

Abstract
The widespread use of nanoparticles (NPs) raises concern over their potential toxicological effects in humans and ecosystems. Here we used transcriptome sequencing (RNA-seq) to evaluate the effects of exposure to four different metal-based NPs, nano-Ag (nAg), nano-TiO2 (nTiO2), nano-ZnO (nZnO), and CdTe/CdS quantum dots (QDs), in the eukaryotic green alga Chlamydomonas reinhardtii. The transcriptome was characterized before and after exposure to each NP type. Specific toxicological effects were inferred from the functions of genes whose transcripts either increased or decreased. Data analysis resulted in important differences and also similarities among the NPs. Elevated levels of transcripts of several marker genes for stress were observed, suggesting that only nZnO caused nonspecific global stress to the cells under environmentally relevant conditions. Genes with photosynthesis-related functions were decreased drastically during exposure to nTiO2 and slightly during exposures to the other NP types. This pattern suggests either toxicological effects in the chloroplast or effects that mimic a transition from low to high light. nAg exposure dramatically elevated the levels of transcripts encoding known or predicted components of the cell wall and the flagella, suggesting that it damages structures exposed to the external milieu. Exposures to nTiO2, nZnO, and QDs elevated the levels of transcripts encoding subunits of the proteasome, suggesting proteasome inhibition, a phenomenon believed to underlie the development and progression of several major diseases, including Alzheimer's disease, and used in chemotherapy against multiple myeloma.
INTRODUCTION
Nanoparticles (NPs) are materials with at least one dimension in the nanoscale (ca. 1 to 100 nm), with resulting size-related physicochemical properties that differ from those of their bulk counterparts. Specifically, their high surface-to-volume ratio generally results in highly reactive and physicochemically dynamic materials. With the increasing use of engineered NPs, there is an enormous uncertainty with respect to their potential environmental impacts, including their toxicological effects. Of special concern is the emerging evidence that numerous NPs can produce reactive oxygen species (ROS), release toxic metals, or react directly with the biological membrane (1, 2). A major key question is whether toxicological effects are due to general properties shared by diverse NP types or whether they are specific to each NP. For example, if toxicological effects are related to the size, shape, or agglomeration of the NP, then different NP compositions would likely exert similar effects (3), whereas distinct toxicological effects would be expected if particle composition controls the interaction with the biological surface (4, 5). An understanding of the toxicological effects of each NP type is critical for any prediction of their immediate and long-term risks for humans and ecosystems.
A powerful approach to determine how an organism responds to a particular abiotic condition is to determine how it changes the expression of its genome. Modern transcriptome methodologies can quantify the expression of most genes in an organism on the basis of the levels of their RNA transcripts in response to an abiotic condition relative to the levels under normal physiological conditions (6). Comparisons of the effects of other specific biotic or abiotic conditions can reveal degrees of similarity or difference. For example, if NP exposure induces a particular stress condition, then this can be recognized by differential expression (DE) in the transcript levels of specific sets of genes. Effects on specific physiological or biochemical processes can be revealed by changes in the transcript levels of genes that are known or predicted to function in them. The transcriptome sequencing (RNA-seq) approach generates a vast inventory of gene transcripts using massive parallel DNA sequencing technologies, bioinformatics, and sequence databases (7). RNA-seq can quantify low-abundance transcripts, which account for approximately 30% of most transcriptomes, and it can identify novel exons and splice junctions. Moreover, in accuracy and precision, RNA-seq is comparable to quantitative real-time PCR (8, 9). Data from a variety of organisms imply that ca. 40% of the variation in the levels of protein accumulation can be explained by knowing mRNA abundances (10, 11). Therefore, while transcriptome profiling does not reveal changes due to effects at the translational, posttranslational, cell biological, or organismal levels, it does provide substantial and detailed information about toxicological responses, which can complement the results of other approaches.
In this study, we characterized the transcriptomic effects of four metal-based NPs using Chlamydomonas reinhardtii. This eukaryotic green alga is widely used as a model organism for metal homeostasis, metal-induced stress responses, and ecotoxicology, and it is increasingly being subjected to transcriptome profiling (12–15). Four NPs were selected due to their extensive use in either manufacturing (nanosilver [nAg], nano-zinc oxide [nZnO], and nano-titanium dioxide [nTiO2]) or medical bioimaging (quantum dots [QDs]; CdTe/CdS). For example, nTiO2 has a worldwide production of up to 10,000 metric tons/year, and about 1,000 tons of nZnO and nAg are produced yearly, whereas QD production does not exceed 10 metric tons/year (16). RNA-seq was used to characterize the transcriptomic effects of four different metal-based NPs in order to determine (i) their physiological effects on C. reinhardtii, (ii) how cells respond to these effects, and (iii) the degrees of similarity of the effects of the different NPs. Emphasis was also placed on (i) characterization of the NPs and their aggregation state under the physicochemical conditions of the experiment, (ii) analysis of the bioaccumulation level of the NPs by C. reinhardtii, and (iii) identification of distinct and similar toxicological effects with the ultimate goal of determining the modes of action (MOA) induced by each NP.
MATERIALS AND METHODS
Characterization of the NPs.
The following NPs were used: (i) nTiO2 (anatase; 99.7% purity; Nanostructured & Amorphous Material, Inc.) with a nominal size of 5 nm, a nearly spherical morphology (17), and density of 3.9 g cm−3 at 20°C. The pH of the zero point of charge (pHzpc) of these NPs is 4.5 to 5.2, and they have an electrophoretic mobility (EPM) of −1.89 ± 0.19 μm cm V−1 s−1 at pH 7.0 and an ionic strength (I) of 0.01 M (adjusted with NaNO3) (17); (ii) nZnO (99.5% purity; Nanostructured & Amorphous Material, Inc.) with a nominal size of 20 nm and a nearly spherical morphology (18), a density of 5.6 g cm−3 at 20°C, a pHzpc of 6.6 to 8.0, an EPM of 0.21 ± 0.18 μm cm V−1 s−1 at pH 7.0, and an I of 0.01 M (adjusted with NaNO3) (70); (iii) polyacrylate-stabilized nAg (Vive Nano, Inc.) at a total concentration of 1.5 × 104 mg liter−1 and with a nominal size of 20 nm and a nearly spherical morphology (19), an EPM of −2.0 ± 0.2 μm cm V−1 s−1 at pH 7.0, and an I of 0.01 M (adjusted with NaNO3) (19); and (iv) polyacrylate-stabilized QDs (CdTe/CdS; 97% purity; CdTe by inductively coupled plasma mass spectrometry from Vive Nano, Inc.) at a total concentration of 2.0 × 104 mg liter−1, as specified by the manufacturer, and with a core of CdTe with a diameter of 3 to 4 nm, a coating of CdS (total nominal size of 6 to 10 nm), and a nearly spherical morphology (18); an EPM of −2.09 ± 0.26 μm cm V−1 s−1 at pH 7.0; and an I of 0.01 M (adjusted with NaNO3) (20).
The diameters of the NPs in the exposure media were quantified by at least one of the following analytical techniques: analytical ultracentrifugation (AUC) (21), fluorescence correlation spectroscopy (FCS) (18, 22), and atomic force microscopy (AFM) (23). The size characterization of the NPs was performed under the exact same conditions as the bioaccumulation experiments, including preequilibration of the dispersions for 24 h. Details on the experimental sizing protocols are provided in the supplemental material.
Algal growth and biouptake.
C. reinhardtii (wild-type strain C137) was inoculated from a week-old Tris-acetate-phosphate (TAP) agar plate (24) into a 4×-diluted TAP solution (I = 10−2 M). Cells were grown in an incubator (Infors) at 20°C under a 12-h light and a 12-h dark regime using fluorescent lighting (80 μmol of photons m−2 s−1) and rotary shaking (100 rpm) to a density of 2 × 106 to 3 × 106 cells ml−1. Cells were then diluted in fresh medium and cultured for 4 more days, allowing them to achieve mid-exponential growth to 1 × 106 to 3 × 106 cells ml−1. The cells were harvested by centrifugation (3,700 × g, 4 min) in 50-ml sterile (polypropylene) centrifuge tubes and resuspended in an experimental medium (pH 7.0) containing 20 mM and 10−5 M Ca [Ca(NO3)2·4H2O] (Sigma). They were subsequently centrifuged, harvested a second time, and again resuspended in the exposure medium. The first centrifugation was to harvest the cells from the exposure medium. The second centrifugation was to wash them as much as possible with the growth medium, in order to avoid changes to the physicochemistry which could change the speciation in the exposure medium. C. reinhardtii was added at a cell density of 1 cm2 ml−1 to five flasks (300 ml) containing the exposure medium and one of the following: control (no NP), 1 mg liter−1 nTiO2, 1 mg liter−1 nZnO, 1 mg liter−1 nAg, or 0.125 mg liter−1 QDs. Preliminary experiments showed that the lower concentration of QDs still induced significant changes at the transcriptomic level (12). Cell densities, sizes, and surface distributions were determined using a Coulter Multisizer 3 particle counter (50-μm orifice; Coulter Electronics). The culture medium and experimental solutions were sterilized (autoclave, filtration through a 0.2-μm-pore-size filter) prior to use, and axenic techniques were maintained for the entire exposure period. Exposure solutions were equilibrated for 24 h in the dark prior to addition of the algae. The exposure experiment was conducted in an algal incubator with orbital shaking (100 rpm) at 20°C under moderate light. Bioaccumulation was examined in short-term experiments (2 h) in order to decrease the likelihood of a significant modification of the exposure conditions due to the production of algal exudates, changes in pH, or biological regulation (25). At the end of the 2-h exposure, bioaccumulation was stopped by adding 5 ml of 10−2 M Na EDTA (Sigma) that was prepared in the exposure medium. The 2-h exposure time was determined on the basis of prior experiments focused on the exposures of C. reinhardtii to Cd (15) and Ni (13). Internalized metal content was determined from filtered (3.0-μm-pore-size nitrocellulose membrane; Millipore), EDTA-washed cells. Filters and algae were digested by heating to 85°C in 0.3 ml of ultrapure HNO3 over 24 h. Metal concentrations were determined by graphite furnace atomic absorption spectrometry (Thermo M Series GF995Z) or by inductively coupled plasma mass spectrometry (Thermo X7). The rest of the exposed cells were harvested at room temperature by centrifugation (3,700 × g, 4 min) in 50-ml sterile (polypropylene) centrifuge tubes and resuspended in the experimental medium (pH 7.0) containing 20 mM HEPES (Sigma) and 10−5 M Ca [Ca(NO3)2·4H2O; Sigma] for washing. They were subsequently centrifuged and harvested a second time. The pellet was transferred to sterilized microcentrifuge tubes, frozen in liquid nitrogen, and stored at −80°C until mRNA extraction.
mRNA preparation.
Total RNA was extracted by adding β-mercaptoethanol (1%, vol/vol) and glass beads (0.5-mm diameter; Thomas Scientific) to each cell pellet, prior to vortexing (4 times, 15 s each time). Between each manipulation, samples were kept frozen on dry ice. Total RNA was isolated by ethanol precipitation and purification using a Qiagen RNeasy kit (Qiagen Inc., Valencia, CA). RNA quality and quantity were estimated by measuring the absorbance at 280 nm and 260 nm and by the analysis of rRNA intactness using an Agilent 2100 BioAnalyzer (Agilent Technologies). RNA quality was considered acceptable when the 28S rRNA/18S rRNA ratio was ≥2. mRNA was isolated by oligo(dT) selection with a PolyATract mRNA isolation kit (Promega) according to the manufacturer's protocol. Samples of 100 ng of purified mRNA extracted from the cells under various conditions were used to prepare paired-end cDNA libraries using a SOLiD whole-transcriptome analysis kit (PN4425680) by following the manufacturer's protocol. These were then submitted for Applied Biosystems SOLiD high-throughput sequencing at the Institut de Recherche en Immunologie et Cancérologie (IRIC) Genomics Core Facility at the University of Montreal.
Mapping of SOLiD.
Processed sequence files from the SOLiD pipeline output were aligned against the Chlre 3.1 version of the C. reinhardtii genome (Joint Genome Institute [26]). The raw 25-mer sequences were aligned to the transcriptome using the MAQ alignment program (27) with a tolerance of up to two mismatches and no deletions or insertions. Reads that mapped to multiple locations were removed from analysis. Approximately 42% of the reads mapped uniquely with a coverage of 4.7.
Estimation of differential gene expression.
Differentially expressed genes were called using the DESeq package (version 1.6.1) (28), an R package implementing a model based on a negative binomial distribution, which was developed in order to cope with biological variance (28). The software was run under R release 2.14.0. Fold changes between conditions (change in expression between the wild type plus each NP and the wild type) were determined using transcript relative abundance (TRA) log2 ratios. Genes were tagged as differentially expressed if they met the following criteria: (i) a TRA log2 ratio of greater than a 2-fold change (≥2 or ≤−2), (ii) a P value of ≤0.05 for differential expression, and (iii) a minimum of 20 reads mapped per gene. Differential expression was estimated for each NP on the basis of the assumption that for most genes there is no true differential expression and that a valid mean-variance relationship can be estimated from treating the control and the treated samples as if they were replicates (28). Using this approach, some overestimation of the variance is expected and will result in an underestimate of the number of genes with DE. Furthermore, data from the SOLiD RNA-seq experiment generated results that were comparable to those produced by Affymetrix exon arrays, even though only a single replicate from each platform was used (29).
GO enrichment analyses.
Gene annotations were retrieved from the JGI C. reinhardtii genome browser (http://genome.jgi-psf.org/Chlre3/Chlre3.home.html) (26). Assignment of Gene Ontology (GO) and KEGG terms was based on JGI annotations and the UCLA Algal Functional Annotation Tool (http://pathways.mcdb.ucla.edu/algal/index.html). Enrichment of differentially regulated genes in KEGG pathways was determined using GOSeq (30), with a false discovery rate of 5% employed as the threshold for enriched KEGG terms.
RESULTS
NP characterization and biouptake.
Our objective was the analysis of the transcriptomic responses to NP exposure that would accurately reflect what the microorganisms might experience in the environment. In order to ensure that the exposure conditions elicited a response while minimizing secondary effects and lethality, conditions were selected by considering prior reports of their toxicological effects for nAg (31), nTiO2 (1), nZnO (32), and QDs (33). The physicochemical properties and effects of the NPs change following their aggregation (17, 34), dissolution (12, 20, 70), or coating with biological substances (e.g., proteins [35]), since these parameters affect the interfacial properties, including the rates of surface-mediated reactions, dissolution, redox reactions, and generation of ROS (36). Therefore, the sizes of the NPs were quantified in the exposure medium (Table 1). Aggregation was minimized by using an exposure medium with a relatively low ionic strength (0.01 M).
Table 1.
Nominal and measured diameters of the TiO2, ZnO, Ag NPs and QDs in the exposure medium quantified by FCS, AFM, and/or AUC
| NP |
da (nm) |
Db (m2 s−1) | |||
|---|---|---|---|---|---|
| Manufacturerc | FCS | AUC | AFM | ||
| TiO2 | 5 | 15–31 | 2–14 | 2.2 × 10−11 | |
| ZnO | 20 | 24–72 | 10–13 | 1.2 × 10−11 | |
| Ag | 1–10 | 0.5–9.0 | 4.8 × 10−11 | ||
| QDs | 6–10 | 3.1–3.2 | 3.9–4.7 | 1.0 × 10−10 | |
d, diameter. For calculation purposes, the average diameter was applied for QD, whereas for nAg, the maximum value from two distinct peaks was used. The signal obtained with the FCS is the diffusion coefficient, so for the oxide particles, the average diffusion coefficient was used; for AUC and AFM, data are particle diameters for nAg and QD, respectively.
D, diffusion coefficient. Diffusion coefficients were used in maximum permeability calculations and were calculated using the Stokes-Einstein equation (see equation S1 in the supplemental material).
Nominal diameters from the manufacturer.
The size range of the nTiO2 determined by FCS was 3 to 6 times larger (15 to 31 nm) than that stated by the manufacturer but consistent with previously reported values obtained under similar conditions (17, 18). AUC results for nTiO2 gave a size range between 2 and 14 nm (mode, 9.1 nm). FCS results showed a large range of diameters (24 to 72 nm) for nZnO, while a smaller size was measured by AUC (10 to 13 nm). AUC results for the nAg showed that these particles were stable under the conditions that were studied; mean values of 0.5 to 9.0 nm were observed (mode range, 1.2 to 6.4 nm). The characterization of the QDs by AFM could be performed only in water and not in the exposure medium due to the formation of a film on the surface of the mica (a consequence of the HEPES buffer presence). Nonetheless, the average diameter (3.9 to 4.7 nm) obtained from the AFM height measurements corresponded well with the diameters (3.1 to 3.2 nm) obtained from sedimentation velocities using AUC. Overall, for the exposures studied as described below, it could be concluded that there was relatively little aggregation of any of the NPs.
C. reinhardtii cells were exposed to each NP type for 2 h and then washed with 10−2 M Na EDTA in order to remove the extracellular metals and NPs. The internalized metal content (Ti, Zn, Ag, and Cd) was measured, and the corresponding permeabilities were calculated (Fig. 1). The maximum permeabilities for each NP type (lines in Fig. 1) were also calculated using Fick's second law for the steady-state, radial diffusion of the NP toward an organism with a radius of 3.5 × 10−4 cm (37) and the diffusion coefficients of each NP (based upon the measured sizes and compiled in Table 1). For nZnO, nAg, and the QDs, the measured permeabilities were significantly less than the calculated maximum permeabilities, implying that the diffusive supply of the NP or its ions was more than enough to sustain the observed biouptake. Somewhat surprisingly, for the only particle that did not show significant dissolution, i.e., nTiO2, the measured and calculated permeabilities were similar (Fig. 1), suggesting that C. reinhardtii was accumulating the particles as fast as they were arriving at the cell surface.
Fig 1.
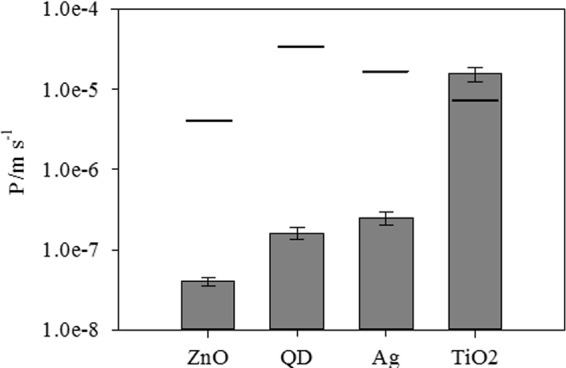
Measured and calculated maximum permeabilities (P) of each of the NPs. The calculations of maximum permeabilities are based upon the radial diffusion of each NP (diffusion coefficients are provided in Table 1) to the surface of a spherical microorganism with a radius of 3.5 μm.
RNA-seq analyses.
Of the 1.2 × 108 total reads obtained using RNA-seq analysis, 5.0 × 107 (42%) mapped uniquely with a maximum of two mismatches and no deletions or insertions. These reads were then used in the downstream analyses (Table 2). Based upon the criteria given above, 96, 156, 141, and 49 upregulated genes were identified for cells exposed to nTiO2, nZnO, nAg, and the QDs, respectively (Table 2). Similarly, 80 downregulated genes were noted for nTiO2, 29 were noted for nZnO, 86 were noted for nAg, and 55 were noted for the QDs. Histograms of the fold changes and scatterplots of the fold change in gene expression for each of the NP libraries are shown in Fig. S1 and S2 in the supplemental material.
Table 2.
Summary of ABI SOLiD RNA-seq mapping results for control strain C137 and NPs
| Treatment | Total no. of expression tags sequenceda | No. (%) of sequence reads mappedb | Transcript abundance |
|
|---|---|---|---|---|
| Increasedc | Decreasedd | |||
| Control | 25,202,539 | 9,417,404 (37) | ||
| TiO2 | 24,896,579 | 11,194,586 (45) | 96 (0.009) | 80 (0.001) |
| ZnO | 19,323,816 | 9,676,538 (50) | 156 (0.0016) | 29 (0.003) |
| Ag | 18,907,352 | 8,895,003 (47) | 141 (0.016) | 86 (0.001) |
| QDs | 29,773,601 | 10,713,366 (36) | 49 (0.005) | 55 (0.005) |
ABI map reads; sequence length, 25 bp; tolerance, 2 errors.
After mapping to the C. reinhardtii genome (version 3.1).
Data represent the number (percentage of total genes in the genome) of upregulated genes determined with the DESeq package (2-fold change in expression, ≥20 reads, P < 0.05).
Data represent the number (percentage of total genes in the genome) of downregulated genes determined with the DESeq package (2-fold change in expression, ≥20 reads, P < 0.05).
As a first step to compare and contrast the transcriptomic effects due to exposure to each of the four NPs, a Venn diagram was generated to display the number of differentially expressed transcripts. It was broken down into sectors according to whether the transcript was similarly altered in the transcriptomes produced in response to one or more of the NPs (Fig. 2a). Figure 2b allows a more quantitative comparison by showing a cluster analysis and a heat map that represents the fold change value under each NP exposure condition for each transcript that was significantly altered in at least one transcriptome produced in response to NPs. These initial analyses revealed that the NPs have specific and different effects, as described below. Common transcriptomic effects between the QDs and nTiO2 were also recorded and are also described below.
Fig 2.

(a) Venn diagram showing the number of upregulated (red) and downregulated (green) transcripts in each transcriptome produced in response to each NP determined with DESeq (≥2-fold change in expression, ≥20 reads, P < 0.05). (b) Heat map of the relative expression of genes with DE following exposure to each of the four NPs. The 363 genes that showed ≥2-fold changes in expression were hierarchically clustered.
Specific effects for a given NP.
Exposure to each of the four NPs resulted in largely different transcriptomic responses in C. reinhardtii. The following percentages of transcripts were elevated in only one transcriptome produced in response to NP (Fig. 2a): (i) for nTiO2, 52% (50/96); (ii) for nZnO, 76% (119/156); (iii) for nAg, 86% (121/141); and (iv) for the QDs, 39% (19/49). The transcripts that decreased in abundance were as follows (Fig. 2a): (i) for nTiO2, 19% (15/80); (ii) for nZnO, 59% (17/29); and (iii) for nAg, 77% (66/86). For the transcriptome produced in response to QDs, all transcripts that decreased in abundance also decreased in a transcriptome produced in response to at least one other NP. Clearly, these four metal-based NPs differ substantially in their effects on C. reinhardtii.
Important differences among the NPs.
The levels of many transcripts were conversely affected by the different NPs. This can be seen in the cluster diagram (Fig. 2b) for the fold change values of many transcripts in four primary hierarchical clusters, labeled A to D. The diagram illustrates the striking fact that the transcriptional response for nAg is the opposite of the responses obtained for the other three NPs. Clusters A and B contain transcripts that were elevated only by the exposure to nAg or nZnO, respectively. Cluster C illustrates the transcripts elevated by exposure to nTiO2, nZnO, and the QDs. Moreover, the fold change values for these transcripts for nTiO2, nZnO, and QDs revealed considerably more decreased than elevated transcripts. Other, opposite effects seen at the transcript level can be noted in the heat map (Fig. 2b). These results further support the finding that these NPs differ in their effects on C. reinhardtii.
Important similarities among the NPs.
Amid these distinct and converse effects of the different NP types, the levels of certain transcripts showed similar changes in transcriptomes produced in response to two or more NPs (Fig. 2a). The most pronounced common effect was the elevated transcription of sets of transcripts: 13 for nTiO2-QDs, 10 for nTiO2-nZnO, and 8 for nAg-nZnO. Subsets of these were enhanced by more than two NP types: 12 by nTiO2-QDs-nZnO and 5 by nAg-nTiO2-nZnO. The transcripts with reduced levels of transcription were 45 for nTiO2-QDs and 6 for nAg-nZnO. Transcription of subsets of these were also reduced by more than two NP types; 5 by nTiO2-QDs-nZnO and 5 by nAg-nTiO2-QDs. Correspondingly, the results of cluster analysis (Fig. 2b) reveal many parallel effects both for transcripts with elevated levels of transcription and for transcripts with reduced levels of transcription. The effects of QDs and nTiO2 were mostly similar, while nZnO had almost the same effects as the other three NPs, as depicted in the tree at the top of Fig. 2b. For example, most transcripts in Cluster A decreased in abundance, while many transcripts in Cluster C were coordinately elevated by nTiO2, QDs, and nZnO. Similarly, parallel increases in the levels of transcripts in cluster B were observed in the transcriptomes produced in response to nAg, nTiO2, and QDs. Each of these cases suggests that those particular NPs have common targets which might lead to similar modes of action and consequences in C. reinhardtii.
Bioinformatic surveys of the known and predicted functions of NP target genes.
In order to identify the biological responses of C. reinhardtii exposed to the NPs, the known and predicted functions of the NP-responsive transcripts were surveyed. First, enrichment of GO analyses was performed with the GOseq program (30) in order to reveal how the known or predicted functions of the NP-responsive transcripts assort into ontologies in three categories: biological processes, molecular functions, and intracellular compartments. Significance was assigned as categories/hits with P values of ≤0.05 (Table 3; Fig. 3). Second, in order to understand the impact of the transcriptomic data at the cellular level, the results of the GO analyses were mapped to specific gene products in the KEGG pathways in C. reinhardtii using KASS software (http://www.genome.jp/tools/kaas) (see Fig. S3 in the supplemental material) (38). Again, assignments with P values of ≤0.05 were considered significant. KEGG analyses revealed the fold change in the levels of specific transcripts, many of which have known or predicted functions listed in their annotations in the JGI v3.1 C. reinhardtii genome sequence database. These results revealed only a few categories, but several were highly significant, as described below.
Table 3.
GO categories that are significantly enriched in differentially expressed genes
| Function and GO accession no. | Rega | Classb | Annotation |
P valuec |
|||
|---|---|---|---|---|---|---|---|
| TiO2 | ZnO | Ag | QDs | ||||
| Ubiquitin proteasome system | |||||||
| GO:0005839 | Up | CC | Proteasome core complex | 9.72 × 10−6 | 9.72 × 10−6 | 1.83 × 10−18 | |
| GO:0006511 | Up | BP | Ubiquitin-dependent protein catabolic process | 2.73 × 10−6 | 2.73 × 10−6 | 5.44 × 10−15 | |
| GO:0004175 | Up | MF | Endopeptidase activity | 1.39 × 10−5 | 1.39 × 10−5 | 1.43 × 10−11 | |
| Redox | |||||||
| GO:0016667 | Up | MF | Oxidoreductase activity | 1.36 × 10−9 | |||
| GO:0015035 | Up | MF | Protein disulfide oxidoreductase activity | 4.68 × 10−6 | |||
| GO:0045454 | Up | BP | Cell redox homeostasis | 2.99 × 10−5 | |||
| Transporters | |||||||
| GO:0022890 | Up | MF | Cation transmembrane transporter activity | 3.69 × 10−4 | |||
| GO:0022804 | Up | MF | Active transmembrane transporter activity | 1.04 × 10−5 | |||
| Protein kinase or phosphatase activity | |||||||
| GO:0004692 | Up | MF | Cyclic GMP-dependent protein kinase activity | 2.15 × 10−4 | |||
| GO:0004723 | Up | MF | Ca-dependent protein Ser/Thr phosphatase activity | 2.54 × 10−4 | |||
| GO:0004724 | Up | MF | Mg-dependent protein Ser/Thr phosphatase activity | 2.54 × 10−4 | |||
| GO:0008420 | Up | MF | Carboxy-terminal domain phosphatase activity | 2.54 × 10−4 | |||
| GO:0008597 | Up | MF | Ca-dependent protein Ser/Thr phosphatase regulator | 2.54 × 10−4 | |||
| Carbohydrate metabolism | |||||||
| GO:0005975 | Up | BP | Carbohydrate metabolic process | 5.22 × 10−3 | |||
| GO:0044262 | Up | BP | Cellular carbohydrate metabolic process | 1.53 × 10−2 | |||
| Photosynthesis | |||||||
| GO:0009765 | Down | BP | Photosynthesis, light harvesting | 3.33 × 10−20 | |||
| GO:0015979 | Down | BP | Photosynthesis | 7.73 × 10−11 | 3.51 × 10−7 | ||
| GO:0009654 | Down | CC | Oxygen-evolving complex | 3.99 × 10−6 | 8.71 × 10−5 | ||
| Protein synthesis | |||||||
| GO:0006412 | Down | BP | Translation | 8.85 × 10−6 | 1.68 × 10−4 | ||
| GO:0005840 | Down | CC | Ribosome | 7.31 × 10−7 | 9.82 × 10−6 | ||
| Tetrapyrrole synthesis | |||||||
| GO:0006779 | Down | BP | Porphyrin biosynthetic process | 2.61 × 10−4 | 7.33 × 10−5 | ||
| GO:0015995 | Down | BP | Chlorophyll biosynthetic process | 2.00 × 10−4 | |||
| GO:0033014 | Down | BP | Tetrapyrrole biosynthetic process | 2.18 × 10−4 | |||
| Other | |||||||
| GO:0005618 | Up | CC | Cell wall | 2.16 × 10−2 | |||
| GO:0006810 | Down | BP | Transport | 1.43 × 10−4 | |||
| GO:0044271 | Down | BP | Cellular nitrogen compound biosynthetic | 3.66 × 10−4 | |||
| GO:0004623 | Down | MF | Phospholipase A2 activity | 4.66 × 10−4 | |||
| GO:0006006 | Down | BP | Glucose metabolic process | 2.64 × 10−5 | |||
Reg, category regulation. Up, upregulation; Down, downregulation.
Class, classification. MF, molecular function; BP, biological process; CC, cellular component.
For GOSeq, significance was set at a P value of <0.05 and a false-discovery rate (FDR [Benjamini-Hochberg]) of <0.05 for up- and downregulation for nTiO2, nZnO, and QDs and downregulation for nAg. For upregulation of nAg, GOSeq significance was set at a P value of <0.03 and an FDR (BH) of <1.
Fig 3.

Major functional categories of transcripts altered by exposure to each NP type for functional categories with ≥5 transcripts whose expression was altered. (A) Number of transcripts whose abundance was elevated; (B) number of transcripts whose abundance was repressed. Transcripts whose annotated functions are not in these categories are grouped as “other,” and those with unknown or highly uncertain functions are categorized as “unknown.”
Proteasome subunit-encoding transcripts elevated by exposure to nTiO2, nZnO, and QDs.
The proteasome is the multisubunit proteolytic complex of the general protein disposal pathway of the cytosol, nucleus, and endoplasmic reticulum, called the ubiquitin proteasome system (UPS). The GO category for the proteasome (map03050) was detected following exposure to nTiO2 (P = 1.3 × 10−30), nZnO (P = 1.2 × 10−16), and QDs (P = 3.8 × 10−8) (Table 3; Fig. 3A), reflecting elevated levels of multiple transcripts encoding different subunits of the proteasome (Table 4; see Fig. S3 in the supplemental material). Of the 36 genes annotated as encoding proteasome subunits in the C. reinhardtii genome database, transcript abundances/levels elevated greater than 2.0 log2 fold were observed for 18 (50%), 12 (33%), and 7 (19%) genes in the transcriptomes produced in response to nTiO2, nZnO, and QDs, respectively (Table 4). The levels of the other proteasome subunit-encoding transcripts increased 1.5 to 2.0 log2 fold in the transcriptomes produced in response to each of these three NPs (as seen for the comparison group in Table 4). In addition, the levels of three transcripts encoding functions potentially related to the proteasome, RB60, CDC48, and UFD1, were elevated by more than 2 log2 fold in the transcriptome produced in response to nZnO (39). In contrast, in the transcriptome produced in response to nAg, only one transcript encoding a proteasome subunit was elevated. While many transcriptome profiling studies have revealed differentially regulated genes encoding proteolysis-related functions, only minor proportions of these transcripts encoded proteasome subunits (40, 41). Therefore, this concerted elevation of proteasome subunit transcripts is not a general stress response and may reflect the specific biological effects of these NP types.
Table 4.
Transcripts encoding proteasome subunits and related functions elevated after exposure to nTiO2, nZnO, and QDs but not to nAg
| Subunit and Augustus v5.0 identifier | Gene | Annotated function | Log2 fold changea |
|||
|---|---|---|---|---|---|---|
| TiO2 | ZnO | Ag | QDs | |||
| Proteasomeb | ||||||
| ≥2 log2 fold increase in expression | ||||||
| 512002 | RPN1 | 26S regulatory subunit | 2.47 | 2.51 | −0.01 | 1.94 |
| 517679 | RPN2 | 26S regulatory subunit | 2.49 | 2.69 | 1.35 | 2.65 |
| 523380 | RPN3 | 26S regulatory subunit | 2.31 | 2.31 | 0.19 | 1.82 |
| 514973 | RPN8 | 26S regulatory subunit | 2.05 | 1.94 | −0.13 | 1.58 |
| 516310 | RPN10 | 26S regulatory subunit | 1.93 | 2.23 | 0.13 | 0.70 |
| 510841 | RPN11 | 26S regulatory subunit | 2.40 | 2.85 | 0.54 | 2.58 |
| 517257 | RPN12 | 26S regulatory subunit | 1.69 | 2.38 | 0.78 | 0.82 |
| 515497 | RPT1 | 26S regulatory subunit | 2.01 | 1.14 | −0.55 | 1.12 |
| 524554 | RPT2 | 26S regulatory subunit | 2.62 | 1.85 | 0.28 | 1.83 |
| 517293 | RPT4 | 26S regulatory subunit | 2.10 | 2.86 | 0.72 | 2.29 |
| 517191 | POA1 | 20S alpha subunit A | 2.53 | 1.89 | −0.13 | 1.87 |
| 525502 | POA2 | 20S alpha subunit B | 2.58 | 2.38 | 0.04 | 2.02 |
| 511244 | POA4 | 20S alpha subunit D | 2.28 | 1.36 | 0.28 | 1.74 |
| 517599 | POA5 | 20S alpha subunit E | 2.09 | 2.09 | −0.81 | 1.87 |
| 523200 | 20S beta subunit 3 | 2.79 | 2.57 | −0.55 | 2.06 | |
| 523449 | 20S beta subunit A1 | 3.05 | 2.53 | 0.45 | 2.40 | |
| 509711 | 20S beta subunit B, type beta 2 | 3.02 | 3.04 | 0.62 | 2.23 | |
| 515181 | 20S beta subunit D, type 2 | 2.50 | 1.98 | −0.55 | 1.47 | |
| 523491 | 20S beta subunit, type 4 | 2.17 | 1.99 | 0.45 | 1.20 | |
| 525170 | 26S regulatory subunit N5 | 2.19 | 1.85 | 0.07 | 1.46 | |
| Comparison group | ||||||
| 518932 | RPN6 | 26S regulatory subunit | 1.22 | 1.69 | 0.39 | 1.00 |
| 514567 | RPN7 | 26S regulatory subunit | −0.05 | 0.72 | −0.17 | 0.19 |
| 515904 | RPN9 | 26S regulatory subunit | 1.29 | 1.07 | −0.84 | 1.41 |
| 510008 | RPT5 | 26S regulatory subunit | 1.94 | 1.76 | −0.02 | 1.29 |
| 521984 | RPT6 | 26S regulatory subunit | 0.14 | 0.40 | −1.78 | 0.17 |
| 519364 | ANK13 | 26S regulatory subunit 10 | −0.12 | 1.72 | 0.68 | 0.12 |
| 509576 | POA3 | 20S alpha subunit C | 1.66 | 0.87 | −0.39 | 0.44 |
| 523979 | POA6 | 20S alpha subunit F | 0.61 | 0.24 | −0.14 | 0.73 |
| 515372 | POA7 | 20S alpha subunit G | 1.85 | 1.04 | 0.52 | 0.42 |
| 522780 | 20S subunit beta 6 | 1.05 | 1.15 | −0.22 | 0.40 | |
| 513499 | 20S beta subunit A2 | 1.83 | 0.82 | −0.18 | 0.35 | |
| 510489 | 20S beta subunit E. type beta 5 | 1.69 | 1.69 | −0.67 | 1.12 | |
| 515638 | Activator subunit 4 | 0.64 | −0.69 | 0.26 | 0.29 | |
| 521702 | Component ECM29 | 1.47 | 0.72 | 0.45 | 0.00 | |
| 526137 | 26S regulatory subunit T3 | 1.91 | 1.24 | 0.23 | 1.54 | |
| Proteasome related | ||||||
| 518726 | RB60 | Protein disulfide isomerase 1 | 1.21 | 2.31 | −0.94 | 0.77 |
| 523252 | CDC48 | Protein degradation/cell cycle | 1.45 | 2.36 | −1.16 | 1.51 |
| 521048 | UFD1 | Ubiquitin fusion degradation | −1.51 | 2.23 | −2.29 | −1.09 |
| 514349 | Proteasome assembly chaperone | 0.47 | 1.31 | 2.26 | 1.29 | |
Boldface data indicate transcripts encoding proteasome subunits that are elevated by ≥2 log2 fold in at least one NP transcriptome.
Data for proteasome subunit transcripts are divided into data for transcripts encoding proteasome subunits that are elevated ≥2 log2 fold in the transcriptome produced into response to at least one NP and the remaining proteasome subunit transcripts for comparison.
Oxidative stress-responsive transcripts elevated by exposure to nZnO.
Metal toxicity is associated with oxidative stress as a result of complex adverse effects of excessive production of ROS (42). Because NPs have been proposed to elicit toxicity by releasing or delivering toxic metal to cells, it seemed likely that the transcriptomes produced in response to NP would have elevated levels of the transcripts that are known to be induced during oxidative stress and other stress conditions. In fact, the transcriptome produced in response to nZnO showed evidence of oxidative stress on the basis of the elevated levels of transcripts of three genes, GSTS1, HSP22C, and HSP70A, which are induced by excess H2O2 or singlet oxygen (1O2), or both ROS types (Table 5) (43). These effects may reflect H2O2-induced oxidative stress because there was no significant change in the level of the GPXH transcript, which is induced only by excess 1O2 (Table 5). For nZnO-regulated genes, the results were highly significant on the basis of the low P values for GO categories relating to redox homeostasis (Table 3). As additional evidence for stress induced by exposure to nZnO, the transcriptome produced in response to nZnO showed elevated levels of the transcripts of MSR1, MSR2, and HSP90. These proteins manage damaged and nonnative proteins that are known to arise during oxidative stress and other stress conditions (44).
Table 5.
Stress-responsive transcripts whose abundance was altered by ≥2 log2 fold in transcriptomes produced in response to at least one NP
| Function and Augustus v5.0 identifier | Gene | Annotated function | Log2 fold changea |
|||
|---|---|---|---|---|---|---|
| TiO2 | ZnO | Ag | QDs | |||
| 143122 | GPXH/5 | Glutathione peroxidase (cytochrome) | −0.36 | 0.92 | −1.44 | −0.01 |
| 193661 | GSTS1 | Glutathione S-transferase | −2.99 | 2.60 | −2.36 | −0.98 |
| 513036 | Glutathione S-transferase | −2.42 | −1.99 | 0.01 | −1.54 | |
| 524323 | HSP22A | Heat shock protein 22A | −3.16 | −1.33 | −3.50 | −1.93 |
| 864288 | HSP22C | Heat shock protein 22C | 0.00 | 3.87 | −1.14 | 0.10 |
| 525480 | HSP70A | Heat shock protein 70A | −0.79 | 2.12 | −3.06 | −0.21 |
| 525808 | HSP90A | Heat shock protein 90A | −0.39 | 1.56 | −2.35 | −0.38 |
| 518563 | HSP90B | Heat shock protein 90B | 4.65 | 4.00 | 0.68 | 1.74 |
| 518085 | CLPB1 | ClpB chaperone, Hsp100 family | −3.36 | 1.37 | −3.11 | −1.59 |
| 524078 | CPN60C | Chaperonin 60C | 0.56 | 1.14 | −2.12 | 0.79 |
| 514347 | MSR1 | Peptide Met sulfoxide reductase | 0.90 | 2.47 | 0.09 | 0.54 |
| 510552 | MSR5 | Peptide Met sulfoxide reductase | −0.58 | 2.51 | −2.18 | −0.12 |
| 525654 | MSRA3 | Peptide Met sulfoxide reductase | −0.43 | 0.87 | −2.03 | −0.08 |
| 511607 | GRX6 | Glutaredoxin, CGFS type | −4.16 | −1.40 | −2.17 | −2.92 |
| 522983 | PRX1 | 2-Cysteine peroxiredoxin | −0.75 | −1.21 | −2.55 | −1.07 |
| 510378 | Dehydroascorbate reductase | −2.04 | 2.25 | −1.35 | −0.55 | |
| 521048 | UFD1 | ERb stress/protein degradation | −1.51 | 2.23 | −2.29 | −1.09 |
| 523252 | CDC48 | ER stress/cell cycle | 1.45 | 2.36 | −1.16 | 1.51 |
| 525728 | Cytochrome P450 | −2.01 | −1.03 | −0.25 | −0.34 | |
| 525343 | LHCSR1 | Stress-related chlorophyll binding protein 1 | −1.13 | −2.52 | −3.68 | −1.11 |
Boldface data indicate transcripts that are elevated or reduced by ≥2 log2 fold in at least one NP transcriptome.
ER, endoplasmic reticulum.
Exposures to nTiO2, nAg, and QDs did not enhance the levels of transcripts encoding stress-related functions (GSTS1, GPXH, HSP22C, and HSP70A). In fact, in many cases, the levels of these transcripts were reduced (Table 5). Moreover, the transcriptomes produced in response to these three NPs also had reduced levels of the transcripts of several other genes that have known or predicted functions related to other stress conditions: oxidative stress (MSRA3, GRX6, PRX1), heat stress (HSP22A, HSP90A, HSP90B, CLPB1, CPN60C), unfolded protein response (CDC48 and Ufd1) (45), and high light-induced stress (LHCSR1) (46). The only exception was the induction of HSP90B by exposure to nTiO2 and, to a lesser extent, to nZnO (Table 5). Evidently, only exposure to nZnO resulted in H2O2-induced oxidative stress, while exposure to the other three NPs did not induce a known stress response at the level of the transcriptome.
Elevated transcripts encoding cell wall components in response to exposure to nAg.
Exposure to nAg dramatically elevated the levels of eight transcripts encoding known or predicted hydroxyproline-rich glycoprotein components of the cell wall (hydroxyproline-rich glycoproteins and pherophorins) and a glycoside hydrolase, which may be involved in cell wall remodeling (Table 6) (47). The transcriptome produced in response to nAg also had elevated levels of 21 transcripts encoding components of the flagella (Table 6) (26). These effects seemed significant, on the basis of the number of genes and their levels of induction, although only marginally higher P values were obtained for the GO category cell wall (Table 3). This discrepancy probably reflects the fact that this category is based on the cell wall of vascular plants, which is composed of cellulose and other carbohydrate polymers. The C. reinhardtii cell wall lacks these polymers and is primarily composed of hydroxyproline-rich glycoproteins, encoded by the transcripts that were elevated during nAg exposure (48). These results suggest that remodeling or repair of the cell wall and flagella occurs in response to the Ag NP-induced damage.
Table 6.
Levels of transcripts encoding proteins that function in the cell wall and in the flagella
| Function and Augustus v5.0 identifier | Gene | Annotated function | Log2 fold changea |
|||
|---|---|---|---|---|---|---|
| TiO2 | ZnO | Ag | QDs | |||
| Cell wall | ||||||
| 517461 | PHC1 | Cell wall pherophorin-C-1 | 0.88 | 0.14 | 3.28 | 0.29 |
| 517016 | PHC19 | Cell wall pherophorin-C19 | 0.21 | −0.28 | 3.48 | −0.88 |
| 512243 | PHC12 | Cell wall pherophorin-C12 | 0.79 | 0.04 | 2.45 | −0.30 |
| 517021 | PHC22 | Cell wall pherophorin-C22 | 1.05 | −0.28 | 4.88 | −0.30 |
| 512446 | GAS28 | Hydroxyproline-rich glycoprotein | 1.87 | 1.00 | 4.67 | 0.48 |
| 512449 | GAS30 | Hydroxyproline-rich glycoprotein | 1.34 | 0.72 | 5.13 | 1.44 |
| 518750 | Hydroxyproline-rich glycoprotein | −1.70 | 1.36 | 2.21 | −1.15 | |
| 517295 | Structural constituent of cell wall | 1.32 | 0.21 | 4.21 | −0.04 | |
| 520047 | Predicted glycoside hydrolase | 0.79 | 0.04 | 3.04 | −1.30 | |
| Flagella | ||||||
| 510365 | AGG3 | Flagellar flavodoxin | 1.34 | 0.46 | 1.78 | 2.20 |
| 523252 | CDC48 | Flagellum associated | 1.45 | 2.36 | −1.16 | 1.51 |
| 518827 | DLC1 | Flagellar dynein light chain | 0.47 | 0.92 | 2.28 | 0.77 |
| 513514 | FAP7 | Flagellum associated | 1.72 | 0.04 | 2.26 | 0.87 |
| 521955 | FAP26 | Flagellum associated | 1.05 | 0.18 | 2.50 | −0.13 |
| 523078 | FAP71 | Flagellum associated | 2.47 | 1.22 | 1.78 | 1.36 |
| 512516 | FAP83 | Flagellum associated | 1.51 | 1.09 | 2.58 | 0.90 |
| 511879 | FAP96 | Flagellum associated | 2.11 | 1.18 | 2.41 | 0.70 |
| 511324 | FAP109 | Flagellum associated | 1.79 | 1.53 | 2.38 | 1.36 |
| 513604 | FAP126 | Flagellum associated | 2.41 | 1.41 | 0.74 | 1.58 |
| 524055 | FAP141 | Flagellum associated | 0.28 | −0.28 | 2.62 | 0.70 |
| 513171 | FAP169 | Flagellum associated | 1.70 | 2.18 | 2.47 | 1.19 |
| 522771 | FAP205 | Flagellum associated | 1.01 | 0.17 | 2.71 | 1.00 |
| 524527 | FAP207 | Flagellum associated | 0.98 | 1.37 | 2.23 | 0.59 |
| 514913 | FAP236 | Flagellum associated | 1.64 | 0.31 | 2.38 | 1.16 |
| 520262 | FAP258 | Flagellum associated | 0.64 | −0.69 | 2.54 | 0.02 |
| 519991 | FAP265 | Flagellum associated | 1.13 | 0.31 | −0.55 | 2.07 |
| 516031 | FAP268 | Flagellum associated | 0.69 | 0.14 | 2.80 | −0.08 |
| 521526 | FAP283 | Flagellum associated | 0.21 | 1.04 | 2.48 | 0.70 |
| 517923 | FAP293 | Flagellum associated | 0.85 | −0.79 | 2.38 | 0.47 |
| 518760 | IFT20 | Intraflagellar transport | 0.95 | 1.65 | 2.22 | 0.70 |
Boldface data indicate transcripts that are elevated or reduced by ≥2 log2 fold in at least one NP transcriptome.
Protein phosphatases, protein kinases, and transporters.
Many transcripts could not be grouped into a function-ontology category with other differentially regulated transcripts by the use of KEGG or GO or manually. In other cases, transcripts could be grouped into categories on the basis of biochemical activity, but the grouping was with genes with no apparent common physiological or cell biological function. For example, among the transcripts with elevated levels of transcription, some encoded predicted protein kinases, phosphatases, and transporters. Similarly, it was not possible to discern a functional relationship between the four GO categories of altered transcripts: protein synthesis, carbohydrate metabolism, protein kinase or phosphatase activity, and transport (Table 3). These groupings may have been fortuitous, reflecting indirect effects of NP exposure and the higher probability that this noise will identify GO categories with major numbers of genes, e.g., transporters (26). For example, transcriptome profiling studies generally reveal the effects on expression of genes that function in protein modification and transmembrane transport of small molecules.
Photosynthesis and related genes downregulated by NP exposure.
A remarkable downregulation in the transcriptome produced in response to nTiO2 was detected for many transcripts with functions related to photosynthesis (Fig. 3B; Table 7). Intermediate effects were detected in the transcriptomes produced in response to nAg and QDs, and slight effects were observed in the transcriptome produced in response to nZnO. These transcripts encode subunits of the oxygen-evolving complex, light-harvesting complex (LHC) proteins, two early light-inducible proteins, two proteins involved in CO2 metabolism, and several enzymes involved in the biosynthesis of tetrapyrroles (both heme and chlorophyll) and the branch that is specific to chlorophyll (49). Finally, although downregulation was observed for transcripts encoding proteins involved in translation, carbohydrate metabolism, metal homeostasis, and amino acid metabolism (Table 7), it not is possible to speculate on any potential significance of these effects.
Table 7.
Transcripts whose abundance decreased by ≥2 log2 fold in transcriptomes produced in response to at least one NPa
| Function and Augustus v5.0 identifier | Gene | Annotated function | Log2 fold changeb |
|||
|---|---|---|---|---|---|---|
| TiO2 | ZnO | Ag | QDs | |||
| Photosynthesis | ||||||
| 519678 | PSBO | O2-evolving complex protein 1 | −1.85 | −0.63 | −0.89 | −2.03 |
| 513916 | PSBP1 | O2-evolving complex protein 2 | −3.13 | −1.21 | −2.11 | −2.75 |
| 523315 | LHCA8 | LHC I protein | −2.25 | −1.11 | −0.71 | −1.36 |
| 523554 | LHCBM4 | LHC II chlorophyll binding | −2.01 | −1.00 | −1.03 | −1.80 |
| 520575 | LHCBM5 | LHC II chlorophyll binding | −2.08 | −1.06 | −1.12 | −1.86 |
| 523580 | LHCBM6 | LHC II chlorophyll binding | −2.05 | −1.04 | −1.10 | −1.81 |
| 523560 | LHCBM8 | LHC II chlorophyll binding | −2.07 | −1.09 | −1.08 | −1.86 |
| 523559 | LHCBM9 | LHC II chlorophyll binding | −2.14 | −0.77 | −1.00 | −1.71 |
| 516517 | LHCB5 | LHC II chlorophyll binding | −2.84 | −0.23 | −1.08 | −2.65 |
| 517104 | FDX5 | Apoferredoxin | −3.50 | 1.60 | −1.29 | −0.90 |
| 519631 | ELI3 | Early light-inducible (LHC like) | −2.28 | −0.76 | −2.64 | −0.54 |
| 518197 | LHL3 | Early light-induced protein | −3.42 | 0.04 | −1.11 | −3.18 |
| Tetrapyrrole and chlorophyll synthesis | ||||||
| 526010 | PPX1 | Protoporphyrinogen oxidase | −3.08 | 0.24 | −1.51 | −2.30 |
| 518465 | UROD2 | Uroporphyrinogen decarboxylase | −1.79 | −0.20 | −1.55 | −2.01 |
| 518660 | CPX1 | Coproporphyrinogen III oxidase | −3.21 | −0.33 | −1.56 | −3.21 |
| 522598 | CHLD | Mg chelatase subunit D | −4.18 | −1.40 | −2.03 | −4.24 |
| 524023 | CHLI1 | Mg chelatase subunit I | −1.79 | 0.74 | −1.06 | −2.12 |
| 511675 | Geranylgeranyl reductase | −2.35 | 0.33 | −0.24 | −2.30 | |
| CO2 metabolism | ||||||
| 522130 | CCP1 | Low-CO2-inducible protein | −0.40 | −0.32 | −3.19 | −0.12 |
| 526413 | CAH3 | Carbonic anhydrase 3 | −0.92 | −1.96 | −2.46 | −0.67 |
| Translation/protein synthesis | ||||||
| 512819 | RPS7 | 80S ribosomal protein S7 | −2.05 | −0.43 | −1.32 | −0.14 |
| 514858 | RPL21 | 80S ribosomal protein L21 | −2.62 | −0.79 | −2.93 | −1.05 |
| 522290 | 7Sb | Signal recognition particle 7S RNA | −0.35 | −1.69 | −2.49 | 0.30 |
| 524211 | GTS2 | Gln-Glu-tRNA synthetase | −2.59 | 0.27 | −0.94 | −2.30 |
| Metal related | ||||||
| 524896 | CRD1 | Cu response defect 1 protein | −2.55 | 0.07 | −1.80 | −2.94 |
| 526016 | HYDA2 | Iron hydrogenase | −2.17 | 1.60 | 0.67 | −2.94 |
| 512933 | NFU2 | Fe-S cluster assembly protein | −1.39 | 0.03 | −2.21 | 0.04 |
| 509912 | CTR2 | CTR type (copper ion transporter) | −2.21 | 0.63 | −0.26 | −0.65 |
| Carbohydrate metabolism | ||||||
| 511538 | PFL1 | Pyruvate-formate lyase | −1.67 | 0.83 | 0.27 | −2.01 |
| 518335 | ADH1 | Alcohol/acetaldehyde dehydrogenase | −1.48 | 0.04 | −0.01 | −2.37 |
| 518684 | Fructose 2,6-bisphosphatase | −0.43 | −0.18 | −2.18 | 0.19 | |
| 520009 | PYK5 | Pyruvate kinase | −1.85 | −0.32 | 0.13 | −2.30 |
| 520975 | PGI1 | Phosphoglucose isomerase | −1.47 | −0.48 | −2.03 | −0.90 |
| 524333 | GWD1 | Alpha-glucan water dikinase | −1.07 | 0.17 | 0.86 | −2.05 |
| 523930 | GTR12 | Glycosyltransferase | −2.66 | −1.90 | −1.88 | −1.59 |
| 526398 | ELG33 | Exostosin-like glycosyltransferase | −4.15 | −1.63 | −1.96 | −3.67 |
| Amino acid metabolism | ||||||
| 510278 | THS1 | Threonine synthase | −2.44 | 0.21 | −1.34 | −3.47 |
| 510279 | AAT1 | Alanine aminotransferase | −0.29 | −0.41 | −2.25 | 0.41 |
| 521080 | METE | B12-independent Met synthase | −3.24 | −1.27 | −3.18 | −3.63 |
| 522837 | METH2 | B12-dependent Met synthase | −1.16 | −0.46 | −0.70 | −2.39 |
See also Table 5 for transcripts with stress-related functions.
Boldface data indicate transcripts that are elevated or reduced by ≥2 log2 fold in at least one NP transcriptome.
DISCUSSION
Our results revealed that each of the four NPs used in this study has both common and distinct effects on the transcriptome of C. reinhardtii and, hence, its physiology. One distinct effect of NP was that only the exposure to nZnO appeared to alter the level of transcription of transcripts with functions in oxidative stress, including three genes that are markers for excess levels of the primary ROS: H2O2 and 1O2 (Table 5). It was somewhat surprising that only nZnO induced oxidative stress since both nAg and the QDs have been shown to release significant concentrations of Ag+ and Cd2+ (12, 50), both of which are toxic to C. reinhardtii (31, 33). The lack of induction of the stress-responsive genes observed (Table 5) (51) suggests a relatively limited toxicity of nTiO2, nAg, and QDs at these concentrations. Exposure to nAg resulted in elevated levels of transcripts encoding proteins of the cell wall and flagella (Table 6), suggesting that, of the four NP types, nAg was the NP that induced the greatest damage to cell structures.
Many NP-specific effects were observed, suggesting that these NPs differ in many of their modes of action in C. reinhardtii (Fig. 2 and 3; Tables 2 to 7). The four transcriptomes that occurred in response to NPs share only one common altered transcript, which argues against a general nanotoxicity in C. reinhardtii (Fig. 2a).
Exposure to nTiO2, nZnO, or QDs dramatically increased the levels of several transcripts encoding subunits of the 26S proteasome (Table 4; see Fig. S3 in the supplemental material). These changes appear to be analogous to the proteasome stress response and the ubiquitin stress response, which alter the subunit composition and biochemical activities of the cellular population of proteasome complexes for as yet unknown reasons (52). Moreover, the data reveal that this is a significant response by comparison to documented transcriptomic responses with known functional significance; more than 80% of the proteome subunits were upregulated 1.5-fold or greater following exposure to at least one particle. It therefore most probably does represent some as yet known remodeling of the proteasome population or enhanced proteasome biogenesis. Global gene expression studies in diverse organisms show similar effects in response to inhibition of the proteasome, e.g., by drugs that specifically target its protease activities, an alkylating agent, and certain metals (53). Together, these results raise the intriguing possibility that exposure to nTiO2, nZnO, or QDs results in proteasome inhibition. Indeed, exposure to certain metal ions [Cd, methyl-Hg, Cu, and As(III)] has been shown to result in proteasome inhibition, enhancing the expression of proteasome subunits in several organisms (54–56). The proteasome has a key role in metal resistance in maize and S. cerevisiae yeast (57). Nonetheless, it is unlikely that the metals released by the NPs in this study were responsible for the elevated levels of the proteasome subunit transcripts. The activation of oxidative stress responses that are generally associated with metal ion toxicity was not observed in the transcriptomes produced in response to nTiO2 and QDs (Tables 3 and 6) (58). Moreover, QDs and the ionic form of their heavy metal component, Cd, have very different transcriptomic effects in C. reinhardtii (12). Notably, Cd exposure did not elevate the levels of transcripts encoding proteasome subunits. Finally, previous studies found that metal exposure induced far fewer proteasome genes than the numbers shown here to be induced by nTiO2, nZnO, and QDs (Table 4) (41, 59). Interestingly, proteasome inhibition in mammalian tumor cells has been reported to be caused by “metal-containing species with asymmetric ligands containing the methylpyridin-amino-methylphenol moiety” (55). These molecules, like other inhibitors of the proteasome, are widely used in cancer chemotherapy (55). This effect on the proteolytic activities of the proteasome is direct and requires specific chemical properties of these molecules and not simply the release of the component metal. Proteasome inhibition is used in chemotherapy of multiple myeloma and has been proposed to underlie the initiation and progression of Alzheimer's disease (54, 60). Further work is required to determine whether nTiO2, nZnO, and QDs inhibit the proteasome and whether they could be used as cancer chemotherapy agents. This induction of genes encoding proteasome transcripts (Table 4) is similar to the coordinated induction of a battery of genes encoding proteasome subunits in the yeast Saccharomyces cerevisiae and mammalian cells (61–64). In addition, exposure of S. cerevisiae to nZnO induced three homologous genes regulated by proteasome inhibition (RB60, CDC48, and UFD1) (65). Two of these genes were also marginally induced during the nTiO2 and QD exposures. These results suggest that C. reinhardtii has a genetic regulatory circuitry to control the rate of proteasome biogenesis which is analogous to the systems in yeast and mammalian cells.
Decreased transcript levels of genes with photosynthesis-related functions were found in the transcriptome produced in response to nTiO2 and, to lesser degrees, in the transcriptomes produced in response to the three other NPs (Table 7). These changes resemble the responses to either changes in light intensity (66, 67) or impaired chloroplast physiology (68, 69) which have been described in plants and algae. For example, C. reinhardtii cells that experienced an increase in light intensity repressed the expression of the genes encoding many of the same genes; several LHCBM genes encoding LHC II subunits; LHL3, one of three LHC II-like genes; and CHLD and CHLI1. These results suggest that nTiO2 has a similar effect in C. reinhardtii as a transition from low to high intensity light or impaired chloroplast physiology, or both conditions.
Supplementary Material
ACKNOWLEDGMENTS
Funding for this work was provided by the Natural Sciences and Engineering Research Council of Canada (NSERC), the Fonds Québécois de la Recherche sur la Nature et les Technologies (FQRNT), and the Fundação para a Ciência e Tecnologia (FCT; Portugal; postdoctoral and Science 2008 IST-CQE3 environmental chemistry assistant researcher positions to R.F.D. and project PTDC/AAC-AMB/111998/2009).
We thank Pierre Chagnon and Patrick Gendron (Genomics Core Facility, University of Montreal) for RNA-seq and Leo Diaz Lozada, an M.Sc. student, for the AUC analysis.
Footnotes
Published ahead of print 31 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00998-13.
REFERENCES
- 1. Chen L, Zhou L, Liu Y, Deng S, Wu H, Wang G. 2012. Toxicological effects of nanometer titanium dioxide (nano-TiO2) on Chlamydomonas reinhardtii. Ecotoxicol. Environ. Saf. 84:155–162 [DOI] [PubMed] [Google Scholar]
- 2. Ivask A, Bondarenko O, Jepihhina N, Kahru A. 2010. Profiling of the reactive oxygen species-related ecotoxicity of CuO, ZnO, TiO2, silver and fullerene nanoparticles using a set of recombinant luminescent Escherichia coli strains: differentiating the impact of particles and solubilised metals. Anal. Bioanal. Chem. 398:701–716 [DOI] [PubMed] [Google Scholar]
- 3. Pujalté I, Passagne I, Brouillaud B, Tréguer M, Durand E, Ohayon-Courtès C, L'Azou B. 2011. Cytotoxicity and oxidative stress induced by different metallic nanoparticles on human kidney cells. Part. Fibre Toxicol. 8:10–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Griffitt TJ, Weil R, Hyndman KA, Denslow ND, Powers K, Taylor D, Barber DS. 2007. Exposure to copper nanoparticles causes injury and acute lethality in zebrafish (Danio rerio). Environ. Sci. Technol. 41:8178–8186 [DOI] [PubMed] [Google Scholar]
- 5. Nair PMG, Choi J. 2011. Characterization of a ribosomal protein L15 cDNA from Chironomus riparius (Diptera; Chironomidae): transcriptional regulation by cadmium and silver nanoparticles. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 159:157–162 [DOI] [PubMed] [Google Scholar]
- 6. Chen M, Zhang M, Borlak J, Tong W. 2012. A decade of toxicogenomic research and its contribution to toxicological science. Toxicol. Sci. 130:217–228 [DOI] [PubMed] [Google Scholar]
- 7. Wang Z, Gerstein M, Snyder M. 2009. RNA-seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10:57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. González-Ballester D, Casero D, Cokus S, Pellegrini M, Merchant SS, Grossman AR. 2010. RNA-seq analysis of sulfur-deprived Chlamydomonas cells reveals aspects of acclimation critical for cell survival. Plant Cell 22:2058–2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miller R, Wu G, Deshpande RR, Vieler A, Gartner K, Li X, Moellering ER, Zäuner S, Cornish AJ, Liu B, Bullard B, Sears BB, Kuo M-H, Hegg EL, Shachar-Hill Y, Shiu S-H, Benning C. 2010. Changes in transcript abundance in Chlamydomonas reinhardtii following nitrogen deprivation predict diversion of metabolism. Plant Physiol. 154:1737–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maier T, Guell M, Serrano L. 2009. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 583:3966–3973 [DOI] [PubMed] [Google Scholar]
- 11. de Sousa Abreu R, Penalva LO, Marcotte E, Vogel C. 2009. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 5:1512–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Domingos RF, Simon DF, Hausser C, Wilkinson KJ. 2011. Bioaccumulation and effects of CdTe/CdS quantum dots on Chlamydomonas reinhardtii—nanoparticles or the free ions? Environ. Sci. Technol. 45:7664–7669 [DOI] [PubMed] [Google Scholar]
- 13. Hutchins CM, Simon DF, Zerges W, Wilkinson KJ. 2010. Transcriptomic signatures in Chlamydomonas reinhardtii as Cd biomarkers in metal mixtures. Aquat. Toxicol. 100:120–127 [DOI] [PubMed] [Google Scholar]
- 14. Jamers A, Blust R, Coen WD. 2009. Omics in algae: paving the way for a systems biological understanding of algal stress phenomena? Aquat. Toxicol. 92:114–121 [DOI] [PubMed] [Google Scholar]
- 15. Simon DF, Descombes P, Zerges W, Wilkinson KJ. 2008. Global expression profiling of Chlamydomonas reinhardtii exposed to trace levels of free cadmium. Environ. Toxicol. Chem. 27:1668–1675 [DOI] [PubMed] [Google Scholar]
- 16. Piccinno F, Gottschalk F, Seeger S, Nowack B. 2012. Industrial production quantities and uses of ten engineered nanomaterials in Europe and the world. J. Nanopart. Res. 14:1109–1120 [Google Scholar]
- 17. Domingos RF, Tufenkji N, Wilkinson KJ. 2009. Aggregation of titanium dioxide nanoparticles: role of a fulvic acid. Environ. Sci. Technol. 43:1282–1286 [DOI] [PubMed] [Google Scholar]
- 18. Domingos RF, Baalousha MA, Ju-Nam Y, Reid MM, Tufenkji N, Lead JR, Leppard GG, Wilkinson KJ. 2009. Characterizing manufactured nanoparticles in the environment: multimethod determination of particle sizes. Environ. Sci. Technol. 43:7277–7284 [DOI] [PubMed] [Google Scholar]
- 19. Peulen TO, Wilkinson KJ. 2011. Diffusion of nanoparticles in a biofilm. Environ. Sci. Technol. 45:3367–3373 [DOI] [PubMed] [Google Scholar]
- 20. Domingos RF, Franco C, Pinheiro JP. Stability of core/shell quantum dots—role of pH and small organic ligands. Environ. Sci. Pollut. Res. Int., in press [DOI] [PubMed] [Google Scholar]
- 21. Carney RP, Kim JY, Qian H, Jin R, Mehenni H, Stellacci F, Bakr OM. 2011. Determination of nanoparticle size distribution together with density or molecular weight by 2D analytical ultracentrifugation. Nat. Commun. 2:335–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Elson EL, Magde D. 1974. Fluorescence correlation spectroscopy. I. Conceptual basis and theory. Biopolymers 13:1–27 [DOI] [PubMed] [Google Scholar]
- 23. Balnois E, Papastavrou G, Wilkinson KJ. 2007. Force microscopy and force measurements of environmental colloids, p 405–467 In Wilkinson KJ, Lead J. (ed), Environmental colloids: behaviour, structure and characterisation, vol 10 John Wiley & Sons, Ltd., Chichester, United Kingdom [Google Scholar]
- 24. Harris ER. 1989. Culture and storage method in the Chlamydomonas sourcebook—a comprehensive guide to biology and laboratory use. Academic Press, San Diego, CA: [DOI] [PubMed] [Google Scholar]
- 25. Kola H, Laglera LM, Parthasarathy N, Wilkinson KJ. 2004. Cadmium adsorption by Chlamydomonas reinhardtii and its interaction with the cell wall proteins. Environ. Chem. 1:172–179 [Google Scholar]
- 26. Merchant SS, Prochnik SE, Vallon O, Harris EH, Karpowicz SJ, Witman GB, Terry A, Salamov A, Fritz-Laylin LK, Maréchal-Drouard L, Marshall WF, Qu L-H, Nelson DR, Sanderfoot AA, Spalding MH, Kapitonov VV, Ren Q, Ferris P, Lindquist E, Shapiro H, Lucas SM, Grimwood J, Schmutz J, Cardol P, Cerutti H, Chanfreau G, Chen CL, Cognat V, Croft MT, Dent R, Dutcher S, Fernández E, Fukuzawa H, González-Ballester D, González-Halphen D, Hallmann A, Hanikenne M, Hippler M, Inwood W, Jabbari K, Kalanon M, Kuras R, Lefebvre PA, Lemaire SD, Lobanov AV, Lohr M, Manuell A, Meier I, Mets L, Mittag M, et al. 2007. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318:245–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H, Ruan J, Durbin R. 2008. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 18:1851–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol. 11:R106–R118. 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bradford JR, Hey Y, Yates T, Li Y, Pepper SD, Miller CJ. 2010. A comparison of massively parallel nucleotide sequencing with oligonucleotide microarrays for global transcription profiling. BMC Genomics 11:282–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Young MD, Wakefield MJ, Smyth GK, Oshlack A. 2010. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11:R14–R26. 10.1186/gb-2010-11-2-r14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Navarro E, Piccapetra F, Wagner B, Marconi F, Kaegi R, Odzak N, Sigg L, Behra R. 2008. Toxicity of silver nanoparticles to Chlamydomonas reinhardtii. Environ. Sci. Technol. 42:8959–8964 [DOI] [PubMed] [Google Scholar]
- 32. Franklin NM, Rogers NJ, Apte SC, Batley CE, Gadd GE, Casey PS. 2007. Comparative toxicity of nanoparticulate ZnO, bulk ZnO, and ZnCl2 to a freshwater microalga (Pseudokirchneriella subcapitata): the importance of particle solubility. Environ. Sci. Technol. 41:8484–8490 [DOI] [PubMed] [Google Scholar]
- 33. Wang J, Zhang X, Chen Y, Sommerfeld M, Hu Q. 2008. Toxicity assessment of manufactured nanomaterials using the unicellular green alga Chlamydomonas reinhardtii. Chemosphere 73:1121–1128 [DOI] [PubMed] [Google Scholar]
- 34. Domingos RF, Peyrot C, Wilkinson KJ. 2010. Aggregation of titanium dioxide nanoparticles: role of calcium and phosphate. Environ. Chem. 7:61–66 [Google Scholar]
- 35. Pasqui D, Golini L, Giovampaola CD, Atrei A, Barbucci R. 2011. Chemical and biological properties of polysaccharide-coated titania nanoparticles: the key role of proteins. Biomacromolecules 11:1243–1249 [DOI] [PubMed] [Google Scholar]
- 36. Fatisson J, Quevedo IR, Wilkinson KJ, Tufenkji N. 2012. Physicochemical characterization of engineered nanoparticles under physiological conditions: effect of culture media components and particle surface coating. Colloids Surf. B Biointerfaces 91:198–204 [DOI] [PubMed] [Google Scholar]
- 37. Wilkinson KJ, Buffle J. 2004. Critical evaluation of physicochemical parameters and processes for modeling the biological uptake of trace metals in environmental (aquatic) systems, p 445–533 In van Leeuwen H, Koester W. (ed), Physicochemical kinetics and transport at chemical-biological interphases. John Wiley & Sons Ltd., Chichester, United Kingdom [Google Scholar]
- 38. Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. 2012. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40:D109–D114. 10.1093/nar/gkr988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stolz A, Hilt W, Buchberger A, Wolf DH. 2011. Cdc48: a power machine in protein degradation. Trends Biochem. Sci. 36:515–523 [DOI] [PubMed] [Google Scholar]
- 40. Castruita M, Casero D, Karpowicz SJ, Kropat J, Vieler A, Hsieh SI, Yan W, Cokus S, Loo JA, Benning C, Pellegrini M, Merchant SS. 2011. Systems biology approach in Chlamydomonas reveals connections between copper nutrition and multiple metabolic steps. Plant Cell 23:1273–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jamers A, Van der Ven K, Moens L, Robbens J, Potters G, Guisez U, Blust R, de Coen W. 2006. Effect of copper exposure on gene expression profiles in Chlamydomonas reinhardtii based on microarray analysis. Aquat. Toxicol. 80:249–260 [DOI] [PubMed] [Google Scholar]
- 42. Sharma SS, Dietz K-J. 2008. The relationship between metal toxicity and cellular redox imbalance. Trends Plant Sci. 14:43–50 [DOI] [PubMed] [Google Scholar]
- 43. Fischer BB, Krieger-Liszkay A, Eggen RIL. 2005. Oxidative stress induced by the photosensitizers neutral red (type I) or rose bengal (type II) in the light causes different molecular responses in Chlamydomonas reinhardtii. Plant Sci. 168:747–759 [Google Scholar]
- 44. Hartl FU, Bracher A, Hayer-Hartl M. 2011. Molecular chaperones in protein folding and proteostasis. Nature 475:324–332 [DOI] [PubMed] [Google Scholar]
- 45. Eisele F, Schafer A, Wolf DH. 2010. Ubiquitylation in the ERAD pathway. Conjugation and deconjugation of ubiquitin family modifiers. Subcell. Biochem. 54:136–148 [DOI] [PubMed] [Google Scholar]
- 46. Mou S, Zhang X, Ye N, Dong M, Liang C, Liang Q, Miao J, Xu D, Zheng Z. 2012. Cloning and expression analysis of two different LhcSR genes involved in stress adaptation in an Antarctic microalga, Chlamydomonas sp. ICE-L. Extremophiles 16:193–203 [DOI] [PubMed] [Google Scholar]
- 47. Baba M, Suzuki I, Shiraiwa Y. 2011. Proteomic analysis of high-CO2-inducible extracellular proteins in the unicellular green alga, Chlamydomonas reinhardtii. Plant Cell Physiol. 52:1302–1314 [DOI] [PubMed] [Google Scholar]
- 48. Harris EH. 2001. Chlamydomonas as a model organism. Annu. Rev. Plant Physiol. Plant Mol. Biol. 52:363–406 [DOI] [PubMed] [Google Scholar]
- 49. Tanaka A. 2007. Photosynthetic activity in winter needles of the evergreen tree Taxus cuspidata at low temperatures. Tree Physiol. 27:641–648 [DOI] [PubMed] [Google Scholar]
- 50. Hadioui M, Leclerc S, Wilkinson KJ. 2013. Multimethod quantification of Ag+ release from nanosilver. Talanta 105:15–19 [DOI] [PubMed] [Google Scholar]
- 51. Bouldin JL, Ingle TM, Sengupta A, Alexander R, Hannigan RE, Buchanan RA. 2008. Aqueous toxicity and food chain transfer of quantum dots in freshwater algae and Ceriodaphnia dubia. Environ. Toxicol. Chem. 27:1958–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hanna J, Finley D. 2007. A proteasome for all occasions. FEBS Lett. 581:2854–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Naujokat C, Fuchs D, Berges C. 2007. Adaptive modification and flexibility of the proteasome system in response to proteasome inhibition. Biochim. Biophys. Acta 1773:1389–1397 [DOI] [PubMed] [Google Scholar]
- 54. Goldberg AL. 2012. Development of proteasome inhibitors as research tools and cancer drugs. J. Cell Biol. 199:583–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Verani CN. 2012. Metal complexes as inhibitors of the 26S proteasome in tumor cells. J. Inorg. Biochem. 106:59–67 [DOI] [PubMed] [Google Scholar]
- 56. Yu X, Robinson JF, Sidhu JS, Hong S, Faustman EM. 2010. A system-based comparison of gene expression reveals alterations in oxidative stress, disruption of ubiquitin-proteasome system and altered cell cycle regulation after exposure to cadmium and methylmercury in mouse embryonic fibroblast. Toxicol. Sci. 114:356–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Forzani C, Lobréaux S, Mari S, Briat J-F, Lebrun N. 2002. Metal resistance in yeast mediated by the expression of a maize 20S proteasome alpha subunit. Gene 293:199–204 [DOI] [PubMed] [Google Scholar]
- 58. Stoiber TL, Shafer MM, Armstrong DE. 22 July 2011. Induction of reactive oxygen species in Chlamydomonas reinhardtii in response to contrasting trace metal exposures. Environ. Toxicol. [Epub ahead of print.] 10.1002/tox.20743 [DOI] [PubMed] [Google Scholar]
- 59. Burton NC, Guilarte TR. 2009. Manganese neurotoxicity: lessons learned from longitudinal studies in nonhuman primates. Environ. Health Perspect. 117:325–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hegde AN, Upadhya SC. 2011. Role of ubiquitin-proteasome-mediated proteolysis in nervous system disease. Biochim. Biophys. Acta 1809:128–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dohmen RJ, Willers I, Marques AJ. 2007. Biting the hand that feeds: Rpn4-dependent feedback regulation of proteasome function. Biochim. Biophys. Acta 1773:1599–1604 [DOI] [PubMed] [Google Scholar]
- 62. Karpov DS, Osipov SA, Preobrazhenskaya OV, Karpov VL. 2008. Rpn4p is a positive and negative transcriptional regulator of the ubiquitin-proteasome system. Mol. Biol. 42:456–462 [PubMed] [Google Scholar]
- 63. Karpov DS, Tyutyaeva VV, Beresten SF, Karpov VL. 2008. Mapping of the Rpn4p regions responsible for transcriptional activation of proteasome genes. Mol. Biol. 42:463–468 [PubMed] [Google Scholar]
- 64. Wang X, Xu H, Ha S-W, Ju D, Xie Y. 2010. Proteasomal degradation of Rpn4 in Saccharomyces cerevisiae is critical for cell viability under stressed conditions. Genetics 184:335–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mannhaupt G, Schnall R, Karpov V, Vetter I, Feldmann H. 1999. Rpn4p acts as a transcription factor by binding to PACE, a nonamer box found upstream of 26S proteasomal and other genes in yeast. FEBS Lett. 450:27–34 [DOI] [PubMed] [Google Scholar]
- 66. Leister D, Wang X, Haberer G, Mayer KFX, Kleine T. 2011. Intracompartmental and intercompartmental transcriptional networks coordinate the expression of genes for organellar functions. Plant Physiol. 157:386–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Teramoto H, Itoh T, Ono T-A. 2004. High-intensity-light-dependent and transient expression of new genes encoding distant relatives of light-harvesting chlorophyll-a/b proteins in Chlamydomonas reinhardtii. Plant Cell Physiol. 45:1221–1232 [DOI] [PubMed] [Google Scholar]
- 68. Jung H-S, Chory J. 2010. Signaling between chloroplasts and the nucleus: can a system biology approach bring clarity to a complex and highly regulated pathway? Plant Physiol. 152:453–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kleine T, Voigt C, Leister D. 2009. Plastid signalling to the nucleus: messengers still lost in the mists? Trends Genet. 25:185–192 [DOI] [PubMed] [Google Scholar]
- 70. Domingos RF, Rafiei Z, Monteiro CE, Khan MAK, Wilkinson KJ. Agglomeration and dissolution of zinc oxide nanoparticles: role of pH, ionic strength and fulvic acid. Environ. Chem., in press [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.