR amplification was carried out in a Veriti thermocycler (Life Technologies, Foster City, CA). PCR amplification cycling parameters were a reverse transcription (RT) step at 53°C for 20 min followed by an enzyme activation step at 95°C for 15 min and then 38 cycles of 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s at a ramp rate of 54%. This was followed by a final elongation step at 72°C for 2 min and a hold at 4°C until hybridization.
Hybridization.
Luminex MagPlex-TAG microspheres (Luminex Corporation) were vortexed and sonicated for repeated 10-s intervals. A bead mixture was prepared by adding 100 μl of each microsphere set to a sterile polystyrene tube (Fisher Scientific, Pittsburgh, PA) (12 by 75 mm) for a total volume of 1.7 ml. The tube was repeatedly vortexed and sonicated, and content was divided into two equal volumes in 1.5-ml microtubes (Fisher Scientific). Tubes were centrifuged at maximum speed (14,000 rpm) in an Eppendorf 5471R centrifuge for 5 min. The supernatant was removed, and beads were resuspended in equal volumes (650 μl each tube) of 1× xTAG Buffer (Luminex Corporation). Microtubes were repeatedly vortexed and sonicated to suspend microspheres prior to consolidation in a tube for storage. Bead mixtures are stable at 2 to 8°C when kept from light exposure.
A volume of 20 μl of MagPlex-TAG bead mixture was dispensed in each well of a V-bottom 96-well polycarbonate plate (Costar, Corning, NY). The PCR product (5 μl) was added to the wells and mixed by pipette. The reporter was prepared by diluting xTAG 0.22 SAPE (Luminex Corporation) at 1:75 in 1× GPP Reporter Buffer (Luminex Corporation). For each reaction, 1 μl SAPE and 74 μl buffer were mixed in a sterile polystyrene tube (12 by 75 mm) and dispensed in a sterile dispensing trough (Fisher, Pittsburgh, PA) followed by addition of 75 μl to each reaction well and pipetting five times to mix. The plate was sealed with a Microseal A film (Bio-Rad, Hercules, CA), and the mixture was allowed to hybridize in the thermocycler for 3 min at 63°C followed by 45 min at 45°C.
xMAP instrument parameters.
An analysis setting for the xMAP technology-based BioPlex 200 system (Bio-Rad) was created using xPONENT 3.1 software (Luminex Corporation). A protocol “batch” was created, and each ASR target was associated with its corresponding microsphere set. The sample size was set to 50 μl, with a minimum of 100 beads per target analyzed. The bead type was set to MagPlex, and the gate opening range was set to 6,000 to 20,000. The plate heater was set to 45°C.
Target detection.
The plate was removed from the thermocycler after hybridization and placed on a retractable 3-plate holder in a BioPlex 200 instrument. The plate seal was removed to uncover the reaction wells, and the sample probe height was adjusted for the plate type. The reactions were read by the instrument, and median fluorescence intensity (MFI) data were exported to TDAS LSM software (version 2.0) for data analysis (Luminex Corporation). A negative threshold was set to 300 MFI or to a value equivalent to a 2× negative-control value for that particular target (whichever was greater). Positive and negative controls were included in each run, with a negative control being the last sample analyzed.
Performance evaluation with spiked isolates.
In order to simulate clinical specimens, reference strains were cultured and plated as described above. A 0.5 McFarland standard suspension was created in phosphate-buffered saline (PBS), and 0.1 ml was added to a stool/lysis buffer matrix as described in the culture isolate section. The tubes were vortexed, and nucleic acid was extracted from these dilutions. Evaluation of the GPP ASRs was carried out on the spiked isolates to determine the performance of the assay with known positive specimens.
LoD analysis with spiked isolates.
Limit of detection (LoD) analysis was carried out by performing serial 1:10 dilutions in triplicate with bacterial, parasite, and viral isolates of known concentration spiked in a stool/lysis buffer matrix. Nucleic acid was extracted and analyzed. LoD was determined as the lowest pathogen concentration detected by GPP ASR in at least two of three replicas.
Performance evaluation on clinical specimens.
Clinical specimens were analyzed using the GPP ASRs, and results were compared to those of reference methods to determine the sensitivity and specificity within a statistical error limit. Specimens were confirmed using single-target real-time PCR assays when available or, if isolates were obtained, by sequence analysis using a 16S sequencing kit and an ABI 3130xl analyzer (Life Technologies, Foster City, CA). Parasite specimens were analyzed using conventional staining and microscopy or real-time PCR, while virus pathogens were confirmed by real-time RT-PCR assays available at the MHDL (Table 1).
Table 1.
Summary of reference or “gold standard” methods
| Target(s) | Reference method available at MHDLb | Reference(s)a |
|---|---|---|
| E. coli | Culture: MAC with sorbitol | 25, 26 |
| E. coli O157:H7 | API20E and ECO157 (slide) + H7 (tube) | 25, 26 |
| Shiga toxins | Meridian Premier EHEC EIA kit (catalog no. 608096); real-time PCR (stx1 + stx2) | 27, 28 |
| Salmonella spp. | Culture: XLD, MAC, SS, BS, BG + GN, Sel; API20E + serology (O and H antigens) | 25, 26 |
| Shigella spp. | Culture: XLD, MAC, SS + GN, Sel; API20E + serotyping | 25, 26 |
| Yersinia spp. | Culture: Yersinia agar at 25°C; API20E, MIDI, MicroSEQ (16S sequencing) | 25, 29 |
| Campylobacter spp. | Culture: Campylobacter agar at 42°C, Gram stain; catalase, oxidase, hippurate, indoxyl acetate, MIDI, MicroSEQ (16S sequencing) | 25, 30 |
| Vibrio cholerae | TCBS and alkaline peptone broth; API20E, Gram stain, oxidase; MIDI, MicroSEQ (16S sequencing) | 25, 26, 31 |
| C. difficile (A/B toxins) | Cepheid Xpert C. difficile | |
| Cryptosporidium spp. | Meridian EIA Crypto/Giardia (catalog no. 250050); microscopy | 32–33 |
| Giardia lamblia | Meridian EIA Crypto/Giardia (catalog no. 250050); microscopy, trichrome stain | 32–34 |
| Entamoeba spp. | Microscopy, trichrome stain, CDC real-time PCR | 27–35, 36 |
| Rotavirus A | CDC real-time RT-PCR | 12 |
| Norovirus | CDC real-time RT-PCR | 36 |
| Adenovirus 40/41 | CDC real-time RT-PCR | 37 |
Additional laboratory-validated methods for confirmations are available at MHDL but not referenced in this table.
MAC, MacConkey agar; XLD, xylose-lysine-deoxycholate agar; SS, Salmonella-Shigella agar; BG, brilliant green agar; GN, Gram-negative broth; MIDI, Sherlock Microbial Identification System (Microbial Identification Inc. [MIDI]); TCBS, thiosulfate-citrate-bile-sucrose agar.
Discrepant analysis.
Stool specimens were concurrently or previously tested with approved gold standard methods appropriate for the pathogen (Table 1). In the case of discrepant or inhibited results, specimens were verified using a comparative method if specimen nucleic acid or stool was available. Inhibited specimen nucleic acid was diluted 1:10 in water, and assays were repeated using GPP ASRs. Any species-level bacterial identification was confirmed using 16S DNA sequencing. In the event of multiple pathogen detection, testing for the additional pathogens was carried out according to the comparative method depending on the availability of the original stool specimen or extracted nucleic acid.
RESULTS
Performance evaluation on spiked isolates.
A total of 45 clinical and reference isolates for bacterial and viral pathogens and 10 negative stool specimens were analyzed using the GPP ASRs. The assay was able to confirm 100% correlation among the methods compared with GPP ASRs for all of the following previously identified pathogens: Salmonella spp. (8/8), Shigella spp. (2/2), Yersinia enterocolitica (1/1), Vibrio cholerae (1/1), Campylobacter jejuni (1/1), C. difficile A/B toxins (2/2), Giardia lamblia (3/3), Cryptosporidium spp. (1/1), adenovirus 40/41 (2/2), norovirus GI (5/5), norovirus GII (5/5), Entamoeba histolytica (4/4), and rotavirus A (1/1). The GPP ASRs identified 8 of 9 toxigenic E. coli isolates, including E. coli O157:H7 (1/1), with the exception being E. coli serotype O26:H11. A new 0.5 McFarland standard of E. coli O26:H11 was processed as described above and retested, with the target being detected. The ASR did not detect Yersinia pseudotuberculosis (0/1), Vibrio parahaemolyticus (0/1), and Campylobacter fetus (0/1). No GI pathogens were detected in previously reported negative stool specimens from healthy patients, and the GPP ASRs confirmed the negative results (10/10). Overall, the GPP ASR performance sensitivity was 100% (confidence interval [CI], 90% to 100%) and specificity was 100% (CI, 99% to 100%).
LoD analysis.
Overall observations from the LoD study indicated that the bacterial LoDs ranged from 102 to 104 CFU/ml depending on the species; virus LoD was 104 to 105 PFU/ml; and parasite LoD was 102 to 103 oocyst/ml. The LoDs of the various analytes are presented in Table S2 in the supplemental material.
Performance evaluation on clinical specimens.
A total of 254 clinical specimens were tested by the GPP ASRs. The stool specimens were tested for one or multiple targets using the conventional and molecular methods available at the MHDL. Concordance to conventional methods was as follows. The test results for adenovirus 40/41 (8/8), rotavirus A (9/9), norovirus GII (5/5), Cryptosporidium spp. (14/14), V. cholerae (3/3), Y. enterocolitica (3/3), E. coli heat-labile enterotoxins (LT)/heat-stable enterotoxins (ST) (5/5), and E. coli O157:H7 (1/1) were in 100% concordance. The results for Giardia lamblia (20/21) were 95% concordant; E. coli stx1/stx2 toxins (28/30) were 94% concordant; Shigella spp. (13/14) were 93% concordant; Salmonella spp. (24/26) were 92% concordant; C. difficile A/B toxins (20/22) were 91% concordant; and Campylobacter jejuni (18/20) were 90% concordant. The E. histolytica target was initially detected in 29 specimens but could be reproduced only in 5 with real-time PCR. Concordance for this target was 100%, but discrepant results are addressed in the subsequent section. The comparative assessment of GPP ASRs, including all targets detected and confirmed by standard methods, was 94.5% (range, 90% to 97%) (n = 175/211). Overall specificity was 99.8% (range, 99.1% to 99.8%). Individual target 95% CI ranges were recorded (Table 2; see data in Table 3).
Table 2.
GPP multiplex reference test method correlation to pathogens detected by gold standard method
| Organism(s) | No. of isolates or clinical specimens detected bya: |
No. of discrepant clinical specimens | |||
|---|---|---|---|---|---|
| Culture isolation, microscopy, or real-time PCR |
GPP multiplex |
||||
| Isolate | Clinical | Isolate | Clinical | ||
| Bacteria | |||||
| Escherichia coli ETEC (LT/ST toxins) | 1 | 5 | 1 | 5 | |
| Escherichia coli STEC (stx1/stx2 toxins) | 7 | 30 | 7 | 28 | 2 |
| Escherichia coli O157:H7 | 1 | 1 | 1 | 1 | |
| Salmonella spp. | 8 | 26 | 8 | 24 | 2 |
| Shigella spp. | 2 | 14 | 2 | 13 | 1 |
| Yersinia enterocolitica | 1 | 3 | 1 | 3 | |
| Yersinia pseudotuberculosis | 1 | 0 | ND | 0 | |
| Vibrio cholerae | 1 | 3 | 1 | 3 | |
| Vibrio parahemolyticus | 1 | 0 | ND | 0 | |
| Campylobacter jejuni | 1 | 20 | 1 | 18 | 2 |
| Campylobacter fetus | 1 | 0 | ND | 0 | |
| C. difficile (A/B toxins) | 2 | 22 | 2 | 20 | 2 |
| Parasites | |||||
| Giardia lamblia | 3 | 21 | 3 | 20 | 1 |
| Cryptosporidium spp. | 1 | 14 | 1 | 14 | |
| Entamoeba histolytica | 4 | 5 | 4 | 29 | 24b |
| Viruses | |||||
| Adenovirus 40/41 | 2 | 8 | 2 | 8 | |
| Rotavirus | 1 | 9 | 1 | 9 | |
| Norovirus GI | 5 | 0 | 5 | ND | |
| Norovirus GII | 5 | 5 | 5 | 5 | |
Data represent comparisons of GPP ASR detection of culture isolates and clinical specimens with conventional and molecular methods performed at MHDL. Bacteria were detected by culture isolation and GPP multiplex analysis, parasites were detected by microscopy and GPP multiplex analysis, and viruses were detected by real-time PCR and GPP multiplex analysis. Isolate, culture or reference isolate; Clinical, clinical specimen; ND, not determined.
The results for 24 specimens were not reproducible with the comparative real-time PCR method.
Table 3.
GPP ASR performance evaluation of clinical specimensa
| Organism(s) | No. of positive samples | No. of negative samples | % sensitivity (95% CI) | % specificity (95% CI) | % PPV (95% CI) | % NPV (95% CI) |
|---|---|---|---|---|---|---|
| Adenovirus 40/41 | 8 | 246 | 100 (60–100) | 100 (98–100) | 100 (60–100) | 100 (98–100) |
| Vibrio cholerae | 3 | 251 | 100 (31–100) | 100 (98–100) | 100 (31–100) | 100 (98–100) |
| Yersinia enterocolytica | 3 | 251 | 100 (31–100) | 100 (98–100) | 100 (31–100) | 100 (98–100) |
| Salmonella spp. | 24 | 228 | 92 (72–99) | 100 (98–100) | 100 (83–100) | 99 (97–99) |
| Shigella spp. | 13 | 240 | 93 (64–99) | 100 (98–100) | 100 (72–100) | 99 (97–99) |
| Campylobacter jejuni | 18 | 233 | 90 (67–98) | 99 (97–99) | 94 (72–99) | 99 (97–99) |
| C. difficile A/B toxins | 20 | 231 | 91 (69–98) | 100 (98–100) | 100 (80–100) | 99 (97–99) |
| ETEC/STECb | 33 | 219 | 94 (79–99) | 100 (98–100) | 100 (87–100) | 100 (87–100) |
| E. coli O157:H7 | 1 | 100 | 100 (55–100) | 100 (95–100) | 100 (55–100) | 100 (95–100) |
| Rotavirus A | 9 | 245 | 100 (63–100) | 100 (98–100) | 100 (63–100) | 100 (98–100) |
| Giardia lamblia | 20 | 232 | 95 (74–99) | 99 (97–99) | 95 (74–99) | 99 (97–99) |
| Entamoeba histolytica | 5 | 201 | 100 (46–100) | 89 (84–93) | 17 (06–36) | 100 (98–100) |
| Cryptosporidium spp. | 14 | 240 | 100 (73–100) | 100 (98–100) | 100 (73–100) | 100 (98–100) |
| Norovirus GII | 5 | 96 | 100 (46–100) | 100 (95–100) | 100 (46–100) | 100 (95–100) |
| Norovirus GI | 0 | 101 | ND | 100 (95–100) | ND | 100 (95–100) |
| Total | 176 | 3,114 | 94.5 (90–97) | 99 (99–100) | 87 (81–91) | 99 (99–100) |
Statistical analysis was based on 95% statistical confidence. PPV, positive predictive value; NVP, negative predictive value; ND, not determined.
Performance data for ETEC and STEC include the targets stx1 and stx2 and LT and ST as presented in Table 2.
Discrepant analysis.
GPP ASRs detected the presence of E. histolytica in 29 referred clinical specimens originally undiagnosed for this parasite (Table 2). Stool specimen microscopy for Entamoeba dispar and E. histolytica did not confirm the presence of viable cell structures. Specimen nucleic acid was analyzed with a real-time PCR assay for detection of Entamoeba histolytica (9). PCR identified 5/29 as E. histolytica. E. histolytica discrepancies in 24 specimens were not resolved by either microscopy or real-time PCR methods.
Coinfection detection and analysis.
There were 31 clinical specimens that were positive for one or more targets in addition to the pathogen identified by the comparative methods. Multiple pathogens were confirmed using culture, microscopy, and PCR methods. The predominant target detected in coinfections was E. coli with 12 coinfections followed by Salmonella spp. (n = 9), Giardia lamblia (n = 4), C. difficile A/B toxins (n = 3), Campylobacter jejuni (n = 2), and rotavirus A (n = 1) (Fig. 1). Of the 31 specimens, only the ETEC/STEC (n = 12) and rotavirus A (n = 1) results were reproducible by real-time PCR methods. All assays of multiple-infection specimens were repeated using GPP ASRs to determine the reproducibility of test results and confirmed with single-target PCR when available (data not shown). The results indicate that all targets were reproducible with GPP ASRs except for one C. difficile A/B toxin-positive specimen, which was considered undetected.
Fig 1.
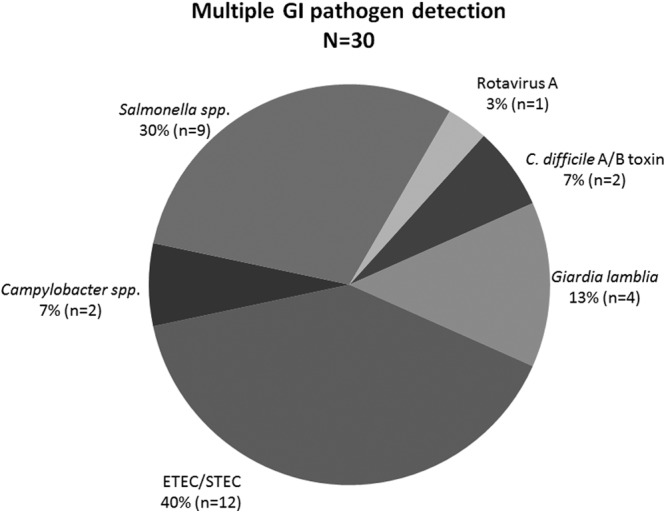
Targets detected in coinfections. GPP ASRs detect multiple GI pathogens undetected in clinical specimens by conventional methods, indicating the potential for multiplex technology in the identification of coinfection in patient stools. The percentage of incidence indicates the most predominant ADD targets detected in single specimens. ETEC and STEC were the most predominant strains detected followed by Salmonella spp. and Giardia lamblia.
DISCUSSION
In this study, we aimed to evaluate the performance of assays using GPP ASRs compared to conventional and molecular methods available to our laboratory for routine clinical diagnostic and public health outbreak responses. We have determined the analytical performance of GPP ASRs by testing reproducibility and sensitivity with confirmed culture isolates. We then proceeded to determine the clinical performance of GPP ASRs using a larger pool of clinical specimens that were concurrently or previously identified by MHDL and other reference laboratories.
We evaluated the performance of GPP ASRs with a panel of pure culture isolates to determine if the assay was able to detect previously confirmed isolates while maintaining a high degree of specificity. Our analytical panel included several E. coli serotypes and Salmonella spp., as well as two Shigella spp. (S. flexneri and S. sonnei), multiple viruses, and parasites. One welcomed highlight is the ability of the ASRs to detect recently emerging norovirus genotypes GI.12 and GII.4 Sydney, which have been widely linked to worldwide norovirus outbreaks in recent months (see Table S1 in the supplemental material) (10, 11). The GPP ASRs were able to detect all serotypes and subspecies in the panel but did not distinguish between them, with the exception of E. coli O157:H7, which was a separate ASR target (Table 2). Additional subtyping and serotyping of food-borne disease-causing agents would be a necessity for a public health laboratory (PHL); however, as a screening tool, detection at a species level of these known pathogens by GPP ASRs can streamline gastrointestinal pathogen detection.
An LoD study from a subset of the culture isolates produced values equivalent if not superior to LoDs reported by comparative methods in our laboratory, except for norovirus genogroup I, where a single-target PCR showed 100-fold-increased sensitivity (12). We found the performance of the GPP ASR to be highly specific and comparable to that of conventional methods while analyzing all 254 specimens for the detection of bacterial, parasitic, and viral ADD pathogens (Table 2). The ASR sensitivity ranged from 100% to 90% for the target organism; however, the aggregate sensitivity value for GPP ASRs was 94.5% (range, 90% to 97%), and specificity was 99.1% (Table 3). During internal validation of the GPP ASR, we noted an inhibition rate of approximately 8%.
Entamoeba histolytica nucleic acid was detected in several specimens that either were not originally analyzed or remained undiagnosed upon microscopic examinations (Table 2). Our laboratory performed microscopy for detection of E. histolytica and E. dispar; however, reduced sensitivity of assays of raw stools that have been frozen or stored for long periods could account for our inability to detect the parasite in clinical specimens. Microscopy as the gold standard for parasite detection often fails to distinguish between multiple parasites, and underdiagnosis is possible (13). We performed a real-time PCR assay for detection of E. histolytica and were able to reproduce 5 of 29 results. With our limited experience with this comparative assay (currently being validated at MHDL), we cannot explain the GPP results in the 24 discrepant specimens. We determined that additional testing is necessary to resolve those discrepant results. Many of the positive E. histolytica specimens were received from patients in Lagos, Nigeria, and from refuge populations in Wisconsin of patients who originated from countries where there may be a higher rate of endemicity of infections by this parasite. The availability of single or multiplex PCR for parasites greatly aids in detection of infections from specimens with very low parasitic load, including those from asymptomatic patient samples, and its implementation along with GPP could greatly improve our testing algorithm (14–16).
While the detection and confirmation of coinfections in initially undiagnosed specimens are of great significance and deserve a separate study, we felt that inclusion of these data in our statistical analysis of clinical specimen detection compared to conventional methods of detection would artificially influence our findings. Future side-by-side evaluations of GPP ASR assays or subsequent similar molecular multiplex assays can better determine the potential added value of multiple detection. A recent study in Germany compared the performance of the xTAG GPP assay (RUO) and the ProGastro SSCS assay (Gen-Probe Incorporated, San Diego, CA) to the performance of conventional microbiology methods. The authors concluded that multiplexing in gastrointestinal infection diagnostics has the potential to reduce the time to the first identification of a pathogen, influence subsequent clinical courses by an earlier start of specific treatments, and avoid false isolation results because of the high degree of sensitivity and specificity of new molecular methods (17).
Studies have further indicated that the introduction of new technology or changes in testing algorithms have dramatically reduced the TAT of clinical testing, improved patient management, and streamlined treatment algorithms, thus resulting in reduced hospital stay durations and costs (18). Based on current practices, identification of ADD pathogens during public health outbreaks using conventional methods is laborious and time-consuming, and molecular methods, such as real-time PCR, are of limited availability and are often specific to a particular pathogen of interest (2, 4, 19). Setting up a screening algorithm that can encompass the diversity of pathogens involved in any possible outbreak is often challenging to do, and if large outbreaks occur, it becomes more difficult to effectively reduce morbidity following delays in initial pathogen identification. For PHLs, surveillance testing and efficient outbreak response are priorities, and GPP ASR-based testing provides a versatile molecular multiplexing option that gives us, as one of the first public health laboratories in the United States to employ this technology, a tool for improving real-time public health laboratory response during GI outbreaks.
The demand for multiplex molecular methods is rapidly outpacing the limited availability of these products, and, judging from recent published studies (20, 21), more laboratories will embrace the advantages of multiplexing as a tool to enhance laboratory testing algorithms. This technology is not without limitations; the initial assay setting and reagent cost can be prohibitive for routine clinical testing and pathogen surveillance purposes. We observed during our limited LoD study that multiplexing up to 20 targets reduces LoD for individual targets compared to single-target conventional or molecular methods. Positive and negative predictive values, however, indicate the potential for this technology. The identification of multiple pathogens, although limited in the current study, indicates that the occurrence of multiple infections in patients is more common than we expected, and clinicians will have to assess the importance of these findings based on clinical criteria. We think the additional pathogen information is not a limitation but represents an opportunity for a reassessment to better understand complex clinical scenarios and will allow efficient treatment regimens that reduce secondary infections and failed treatments. GPP ASR assay performance was determined using both Cary-Blair and raw stool samples. While we did not perform a comparative study to determine differences in detection of pathogens from those matrices, the multiplexing approach using GPP worked well with both. There were inhibition challenges encountered while using Zn-PVA or formalin-fixed stools (data not shown), which is also an inherent challenge for nucleic acid-based testing (22–24).
In conclusion, our study determined the performance of the Luminex GPP ASR and demonstrated that it could be suitable as a primary screening tool for enteric bacteria, viruses, and parasites. We also showed that the sensitivity of assays using GPP ASRs was equivalent to or better than that of conventional and molecular test methods currently employed by clinical and public health laboratories. We think that the versatility of this tool can be used for streamlining real-time detection algorithms in detection of ADD-causing organisms. The reduced TAT will also help physicians to improve patient care and minimize hospital stays for acute and chronic GI cases. Our results indicated that multiplex pathogen screening using GPP ASRs provides an enormous range of information that is highly reliable, accurate, and actionable. With 94.5% sensitivity and 99% specificity, we have found the 19 GI pathogen target assay to be an extraordinary tool for rapid screening of stool specimens during food-borne and other GI outbreak investigations. The same-day TAT will allow PHLs and epidemiologists to identify the most likely pathogens involved in the outbreak, as well as to track possible sources of those outbreaks. Recent FDA clearance of the xTAG GPP in vitro diagnostic (IVD) assay kit could also minimize clinical and public health laboratory challenges for complex method validation, verification, and billing for those laboratories that might not have the resources or regulatory freedom to develop ASR-based LDTs.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the assistance from our public health nurses, food inspectors, and epidemiologists of the City of Milwaukee Health Department Division of Disease Control and Environmental Health (DCEH) for enteric and food-borne outbreak investigations. We thank Jody Lokken, Manjeet Khubbar, Bradley Krause, and David Bina at the MHDL for their technical assistance in stool culture, PCR, and nucleotide sequence analysis. We appreciate the support of the Wisconsin State Laboratory of Hygiene, Marshfield clinic, University of Lagos, Nigeria, and the CDC Enteric Disease Laboratory Branch for sharing clinical stool specimens and isolates and for additional confirmatory testing for GI pathogens. We thank Sherry Dunbar at Luminex Corporation and Julie Becker at MHDL for their critical review of the manuscript.
Footnotes
Published ahead of print 12 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.00896-13.
REFERENCES
- 1.Kaptan G, Fischhoff B. 2011. Diagnosing food-borne illness: a behavioral analysis of barriers to testing. J. Public Health Policy 32:60–72 [DOI] [PubMed] [Google Scholar]
- 2.Cunningham SA, Sloan LM, Nyre LM, Vetter EA, Mandrekar J, Patel R. 2010. Three-hour molecular detection of Campylobacter, Salmonella, Yersinia, and Shigella species in feces with accuracy as high as that of culture. J. Clin. Microbiol. 48:2929–2933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tauxe RV. 1997. Emerging foodborne diseases: an evolving public health challenge. Emerg. Infect. Dis. 3:425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolffs P, Bruggerman C, Well G, Van Loo I. 2011. Replacing traditional diagnostics of fecal viral pathogens by a comprehensive panel of real-time PCR. J. Clin. Microbiol. 49:1926–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quiroz ES, Bern C, MacArthur JR, Xiao L, Fletcher M, Arrowood MJ, Shay DK, Levy ME, Glass RI, Lal A. 2000. An outbreak of cryptosporidiosis linked to a foodhandler. J. Infect. Dis. 181:695–700 [DOI] [PubMed] [Google Scholar]
- 6.Barwick RS, Levy DA, Craun GF, Beach MJ, Calderon RL. 2000. Surveillance for waterborne-disease outbreaks—United States, 1997–1998. MMWR CDC Surveill Summ. 49:1–21 [PubMed] [Google Scholar]
- 7.Johnston S, Ballard M, Beach M, Causer L, Willkins P. 2003. Evaluation of three commercial assays for detection of Giardia and Cryptosporidium organisms in fecal specimens. J. Clin. Microbiol. 41:623–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones TF, Imhoff B, Samuel M, Mshar P, McCombs KG, Hawkins M, Deneen V, Cambridge M, Olsen SJ; Emerging Infections Program FoodNet Working Group 2004. Limitations to successful investigation and reporting of foodborne outbreaks: an analysis of foodborne disease outbreaks in FoodNet catchment areas, 1998–1999. Clin. Infect. Dis. 38(Suppl 3):S297–S302 [DOI] [PubMed] [Google Scholar]
- 9.Verweij JJ, Blange RA, Templeton K, Schinkel J, Brienen EAT, van Rooyen MAA, van Lieshout L, Polderman AM. 2004. Simultaneous detection of Entamoeba histolytica, Giardia lamblia, and Cryptosporidium parvum in fecal samples by using multiplex real-time PCR. J. Clin. Microbiol. 42:1220–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vega E, Vinjé J. 2011. Novel GII.12 norovirus strain, United States, 2009–2010. Emerg. Infect. Dis. 17:1516–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barclay L, Wikswo M, Gregoricus N, Vinjé J. 2013. Notes from the field: emergence of new norovirus strain GII.4 Sydney—United States. MMWR Morb. Mortal Wkly. Rep. 62:55. [PMC free article] [PubMed] [Google Scholar]
- 12.Freeman MM, Kerin T, Hull J, McCaustland K, Gentsch J. 2008. Enhancement of detection and quantification of rotavirus in stool using a modified real-time RT-PCR assay. J. Med. Virol. 80:1489–1496 [DOI] [PubMed] [Google Scholar]
- 13.Taniuchi M, Verweij J, Noor Z, Sobuz S, Lieshout L, Petri WA, Haque R, Houpt ER. 2011. High throughput multiplex PCR and probe-based detection with Luminex beads for seven intestinal parasites. Am. J. Trop. Med. Hyg. 84:332–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forbes B, Sahm D, Weissfeld AS. 1998. Laboratory methods for diagnosis of parasitic infections, p 784–787 In Bailey & Scott's diagnostic microbiology, 10th ed. Mosby, St. Louis, MO [Google Scholar]
- 15.Roy S, Kabir M, Mondal D, Ali IK, Petri WA, Jr, Haque R. 2005. Real-time-PCR assay for diagnosis of Entamoeba histolytica infection. J. Clin. Microbiol. 43:2168–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mens P, Spieker N, Omar S, Heijnen M, Schallig H, Kager PA. 2007. Is molecular biology the best alternative for diagnosis of malaria to microscopy? A comparison between microscopy, antigen detection and molecular tests in rural Kenya and urban Tanzania. Trop. Med. Int. Health 12:238–244 [DOI] [PubMed] [Google Scholar]
- 17.Kahalu P, Maleki M, Schilgden V, Schulz C, Winterfield I, Messler S, Mattner F, Schildgen O. 2013. Utility of two novel multiplexing assays for the detection of gastrointestinal pathogens—a first experience. Springerplus 2:106. 10.1186/2193-1801-2-106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barenfanger J, Drake C, Kacich G. 1999. Clinical and financial benefits of rapid bacterial identification and antimicrobial susceptibility testing. J. Clin. Microbiol. 37:1415–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Logan C, O'Leary JJ, O'Sullivan N. 2006. Real-time reverse transcription-PCR for detection of rotavirus and adenovirus as causative agents of acute viral gastroenteritis in children. J. Clin. Microbiol. 44:3189–3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Xu ZQ, Zhang Q, Jin M, Yu JM, Li JS, Liu N, Cui SX, Kong XY, Wang H, Li HY, Cheng WX, Ma XJ, Duan ZJ. 2012. Simultaneous detection of seven enteric viruses associated with acute gastroenteritis by a multiplexed Luminex-based assay. J. Clin. Microbiol. 50:2384–2389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Babady N, Miranda E, Gilhuley KA. 2011. Evaluation of Luminex xTAG fungal analyte-specific reagents for rapid identification for clinically relevant fungi. J. Clin. Microbiol. 49:3777–3782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J, Scholten T. 1977. A fixative for intestinal parasites permitting the use of concentration and permanent staining procedures. Am. J. Clin. Pathol. 67:300–304 [DOI] [PubMed] [Google Scholar]
- 23.Goldsworthy SM, Stockton PS, Trempus CS, Foley JF, Maronpot RR. 1999. Effect of fixation on RNA extraction and amplification from laser capture microdissected tissue. Mol. Carcinog. 25:86–91 [PubMed] [Google Scholar]
- 24.Okello J, Zurek J, Devault A, Kuch M, Okwi A, Sewankambo NK, Bimenya GS, Poinar D, Poinar H. 2010. Comparison of methods in the recovery of nucleic acids from archival formalin-fixed paraffin-embedded autopsy tissues. Anal. Biochem. 400:110–117 [DOI] [PubMed] [Google Scholar]
- 25.Carroll KC, Weinstein MP. 2007. Manual and automated systems for detection and identification of microorganisms, p 199–204 In Murray PR, Baron EJ, Jorgensen JH, Landry ML, Pfaller MA. (ed), Manual of clinical microbiology, 9th ed. ASM Press, Washington, DC [Google Scholar]
- 26.Nataro JP, Bopp CA, Fields PL, Kaper JB, Stockbene NA. 2007. Escherichia, Shigella, and Salmonella, p 670–682 In Murray PR, Baron EJ, Jorgensen JH, Landry ML, Pfaller MA. (ed), Manual of clinical microbiology, 9th ed. ASM Press, Washington, DC [Google Scholar]
- 27.Kehl KS, Havens P, Behnke CE, Acheson DW. 1997. Evaluation of the premier EHEC assay for detection of Shiga toxin-producing Escherichia coli. J. Clin. Microbiol. 35:2051–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jinneman KC, Yoshitomi KJ, Weagant SD. 2003. Multiplex real-time PCR method to identify Shiga toxin genes stx1 and stx2 and Escherichia coli O157:H7/H- serotype. Appl. Environ. Microbiol. 69:6327–6333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wanger A. 2007. Yersinia, p 692–693 In Murray PR, Baron EJ, Jorgensen JH, Landry ML, Pfaller MA. (ed), Manual of clinical microbiology, 9th ed. ASM Press, Washington, DC [Google Scholar]
- 30.Fitzgerald C, Nachamki I. 2007. Campylobacter and Arcobacter, p 933–940 In Murray PR, Baron EJ, Jorgensen JH, Landry ML, Pfaller MA. (ed), Manual of clinical microbiology, 9th ed. ASM Press, Washington, DC [Google Scholar]
- 31.Tison DL. 1999. Vibrio, p 500–502 In Murray PR, Baron EJ, Jorgensen JH, Landry ML, Pfaller MA. (ed), Manual of clinical microbiology, 9th ed. ASM Press, Washington, DC [Google Scholar]
- 32.Garcia LS. 2001. Macroscopic and microscopic evaluation of fecal specimens, p 746–764 In Diagnostic Medical Parasitology, 4th ed. ASM Press, Washington, DC [Google Scholar]
- 33.Garcia LS, Brewer TC, Brucher DA. 1987. Fluorescence detection of Cryptosporidium oocyst in human fecal specimens by using monoclonal antibodies. J. Clin. Microbiol. 25:119–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharp SE. 2007. Reagents, stains, and media: parasitology, p 216–217 In Murray PR, Baron EJ, Jorgensen JH, Landry ML, Pfaller MA. (ed), Manual of clinical microbiology, 9th ed. ASM Press, Washington, DC [Google Scholar]
- 35.Garcia LS, Shimizu RY, Palmer JC. 1999. Algorithm for detection and identification of parasites, p 1336–1342 In Murray PR, Baron EJ, Pfaller MA, Tenover FC, Yolken RH. (ed), Manual of clinical microbiology, 7th ed. ASM Press, Washington, DC [Google Scholar]
- 36.Trujillo A, McCaustland K, Zheng D, Hadley L, Vaughn G, Adams S, Ando T, Glass R, Monroe S. 2006. Use of TaqMan real-time reverse transcription PCR for rapid detection, quantification, and typing of noroviruses. J. Clin. Microbiol. 44:1405–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarantis H, Johnson G, Brown M, Petric M, Tellier R. 2004. Comprehensive detection and serotyping of human adenoviruses by PCR and sequencing. J. Clin. Microbiol. 42:3963–3969 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.