

Abstract
Salmonella enterica is a globally significant bacterial food-borne pathogen that utilizes a variety of carbon sources. We report here that Salmonella enterica subsp. enterica serovar Typhimurium (S. Typhimurium) uses d-glucosaminate (2-amino-2-deoxy-d-gluconic acid) as a carbon and nitrogen source via a previously uncharacterized mannose family phosphotransferase system (PTS) permease, and we designate the genes encoding the permease dgaABCD (d-glucosaminate PTS permease components EIIA, EIIB, EIIC, and EIID). Two other genes in the dga operon (dgaE and dgaF) were required for wild-type growth of S. Typhimurium with d-glucosaminate. Transcription of dgaABCDEF was dependent on RpoN (σ54) and an RpoN-dependent activator gene we designate dgaR. Introduction of a plasmid bearing dgaABCDEF under the control of the lac promoter into Escherichia coli strains DH5α, BL21, and JM101 allowed these strains to grow on minimal medium containing d-glucosaminate as the sole carbon and nitrogen source. Biochemical and genetic data support a catabolic pathway in which d-glucosaminate, as it is transported across the cell membrane, is phosphorylated at the C-6 position by DgaABCD. DgaE converts the resulting d-glucosaminate-6-phosphate to 2-keto-3-deoxygluconate 6-phosphate (KDGP), which is subsequently cleaved by the aldolase DgaF to form glyceraldehyde-3-phosphate and pyruvate. DgaF catalyzes the same reaction as that catalyzed by Eda, a KDGP aldolase in the Entner-Doudoroff pathway, and the two enzymes can substitute for each other in their respective pathways. Examination of the Integrated Microbial Genomes database revealed that orthologs of the dga genes are largely restricted to certain enteric bacteria and a few species in the phylum Firmicutes.
INTRODUCTION
Salmonella is an enteropathogen that, depending on the serovar, causes gastroenteritis and/or fatal systemic disease in a variety of mammals, including humans, cattle, and swine. Over 2,500 serovars of Salmonella have been identified to date (1). While a considerable amount of work has focused on elucidating the mechanisms by which Salmonella pathogenicity genes promote infection (2–6), until recently little attention has been paid to understanding the nutritional and metabolic requirements of Salmonella during colonization (7, 8). Several recent papers have used “-omics”-driven (i.e., genomics, transcriptomics, proteomics, and metabolomics) systems biology approaches to investigate potential contributions of metabolic processes to Salmonella colonization and/or virulence (9–13). The utility of such approaches, however, is limited by gaps in our knowledge of metabolic pathways in Salmonella and by dubious or uninformative gene annotations.
Salmonella is catabolically robust and capable of growing free-living as well as within a variety of animal hosts. Gutnick and coworkers tested roughly 600 compounds and found that ∼90 of these compounds serve as carbon and/or nitrogen sources for Salmonella enterica serovar Typhimurium (S. Typhimurium) LT2 (14). Using a high-throughput phenotype microarray system from Biolog (Hayward, CA), AbuOun and coworkers (9) reported that S. Typhimurium strains LT2 and DT104 utilized, to various degrees, 91 of the 195 carbon sources in the phenotype microarray plates and 34 of the 95 nitrogen sources; an additional 6 carbon sources and 6 nitrogen sources were utilized by one or the other of the two strains. The same study (9) identified three carbon sources (d-glucosaminate, d-psicose, and d-tartrate) utilized by the Salmonella strains but not by Escherichia coli MG1655, but there was no information regarding their catabolism. Such results highlight the need for the continued investigation of metabolic pathways in Salmonella.
Phosphoenolpyruvate-dependent sugar phosphotransferase systems (PTS) are responsible for the transport of many of the carbohydrates utilized by Salmonella. S. Typhimurium LT2 possesses over 20 different sugar PTS permeases, but not all of these PTS permeases are present in other S. Typhimurium strains. For example, S. Typhimurium 14028s (the strain used in the present study) lacks three of the PTS found in S. Typhimurium LT2: a galactitol-specific PTS, a d-tagatose-specific PTS, and a PTS of unknown function (15, 16). All S. Typhimurium strains whose genomes have been sequenced to date, however, possess a mannose PTS permease (ManXYZ) and three additional mannose family PTS permeases of unknown function. PTS-mediated sugar transport involves a cascade of phospho-transfer reactions that culminates in the phosphorylation of the sugar as it is transported across the cell membrane (Fig. 1A). Enzyme I (EI) and HPr are cytoplasmic proteins that serve as common elements for transport of most PTS sugars, while enzyme II (EII) complexes or PTS permeases are associated with the cell membrane and are specific for a particular sugar or group of structurally similar sugars (17). PTS permeases are organized into several families based on shared structural features. All PTS permeases contain three domains, designated EIIA, EIIB, and EIIC, while mannose family PTS permeases contain a fourth domain, designated EIID (18, 19). These domains are arranged in a single polypeptide or within multiple polypeptides (20). Escherichia coli ManXYZ, the best-characterized mannose family PTS permease, transports d-mannose and related hexoses with different substituents at the C-2 position (21).
Fig 1.

Mannose family PTS-mediated carbohydrate transport and the S. Typhimurium dga locus. (A) Phosphoenolpyruvate (PEP) serves as the phospho-donor for a phosphorylation cascade that is formed by the general PTS proteins EI and HPr and an EII complex (PTS permease) which is specific for a given sugar(s). Mannose family PTS permeases consist of four domains (A, B, C, and D) arranged on one to four polypeptides. EIIC and EIID are integral membrane proteins, while EIIA and EIIB are soluble cytoplasmic proteins if not part of a polypeptide that includes EIIC or EIID. As the carbohydrate is transported across the membrane, it is phosphorylated by the EII complex. (B) The EIIA, EIIB, EIIC, and EIID components of the d-glucosaminate PTS permease are encoded by dgaA, dgaB, dgaC, and dgaD, respectively. The other genes in the operon are dgaE, which encodes a dehydratase that converts d-glucosaminate-6-phosphate to 2-keto-3-deoxy-6-phosphogluconate (KDGP), and dgaF, which encodes a KDGP aldolase. DgaR is an RpoN-dependent activator required for expression of dgaABCDEF. Located 138 bp upstream of the predicted translational start site of dgaA is a sequence (5′-TGGCACAACCTTTGCT-3′) that matches the RpoN-dependent promoter consensus sequence. Genes that flank the dga locus in S. Typhimurium 14028s are indicated in gray. The location of the enhancer for the dgaABCDEF operon (i.e., DgaR-binding site) is not known.
Genes encoding the three uncharacterized mannose family PTS permeases in S. Typhimurium were predicted from bioinformatic analysis to be under the control of the alternative sigma factor RpoN (σ54) (22). Initiation of transcription by the σ54-RNA polymerase holoenzyme (σ54-holoenzyme) requires an activator which stimulates the isomerization of a closed complex between σ54-holoenzyme and the promoter to an open complex that is competent to initiate transcription (23, 24). Genes encoding RpoN-dependent activators are often closely associated with the genes they regulate, and juxtaposed to each of the PTS permease operons is a gene encoding an RpoN-dependent activator.
We report here that d-glucosaminate (2-amino-2-deoxy-d-gluconic acid) is a substrate for the mannose family PTS permease encoded by the genes with locus numbers STM14_4545 to STM14_4548 in S. Typhimurium 14028s (Fig. 1B), and we designate these genes dgaABCD (d-glucosaminate PTS permease components EIIA, EIIB, EIIC, and EIID). Deleting the gene encoding a predicted RpoN-dependent activator located upstream of the dgaABCDEF operon (STM14_4550) prevented S. Typhimurium 14028s from utilizing d-glucosaminate, and we designate this gene dgaR. Two additional genes in the dgaABCD operon were found to be involved in the catabolism of d-glucosaminate, and we designate these genes dgaE and dgaF. Genetic and biochemical data were consistent with the hypothesis that d-glucosaminate is catabolized to pyruvate plus glyceraldehyde-3-phosphate by the products of the dgaABCDEF operon.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and reagents.
The S. Typhimurium strains and plasmids used in this study are listed in Tables S1 and S2 in the supplemental material, respectively. E. coli DH5α was used for routine cloning procedures. Strains were maintained in Luria-Bertani (LB) broth or agar supplemented as needed with 100 μg ml−1 ampicillin or 50 μg ml−1 kanamycin. Growth of S. Typhimurium on various carbon or nitrogen sources was tested using a version of a minimal medium developed by Neidhardt and coworkers (25) and modified by Maloy et al. (26). The minimal medium was buffered with 40 mM 3-(N-morpholino)propanesulfonic acid and 4 mM Tricine, with the pH adjusted to 7.4 with NaOH. Additional components of the basic minimal medium were 20 mM NaHCO3, 0.88 mM K2HPO4, 50 mM NaCl, 10 μM FeSO4 · 7H2O, 0.55 mM MgSO4 · 7H2O, 0.4 μM H3BO3, 0.5 μM CaCl2 · 2H2O, 0.03 μM CoCl2 · 6H2O, 0.01 μM CuSO4 · 5H2O, 0.08 μM MnCl2 · 4H2O, and 0.01 μM ZnSO4 · 7H2O. Various carbon and nitrogen sources were added to the concentrations indicated.
Determination of growth rates and Ks.
S. Typhimurium strains were grown aerobically at 37°C in minimal medium, and growth of cell cultures was monitored using a Klett colorimeter (model 900-3) with a green (520 to 580 nm) glass filter. Generation (g) times were calculated from the equation g = ln(2)/b, where b is the best-fit constant from a plot of log values of Klett units versus incubation times. The Michaelis or substrate concentration at half-maximal growth rate (Kμ) for d-glucosaminate was estimated by growing cells in minimal medium containing 20 mM l-arabinose as the primary carbon source and d-glucosaminate at concentrations ranging from 0 to 20 mM as the sole nitrogen source. Cells were cultured in sterile 96-well plates (0.2 ml medium per well) at 37°C in a BioTek ELx808 absorbance microplate reader (BioTek, Winooski, VT). Each culture condition was performed with three biological replicates. Absorbance was measured every 30 min for 24 h. Generation times for each condition were calculated as described above, after plotting log values of absorbance versus incubation times. The Ks value for d-glucosaminate was estimated from a double-reciprocal plot (Lineweaver-Burk plot) of g values versus d-glucosaminate concentrations, using the SigmaPlot 12.5 software package.
A modification of the auxanographic technique described by Gutnick and coworkers (14) was used to test the ability of compounds to support the growth of S. Typhimurium. S. Typhimurium strains were grown overnight in nutrient broth and then diluted 20-fold in minimal salts-soft agar, containing 1.0 g K2SO4, 13.5 g K2HPO4, 4.7 g KH2PO4, 0.1 g MgSO4 · 7H2O, and 6.0 g agar per liter, and the mixture was poured into sterile petri dishes and allowed to solidify. For testing of potential carbon sources, ammonium chloride was included in the soft agar, at a final concentration of 13 mM. For testing of potential nitrogen sources, l-arabinose was included in the soft agar, at a final concentration of 20 mM. Compounds tested to support growth of S. Typhimurium were sterilized by irradiation with short-wave (254 nm) UV light for 2 min, using a handheld UV lamp. Approximately 2 mg of each compound was placed on the surface of the solidified agar, and bacterial growth in the agar was recorded after incubating the plates for 2 days at 37°C.
Chemical syntheses.
d-Glucosaminic acid was purchased from Toronto Research Chemicals or synthesized by the oxidation of d-glucosamine hydrochloride (Sigma-Aldrich) with sodium chlorite (27). d-Glucosamine hydrochloride (2.16 g; 10 mM) was dissolved in 80 ml water, after which 10 ml of 0.5 M NaH2PO4, 1.08 g NaClO2, and 1.28 ml 30% H2O2 were added to the solution. The solution was stirred at room temperature for 48 h. The solution was decolorized by the addition of a small amount of Na2S2O5, and the pH of the solution was adjusted to 7 by the dropwise addition of 1 M NaOH (the final volume of 1 M NaOH was ∼20 ml). The solution was applied to a Dowex 50-X8 column (H+; 100-ml bed volume), and the column was washed with water until the pH of the washings was neutral. The amino acid was then eluted with 2 M NH3, and the total eluate (∼100 ml) was evaporated in vacuo, which resulted in a white solid. The residue was dissolved in 13 ml hot water. The resulting solution was diluted with 13 ml ethanol, cooled to room temperature, and then allowed to stand at 4°C overnight. The precipitate was filtered, washed with ethanol, and dried. The final product was fluffy white crystals, with a final yield of 0.808 g (41.4%). The 1H nuclear magnetic resonance (NMR) spectrum of the final material in D2O was identical to a reference spectrum.
d-Galactosaminic acid was synthesized from d-galactosamine hydrochloride (Sigma-Aldrich) (0.108 g; 0.5 mM), using a scaled-down version of the procedure described above for the synthesis of d-glucosaminic acid from d-glucosamine hydrochloride. The final yield of d-galactosaminic acid was 0.0457 g (48.7%). l-Mannosaminic acid was synthesized from d-mannosamine hydrochloride (Sigma-Aldrich) (0.108 g; 0.5 mM) in the same manner, except that the product obtained following evaporation of the aqueous NH3 was suspended in 95% ethanol, filtered, and dried in vacuo, to give 0.039 g (41.6% yield) of a pale yellow solid.
d-Glucosaminic acid 6-phosphate was synthesized from d-glucosamine-6-phosphate (50 mg; 0.193 mmol) dissolved in 1.5 ml water, to which 22.5 mg NaClO2 was added, followed by 24.7 μl 30% H2O2 and 115.8 μl 0.5 M NaH2PO4. The reaction mixture was left stirring at room temperature for 48 h. The remaining oxidant was neutralized with a little Na2S2O5, and then the pH was brought up to ∼7 with a few drops of 10% NaHCO3 and the solution was stirred for two more hours. The solution was then loaded on a Dowex 50 column (5 ml; H+ form) and washed with water until the pH of the washings was neutral. The amino acid product was eluted with 1 M NH3, and the eluate was evaporated in vacuo. The white solid was dissolved in ∼1 ml water, and 40 mg Ba(OAc)2 was added, followed by 4 ml ethanol. A flocculent white precipitate formed immediately, and the solution was placed at 4°C to complete precipitation. The white precipitate was collected by centrifugation, the supernatant was discarded, and the precipitate was resuspended in ∼2 ml ethanol and collected by centrifugation. Drying the precipitate in vacuo over P2O5 gave 14.5 mg (27.4% yield) of an amber solid. The product had the expected mass as analyzed by electrospray ionization mass spectrometry: m/z = 274 (M − 1)−.
Construction of mutant strains.
The S. Typhimurium mutants indicated in Table S1 in the supplemental material were constructed using the λ Red recombineering method as described previously (28). Plasmid pKD4, which contains a kanamycin resistance (kan) cassette flanked by two Flp recognition targets (FRT sites), was used as a PCR template to create amplicons which contained sequences at their ends that were homologous to the genes targeted for deletion. Primer sets used to generate the amplicons for the targeted mutagenesis are listed in Table S3. Amplified DNA was introduced by electroporation into S. Typhimurium 14028s bearing plasmid pKD46, which carries the λ phage recombinase genes under the control of the araBAD promoter. Genomic DNAs isolated from resulting kanamycin-resistant colonies were checked by PCR to confirm that the target gene had been knocked out, using PCR primers that flanked the target deletion gene (primers are listed in Table S3). Mutant alleles in which the target genes had been replaced with the kan cassette were moved into a clean genetic background by transduction using P22 HT int. The kan cassette was then removed by introducing plasmid pCP20 into the mutant strains. Plasmid pCP20 expresses the Flp recombinase, which recognized the FRT sites and excised the kan cassette. Loss of the kan cassette was confirmed by susceptibility to kanamycin and by PCR using the same flanking primers (listed in Table S3). S. Typhimurium 14028s strains with disruptions in ptsG or crp were constructed using P22 HT int to transduce mutant alleles from strains DM12321 [met+ ptsG4152::Tn10d(Tcr)] and JE16465 (met205 ara-9 crp891::kan+), which are derivatives of S. Typhimurium LT2 that were kindly provided by Diana Downs and Jorge Escalante-Semerena.
Construction of plasmids.
The dgaABCDEF operon and dgaF were amplified by PCR from S. Typhimurium 14028s genomic DNA by use of Phusion High-Fidelity DNA polymerase (New England BioLabs) and the primer sets indicated in Table S3 in the supplemental material. The PCR primer sets introduced BglII and HindIII sites for subsequent cloning into the vector pLAC22 (29). In the resulting plasmids, designated pLAC22+dgaABCDEF and pLAC22+dgaF, dgaABCDEF and dgaF were placed under the control of the lac promoter/operator. Plasmids for overexpression of DgaE and DgaF were constructed by amplifying dgaE and dgaF from S. Typhimurium 14028s genomic DNA by a PCR using Phusion High-Fidelity DNA polymerase and the primer sets indicated in Table S2. A's were added to the 3′ ends of the amplicons by use of Taq DNA polymerase (Thermo Fisher Scientific). The amplicons were cloned into pCR2.1-TOPO (Invitrogen), and the sequences of the cloned fragments were confirmed by DNA sequencing (Genewiz). NdeI and HindIII sites introduced by the primer sets were used to clone dgaE and dgaF into the expression vector pET21a (Novagen). The resulting plasmids, designated pET21a+dgaE and pET21a+dgaF, expressed the native DgaE and DgaF proteins (i.e., lacking any tag to facilitate purification).
For all transformations involving S. Typhimurium 14028s, DNA was introduced into the bacterium by electroporation at 2.4 kV, 25 μF, and 400 Ω. Following electroporation, cells were allowed to recover in SOC broth (0.5% yeast extract, 2% tryptone, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, and 20 mM glucose) for 1 h at 37°C. Chemically competent cells prepared using the calcium chloride method were used for transformation of all E. coli strains, and cells were allowed to recover in LB broth for 1 h at 37°C following the heat shock step.
Biolog phenotype microarray plate assays.
S. Typhimurium strains were grown on Biolog universal growth (BUG) agar supplemented with 5% sheep blood at 37°C for 24 h. Cells were resuspended in IF-O+dye solution (Biolog) to a final cell density of 85% transmittance, which was determined by use of a spectrophotometer. The cell suspension was used to inoculate Biolog PM1 and PM2 plates. Before inoculating the Biolog PM3 plates, sodium succinate and ferric citrate were added to the cell suspension, to final concentrations of 20 mM and 2 μM, respectively. Plates were incubated in an OmniLog PM system at 37°C for 48 h and scanned every 15 min. Collected data were analyzed with OmniLog software.
Purification of DgaE.
Plasmid pET21a+dgaE was transformed into E. coli BL21(DE3λ) containing the plasmid pLysE (30). E. coli BL21(DE3λ) expresses T7 RNA polymerase, which is needed for transcription of dgaE from pET21a+dgaE. The cellular extract from 1 liter of cells cultured in rich autoinduction medium (31) was prepared by sonication for 4 min at 1-min intervals, followed by centrifugation for 90 min at 2,500 × g. The supernatant was applied to a Ni affinity column (10 ml) and washed with 0.05 M potassium phosphate, pH 7.0, 0.1 mM pyridoxyl-5′-phosphate (PLP), 0.3 M NaCl until the absorbance at 280 nm was back to baseline. The column was then eluted with 100 ml of wash buffer containing 0.2 M imidazole. Peak protein fractions were pooled, concentrated in a centrifugal concentrator (YM-30 membrane), and then dialyzed overnight against 500 ml of 0.05 M potassium phosphate, pH 7.0, 0.1 mM PLP.
Identification of DgaE reaction product.
Solutions containing 0.1 M NH4HCO3 and 1 mM d-glucosaminic acid-6-phosphate were prepared in a total volume of 200 μl. To one solution, 5 μl purified DgaE (24.4 mg/ml) was added, and the solutions were allowed to stand at room temperature for 48 h. The solutions were then analyzed by electrospray ionization mass spectrometry.
Coupled assay for DgaE and DgaF.
Reaction mixtures contained 0.1 M potassium phosphate, pH 7.0, 1 mM d-glucosaminic acid-6-phosphate, 40 μM PLP, 0.2 mM NADH, 10 μg lactate dehydrogenase (Sigma-Aldrich), and 10 to 20 μg of crude cell extract containing DgaF. Cell extracts containing DgaF were prepared from E. coli BL21(DE3λ) containing plasmids pET21a+dgaF and pLysE. Cells were grown overnight in rich autoinduction medium, harvested by centrifugation, and lysed by sonication for 4 min at 1-min intervals, and the resulting crude cell extract was clarified by centrifugation for 90 min at 2,500 × g. The coupled enzyme reactions were performed at 37°C, and the absorbance decrease at 340 nm (Δε = 6,220 M−1 cm−1) was recorded.
qRT-PCR assays.
Transcript levels of dgaAB in wild-type S. Typhimurium 14028s cultured in minimal medium that contained d-glucose, d-glucosaminate, d-gluconate, or l-arabinose (final concentration of 20 mM for each sugar) as the sole carbon source were assessed by quantitative reverse transcription-PCR (qRT-PCR). One milliliter of cell culture grown to mid-log phase was used for RNA extraction. RNA was isolated using an Aurum Total RNA minikit (Bio-Rad) and treated with an additional DNase treatment by use of a Turbo DNA-free kit (Ambion). cDNA was synthesized using an iScript cDNA synthesis kit (Bio-Rad), after which qRT-PCR was carried out using an iCycler iQ system (Bio-Rad). Primer specificity was confirmed by PCR, using genomic DNA as the template. Three biological samples and three technical replicates were assessed for expression of dgaAB and were normalized to levels of rpoD, an internal control for S. Typhimurium gene expression. All reaction mixtures totaled 20 μl and included 10 μl of iQ SYBR green Supermix (Bio-Rad), 5 μl of 100-fold-diluted cDNA from each synthesis, and 200 nM (each) primers specific for either dgaAB or rpoD. Gene expression levels were determined by using the 2−ΔΔCT equation (32).
Computational analysis.
Complete genomes of Salmonella enterica serovar Typhimurium 14028s, Escherichia coli IAI1, Enterobacter aerogenes KCTC 2190, and Enterococcus faecalis 62 were downloaded from the NCBI FTP server (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/ ). Sequences of the dga loci were extracted from the complete genomes. The dga loci span the genes STM14_4550 through STM14_4543, ECIAI1_3068 through ECIAI1_3062, EAE_10150 through EAE_10120, and EF62_0067 through EF62_0061, respectively. The dga loci and their host genomes were compared in terms of their overall G+C content, G+C content at codon site 3 (S3), and relative abundance of the tetranucleotide CTAG (τ*CTAG). CTAG is strongly suppressed in Gammaproteobacteria but generally not in Firmicutes, and any laterally transferred DNA segment between Gammaproteobacteria and Firmicutes could therefore exhibit anomalous CTAG representation with respect to the bulk of the host genome (33, 34). Relative abundance measures an excess or deficit of a given oligonucleotide relative to the expected occurrence, based on known frequencies of all embedded shorter oligonucleotides. For a tetranucleotide XYZW, the relative abundance, τ*XYZW, is assessed as follows:
where f∗XYZW denotes the symmetrized frequency of the tetranucleotide XYZW, f∗XY denotes the symmetrized frequency of the dinucleotide XY, etc. “N” stands for any nucleotide. For double-stranded DNA, symmetrized frequencies are calculated from the nucleotide sequence at hand concatenated with its inverted complement. Relative abundance values of ∼1 signify that the tetranucleotide occurs approximately as expected, values of <1 indicate that it occurs less frequently than expected, and values of >1 indicate that it occurs more than expected. Values between 0.78 and 1.23 are considered in the “normal range” (33, 35).
(i) δ∗ differences.
The relative abundance of a dinucleotide XY is defined as follows: ρ∗XY = f∗XY/f∗Xf∗Y. The vector of the 16 dinucleotide relative abundances constitutes a genome signature (36, 37). A difference between two nucleotide sequences, A and B, can subsequently be defined as follows:
where ρ∗XY(A) signifies the relative abundance of dinucleotide XY in sequence A and ρ∗XY(B) signifies the relative abundance of dinucleotide XY in sequence B. To avoid statistical artifacts in comparing sequences of vastly different sizes, it is reasonable to divide the compared sequences into disjointed samples of approximately equal sizes and to use the mean distance among all pairwise comparisons between different samples as a measure of dissimilarity of the analyzed sequences. In this work, we used the sample size 7,000 bp, which is similar to the size of the dga locus. The software for genome signature comparisons and assessments of oligonucleotide relative abundances is available at http://www.cmbl.uga.edu/software.html.
RESULTS
A mannose family PTS permease in S. Typhimurium is required for utilization of d-glucosaminate.
Functions of three previously uncharacterized mannose family PTS permeases in S. Typhimurium 14028s were examined by deleting the genes encoding the EIIA and EIIB components of each of the PTS permeases and analyzing the phenotypes of the resulting mutants. Each of the PTS operons has a predicted RpoN-dependent promoter and a gene encoding a putative RpoN-dependent activator immediately downstream or upstream of the operon, suggesting that transcription of the PTS genes is dependent on σ54-holoenzyme (22). Genes encoding the activators were similarly deleted and the phenotypes of the resulting mutants analyzed. Given the broad substrate specificity of E. coli ManXYZ (21), we thought that there may be overlap in the substrates of the PTS permeases. Therefore, strains were constructed in which genes encoding all three PTS permeases or activators were deleted.
Phenotypes of the single and triple mutants were analyzed using Biolog phenotype microarrays. Two carbon source plates (PM1 and PM2) and a nitrogen source plate (PM3) were used for the analysis, which allowed us to assess the ability of the mutant strains to metabolize 190 different carbon sources and 95 different nitrogen sources (compounds included on these plates are listed at http://www.biolog.com/pdf/pm_lit/PM1-PM10.pdf). Mutant strains that lacked all three PTS permeases, all three activators, dgaAB, or dgaR were deficient in metabolizing d-glucosaminate compared to the parental strain. This was the only phenotypic difference observed consistently between the parental strain and the mutant strains.
The ΔdgaAB and ΔdgaR mutants were examined for the ability to grow aerobically in a minimal medium with d-glucosaminate as the sole carbon source. Wild-type S. Typhimurium 14028s grew in minimal medium with a doubling time of 91 ± 3 min. Cells grew faster when ammonium chloride was omitted from the minimal medium and d-glucosaminate was the sole carbon and nitrogen source (80 ± 2 min). The ΔdgaAB and ΔdgaR mutants failed to grow with d-glucosaminate as the sole carbon source, indicating that the dga locus is responsible for d-glucosaminate utilization.
Consistent with the prediction that transcription of dgaABCD is dependent on RpoN, a ΔrpoN mutant failed to grow in a d-glucosaminate minimal medium supplemented with 5 mM l-glutamine. l-Glutamine was included in the medium because deletion of rpoN in S. Typhimurium results in glutamine auxotrophy (38). l-Glutamine is a poor nitrogen source and does not support robust growth of S. Typhimurium at the concentration used here. An S. Typhimurium ΔptsH (encodes HPr) mutant also failed to grow in the d-glucosaminate minimal medium, indicating that the general PTS component HPr is required for d-glucosaminate utilization.
To see if S. Typhimurium 14028s employs DgaABCD to utilize sugars which are structurally similar to d-glucosaminate, we used an auxanographic method to assess the ability of S. Typhimurium 14028s strains to utilize various compounds as carbon or nitrogen sources. The ΔdgaAB mutant was able to grow with d-gluconate, d-glucosamine, or N-acetylglucosamine as a carbon source, which was expected, as there are known transporters of these compounds in S. Typhimurium. Wild-type S. Typhimurium 14028s failed to grow with d-galactosaminate or l-mannosaminate as a carbon or nitrogen source, indicating that S. Typhimurium is unable to transport or metabolize these compounds.
d-Glucosaminate supports robust growth of S. Typhimurium as a nitrogen source.
To determine the efficacy of d-glucosaminate as a nitrogen source for S. Typhimurium, we compared the growth of S. Typhimurium 14028s in minimal media that contained d-glucosaminate as the sole nitrogen source and various primary carbon sources. For these experiments, we compared the ability of S. Typhimurium to utilize d-glucosaminate versus a good nitrogen source (ammonium chloride) and a poor nitrogen source (l-arginine). Primary carbon sources that were tested included three PTS sugars (d-glucose, d-mannose, and d-fructose) and a non-PTS sugar (l-arabinose). Transport of d-glucose and d-mannose involves EI and HPr, while the d-fructose transport system relies on its own HPr-like and EI-like activities (39, 40). Cultures of S. Typhimurium 14028s were grown overnight in minimal media with various combinations of nitrogen sources and sugars. The overnight cultures were diluted into fresh medium, and doubling times during exponential growth were measured for each culture condition.
S. Typhimurium failed to grow in a minimal medium containing glucose and d-glucosaminate as the sole nitrogen source. S. Typhimurium was able to grow in a minimal medium that contained glucose as the primary carbon source and both d-glucosaminate and ammonium as nitrogen sources. The doubling time of S. Typhimurium in glucose minimal medium containing both d-glucosaminate and ammonium was similar to that of S. Typhimurium grown in glucose minimal medium containing ammonium as the sole nitrogen source (44 ± 4 min versus 45 ± 3 min, respectively). These findings indicate that the failure of S. Typhimurium to grow in glucose minimal medium containing d-glucosaminate as the nitrogen source results from glucose inhibiting utilization of d-glucosaminate.
In minimal medium that contained l-arabinose or d-fructose as the primary carbon source, d-glucosaminate as the nitrogen source supported growth rates of S. Typhimurium that were comparable to those with ammonium chloride (Table 1). Compared to growth in minimal medium containing d-mannose and ammonium chloride, S. Typhimurium grew significantly slower in medium containing d-mannose and d-glucosaminate. The lower growth rate of S. Typhimurium in minimal medium with d-glucosaminate plus d-mannose than in minimal medium with d-glucosaminate plus l-arabinose or d-fructose may have been due to competition between the mannose PTS and d-glucosaminate PTS for phosphorylated HPr (P∼HPr). For all of the sugars tested, d-glucosaminate served as a significantly better nitrogen source than l-arginine and was comparable to ammonium chloride in its efficacy of promoting rapid growth of S. Typhimurium.
Table 1.
Growth rates of S. Typhimurium 14028s grown in minimal medium with various combinations of carbon and nitrogen sources
| Nitrogen source | Doubling time (min) on carbon source |
||
|---|---|---|---|
| l-Arabinose | d-Mannose | d-Fructose | |
| NH4Cl | 44 ± 2 | 39 ± 4 | 43 ± 2 |
| d-Glucosaminate | 43 ± 4 | 50 ± 1 | 45 ± 2 |
| l-Arginine | 100 ± 5 | 130 ± 9 | 110 ± 5 |
To determine the affinity of the Dga PTS for d-glucosaminate, the half-saturation constant (Ks) for d-glucosaminate was estimated. The Ks for d-glucosaminate was estimated by measuring the growth rates of S. Typhimurium grown in minimal media with l-arabinose as the primary carbon source and various amounts of d-glucosaminate as the sole nitrogen source. The estimated Ks was 0.098 ± 0.02 mM d-glucosaminate, indicating that d-glucosaminate is a physiologically relevant substrate for the Dga PTS.
Two genes downstream of dgaABCD function in catabolism of d-glucosaminate.
The two genes located immediately downstream of dgaABCD (STM14_4544 and STM14_4543) were deleted, and the phenotypes of the resulting mutants were analyzed. The STM14_4544 deletion mutant failed to grow in d-glucosaminate-containing minimal medium, indicating that this gene is essential for catabolism of d-glucosaminate, so we named it dgaE. DgaE belongs to the fold type I or aspartate aminotransferase (AAT) superfamily of PLP-dependent enzymes (41). In reactions catalyzed by AAT superfamily members, PLP combines with an α-amino acid to form a Schiff base intermediate. Depending on the enzyme, the Schiff base serves as a substrate for transamination, racemization, decarboxylation, or one of a variety of side chain reactions.
Based on results described below which suggested that STM14_4543 encodes a keto-3-deoxygluconate 6-phosphate (KDGP) aldolase, we postulated that d-glucosaminate is phosphorylated at the C-6 position as it is transported across the cell membrane and that the resulting d-glucosaminate 6-phosphate is converted to KDGP by DgaE (Fig. 2). Consistent with this hypothesis, incubation of purified DgaE with d-glucosaminic acid 6-phosphate resulted in a new peak with m/z = 257, demonstrating the net loss of 17 atomic mass units (amu) (NH3) from the molecule. Incubation in the absence of DgaE did not show the peak at 257, but only the d-glucosaminic acid 6-phosphate peak at m/z = 274. The mass of the product is consistent with the formation of KDGP from d-glucosaminic acid 6-phosphate, indicating that DgaE functions as a glucosaminic acid 6-phosphate dehydratase.
Fig 2.
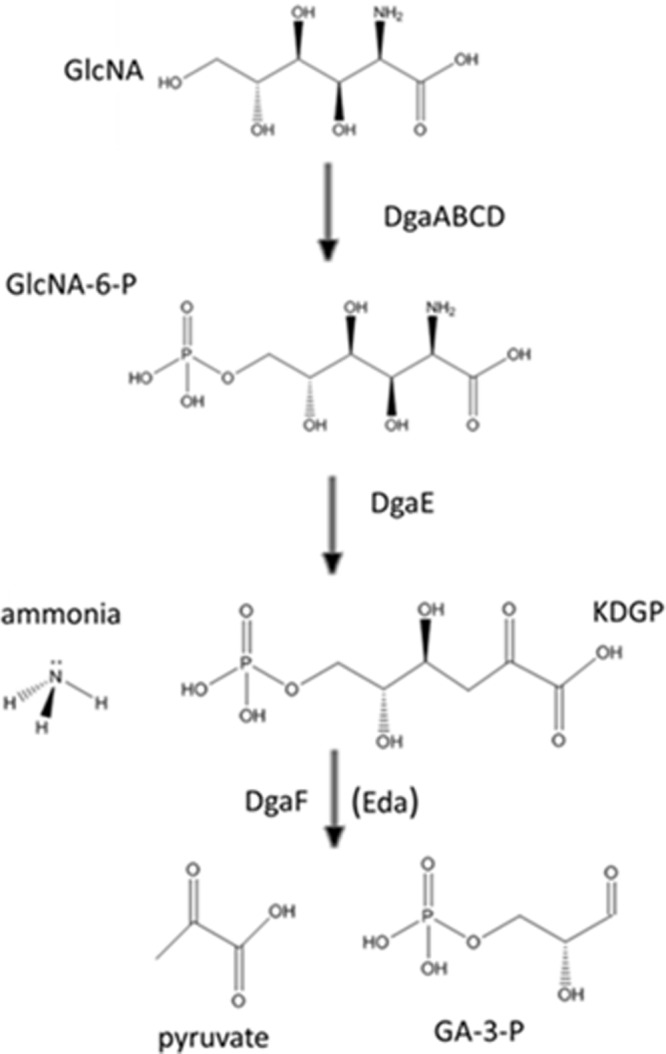
Proposed pathway for d-glucosaminate catabolism in S. Typhimurium. As d-glucosaminate (GlcNA) is transported across the cell membrane, it is phosphorylated at the C-6 position by the DgaABCD permease. DgaE is a d-glucosaminate-6-phosphate (GlcNA-6-P) dehydratase that converts GlcNA-6-P to KDGP. KDGP is subsequently cleaved by the aldolase DgaF to form glyceraldehyde-3-phosphate (GA-3-P) and pyruvate. KDGP can also be cleaved by the Entner-Doudoroff aldolase (Eda).
In contrast to the ΔdgaE mutant, the STM14_4543 deletion mutant grew in the d-glucosaminate minimal medium, although the growth rate of the mutant was significantly lower (doubling time of 100 ± 4 min for the mutant versus 80 ± 2 min for the wild type). These data indicated that STM14_4543 is not essential for d-glucosaminate catabolism but is required for wild-type growth on d-glucosaminate, so we designated the gene dgaF.
DgaF belongs to the DUF1341 superfamily. Some members of this family are annotated KDGP aldolases (Entner-Doudoroff aldolases [Eda]) or 4-hydroxy-2-oxoglutarate aldolases, although it is not clear if there is experimental evidence to support these annotations. Based on the above observations indicating that DgaE converts glucosaminic acid 6-phosphate to KDGP, we postulated that DgaF is a KDGP aldolase and that Eda can substitute for DgaF in d-glucosaminate catabolism. Deletion of eda had no effect on growth of S. Typhimurium 14028s in the d-glucosaminate minimal medium, as the Δeda mutant had a doubling time of 74 ± 1 min, compared to 80 ± 2 min for the wild type. A strain lacking both dgaF and eda, however, failed to grow in the d-glucosaminate minimal medium, indicating that Eda can indeed substitute for DgaF in the catabolism of d-glucosaminate. Conversely, DgaF was able to substitute for Eda. Eda is required for growth of S. Typhimurium with d-glucuronate or d-gluconate as a carbon source. Introduction of a plasmid-borne copy of dgaF under the control of the lac promoter into the Δeda mutant restored the ability of the mutant to grow on a minimal medium with d-glucuronate or d-gluconate as the carbon source (data not shown).
These combined genetic and biochemical data indicated that DgaE and DgaF function together to convert d-glucosaminic acid 6-phosphate to pyruvate plus glyeraldehyde-3-phosphate (Fig. 2). Consistent with this hypothesis, addition of DgaE to reaction mixtures containing d-glucosaminic acid 6-phosphate, a cellular extract containing DgaF, lactate dehydrogenase, and NADH resulted in the DgaE-dependent consumption of NADH, demonstrating that pyruvate is formed from d-glucosaminic acid 6-phosphate by the combined action of DgaE and DgaF. The specific activity of DgaE in the coupled assay was 6.2 μmol/min-mg.
The dgaABCDEF operon is sufficient for d-glucosaminate utilization in E. coli.
The S. Typhimurium dgaABCDEF operon was cloned into an expression vector and placed under the control of an inducible lac promoter (plasmid pLAC22+dgaABCDEF). The dgaABCDEF expression vector restored the ability of the S. Typhimurium ΔdgaAB and ΔdgaR mutants to grow on the d-glucosaminate minimal medium if IPTG (isopropyl-β-d-thiogalactopyranoside) was included in the medium to induce expression of the dga genes. E. coli strains BL21, DH5α, and JM101 are typical of most E. coli strains whose genomes have been sequenced to date in that they lack the dga locus. These three E. coli strains failed to utilize d-glucosaminate as a carbon or nitrogen source. Introduction of the dgaABCDEF expression vector into the strains, however, allowed them to grow in the d-glucosaminate minimal medium if IPTG was included in the medium to induce expression of the dga genes. E. coli IAI1 is a strain that possesses the dga locus, and we found that it is capable of growing in minimal medium with d-glucosaminate as the sole carbon and nitrogen source. There does not appear to be any obvious trait (e.g., commensal versus pathogen) that distinguishes E. coli strains which possess the dga locus from those that lack the locus.
d-Glucosaminate induces expression of the dgaABCDEF operon.
DgaR contains a PTS regulation domain (PRD), which is a phosphorylation target of certain PTS proteins (17, 42). Bacillus subtilis LevR, the best-characterized PRD-containing RpoN-dependent activator, stimulates transcription of the levanase operon, which encodes a fructose-specific PTS permease (43). Phosphorylation of a specific PRD histidine residue by P∼HPr stimulates LevR activity, while phosphorylation of a different PRD histidine residue by the phosphorylated EIIB component (P∼LevE) of the levanase PTS inhibits LevR activity (44). The negative regulation of LevR is the basis for induction of the levanase operon by fructose, because in the absence of fructose in the medium, P∼LevE predominates and phosphorylates LevR to inhibit transcription of the levanase operon.
Given that fructose induces expression of the B. subtilis lev operon, we postulated that d-glucosaminate similarly induces expression of the dga operon. To test this hypothesis, qRT-PCR was used to measure dgaAB transcript levels in S. Typhimurium 14028s grown in minimal medium containing d-glucosaminate, d-glucose, d-gluconate, or l-arabinose as the sole carbon source. For these assays, dgaAB transcript levels were normalized to rpoD transcript levels. Levels of dgaAB transcripts were ∼1,000-fold higher in cultures grown with d-glucosaminate than in cultures grown with the other carbohydrates tested (Fig. 3), indicating that d-glucosaminate induces expression of the dga operon.
Fig 3.
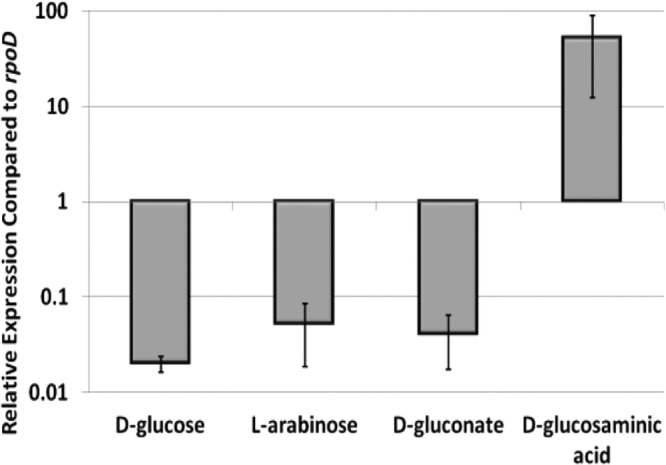
Relative levels of dgaAB transcripts in S. Typhimurium grown on minimal medium containing different carbon sources. Cultures were grown to mid-log phase in minimal medium containing 20 mM d-glucose, l-arabinose, d-gluconate, or d-glucosaminate as the carbon source. cDNAs were prepared from RNAs isolated from the cultures and quantified by qRT-PCR. Levels of dgaAB transcripts were compared to rpoD transcript levels. Average values for three sample replicates of three biological replicates are shown. Error bars indicate standard deviations. Levels of dgaAB transcripts from cultures grown with d-glucosaminate differed significantly from those of the other cultures, as assessed using the Student t test (P < 0.03).
Glucose inhibition of d-glucosaminate utilization is mediated by the glucose PTS.
We wished to determine the basis for glucose inhibition of d-glucosaminate utilization. One possible explanation is that transcription of dgaR or dgaABCDEF is dependent on the cyclic AMP (cAMP) receptor protein (CRP) and that glucose inhibits dga expression by depressing cAMP levels inside the cell. To address this hypothesis, we constructed a crp::kan+ mutant in S. Typhimurium 14028s and tested its ability to utilize d-glucosaminate. The crp mutant grew as well as the parental strain in minimal medium with d-glucosaminate as the sole carbon and nitrogen source (doubling times of the crp mutant and the wild type were 76 ± 4 min and 80 ± 2 min, respectively), indicating that CRP is not required for transcription of dgaR or dgaABCDEF.
To investigate further the reason that S. Typhimurium failed to grow in a minimal medium containing glucose as the primary carbon source and d-glucosaminate as the sole nitrogen source, we introduced a ptsG::Tn10d(Tcr) mutation into S. Typhimurium 14028s to determine if the glucose-specific PTS was involved in glucose inhibition of d-glucosaminate utilization (ptsG encodes the EIIBC component of the glucose-specific PTS). ManXYZ transports glucose efficiently, and in the absence of the glucose-specific PTS, it is able to support rapid growth with glucose (45, 46). The ptsG mutant grew with a doubling time of 46 ± 4 min in a minimal medium containing 20 mM glucose as the primary carbon source and 5 mM d-glucosaminate as the sole nitrogen source. These data indicate that glucose does not directly inhibit transport or utilization of d-glucosaminate and that the glucose inhibition of d-glucosaminate utilization is mediated through the glucose-specific PTS. We postulate that the glucose-specific PTS inhibits transcription of dgaABCDEF by diverting P∼HPr to prevent phosphorylation of DgaR and thereby block its activation.
Phylogenetic distribution of the dga locus.
Bacterial genomes in the Integrated Microbial Genomes (IMG) database (http://img.jgi.doe.gov) were searched for potential orthologs of the dga genes, using the synteny of dga genes to facilitate the identification of orthologs (data for the analysis were collected on 20 February 2013). The IMG Conserved Neighborhood tool, which searches for orthologs in user-selected genomes and displays neighborhoods of similarly sized orthologs (47), was used for the analysis. Each dga gene from S. Typhimurium 14028s was compared with the genomes in the IMG database, and the resulting gene ortholog neighborhoods were examined for colocalization of the dga genes. Bacterial genomes that possessed the dga locus according to these criteria were limited to some members of seven genera (Salmonella, Escherichia, Citrobacter, Enterobacter, Hafnia, Klebsiella, and Serratia) within the family Enterobacteriaceae in the subphylum Gammaproteobacteria, one member of the subphylum Betaproteobacteria (Chitiniphilus shinanonensis DSM 23277), and some members of the genera Enterococcus and Lactobacillus, in the phylum Firmicutes (see Table S4 in the supplemental material). Collinsella tanakaei YIT 12063, a member of the phylum Actinobacteria, possesses orthologs of the dgaABCDEF genes but lacks homologs of dgaR and rpoN, indicating that expression of the dga orthologs in this bacterium is regulated in a manner that differs from that of S. Typhimurium.
For most of the Salmonella genome sequences in the IMG database, we were able to unambiguously identify complete dga loci (68 of 86 sequences). Most of the S. Typhi genomes in the database are draft sequences that consist of numerous and short contigs, and while these genomes possess at least some of the dga orthologs, we were unable to ascertain if they contained complete sets of dga genes. The three finished S. Typhi genomes (S. Typhi CT18, S. Typhi Ty2, and S. Typhi P-stx-12) and one draft genome sequence (S. Typhi E98-3139) in the database, however, allowed us to unambiguously assess the content of dga genes in these strains. All four of these S. Typhi strains possess dgaABCDEF orthologs but contain the same apparent frameshift mutation (G inserted between nucleotides 207 and 208) in dgaR, suggesting that they lack a functional DgaR protein. Sequences which matched the RpoN-dependent promoter consensus sequence generated by Barrios and coworkers (48) were found upstream of dgaA for all of the Salmonella strains (see Table S4 in the supplemental material).
All of the Enterococcus faecalis genomes in the IMG database contain at least some of the dga orthologs. Many of the E. faecalis strains lack full-length versions of either or both dgaE and dgaF within the dga operon but possess dgaEF orthologs at a separate locus. We postulate that this altered gene arrangement resulted from a gene duplication and subsequent deletion of the dgaEF genes at the dga locus, since some E. faecalis strains (e.g., AR01/DG) possess two full-length copies of dgaEF. In the case of E. faecalis AR01/DG, the DgaF paralogs share 100% amino acid identity over their entire length, while the DgaE paralogs share 98% amino acid identity over their entire length.
Two of the eight Lactobacillus rhamnosus sequenced genomes contain the dga locus (strains LRHMDP2 and LRHMDP3). In both of these strains, rpoN is situated between the dgaA and dgaR homologs. The six L. rhamnosus strains which lack the dga locus do not possess rpoN in this region or elsewhere in their genomes. Thus, L. rhamnosus strains LRHMDP2 and LRHMDP3 appear to have acquired the dga locus, together with the gene encoding the σ factor required for transcription of the dga operon, through lateral gene transfer.
RpoN-dependent PTS operons are more prevalent in Firmicutes than in Proteobacteria, so we wished to test the hypothesis that enterobacteria acquired the dga genes from E. faecalis. To test this hypothesis, dga orthologs of four bacterial species for which complete genome sequences are available were compared in terms of G+C content, G+C at codon site 3, and suppression of the CTAG tetranucleotide. These comparisons showed that the dga loci match the properties of their respective host genomes, arguing against recent lateral transfer of the dga locus between E. faecalis and other enterobacteria (see Table S5 in the supplemental material). Genome signature comparisons of the genomes and dga loci revealed that E. faecalis is an outlier among the four genomes and that the dga loci feature signatures similar to those of their host genomes (see Table S6). Moreover, BLAST comparisons among orthologous genes in the four dga loci showed that for each of the six genes, the three enterobacterial orthologs are invariably more similar to each other than any is to the E. faecalis ortholog (see Table S7). Taken together, these data fail to support the hypothesis that the dga genes were transferred from E. faecalis to the enteric bacteria or vice versa.
DISCUSSION
We show here that S. Typhimurium uses a mannose family PTS permease encoded by dgaABCD, together with a novel d-glucosaminic acid 6-phosphate dehydratase (DgaE) and a redundant KDGP aldolase (DgaF), to transport and catabolize d-glucosaminate (Fig. 2). The proposed DgaE activity is similar to that of d-glucosaminate dehydratase, a PLP-dependent enzyme that has been purified from Agrobacterium radiobacter and Pseudomonas fluorescens and that converts d-glucosaminate to 2-keto-3-deoxygluconate (49–52). We could not compare the sequence of DgaE with sequences of the A. radiobacter and P. fluorescens enzymes because the sequences of these enzymes have not been reported. DgaE does share 40% amino acid identity (59% similarity) over its entire length with a predicted protein present in most of the Agrobacterium strains whose genomes have been sequenced, and it is possible that the Agrobacterium DgaE homolog is a d-glucosaminate dehydratase. The only P. fluorescens protein that shares significant homology with DgaE is a selenocysteine synthase, but this sequence homology is considerably less than that of the Agrobacterium DgaE homolog (24% identity and 42% similarity over 287 of 369 amino acid residues).
It is not obvious where Salmonella might encounter d-glucosaminate in nature. Given that many of the bacteria that possess the d-glucosaminate PTS can colonize animal intestinal tracts, it is possible that Salmonella encounters d-glucosaminate in such environments. We are unaware, though, of any reports of d-glucosaminate in the intestinal tract contents of any animal. There have been numerous studies in which the Salmonella transcriptome has been analyzed by use of bacteria grown in vivo (e.g., during infection of macrophage-like cells or epithelial cells or isolated from the intestinal tract), cultured under different conditions, or exposed to various chemical stimuli (53–61). In addition, there are several reports on how the loss of known global regulators affects the Salmonella transcriptome (62–64). A recent study used proteomic, mutant phenotyping, and computational approaches to investigate Salmonella nutrition in a mouse typhoid fever model (65). None of these studies, however, identified conditions that resulted in upregulation of the dga operon or offered clues as to where Salmonella might encounter d-glucosaminate.
It is possible that S. Typhimurium obtains d-glucosaminate from other microorganisms. d-Glucosaminate is a component of Rhizobium leguminosarum lipid A (66), and S. Typhimurium could obtain d-glucosaminate from bacteria that produce it for the biosynthesis of lipid A or other macromolecules. Another possible source of d-glucosaminate is from the oxidation of d-glucosamine by glucose oxidase. E. coli glucose oxidase effectively converts d-glucosamine to d-glucosaminate, with a catalytic efficiency for d-glucosamine that is about half that for d-glucose (67). S. Typhimurium possesses a glucose oxidase and could use the enzyme to convert d-glucosamine to d-glucosaminate. The benefit of such a scheme is not obvious, since d-glucosamine can be transported directly by the mannose and glucose PTS (19, 68). Moreover, glucose oxidase requires the cofactor pyrroloquinoline quinone (PQQ), but S. Typhimurium is unable to synthesize PQQ (69). In nature, S. Typhimurium presumably obtains PQQ from the environment to activate glucose oxidase. It is possible that S. Typhimurium uses glucose oxidase as a strategy to compete for limiting nutrients. For example, if S. Typhimurium obtained PQQ from associated bacteria in the environment, it could use its glucose oxidase to convert sugars such as d-glucosamine to compounds that it could use but its neighbors could not. Alternatively, S. Typhimurium may obtain d-glucosaminate generated from d-glucosamine as an unwanted side reaction of the glucose oxidases of other bacteria. Since glucose oxidase is located on the periplasmic side of the cell membrane in Gram-negative bacteria, d-glucosaminate formed from the oxidation of d-glucosamine could diffuse into the surrounding area and be available for S. Typhimurium (14). Regardless of the source of d-glucosaminate, the d-glucosaminate PTS is likely a scavenging system that allows S. Typhimurium to compete for limiting nutrients in one or more of its native environments.
Supplementary Material
ACKNOWLEDGMENTS
We thank Anna Karls, Margie Lee, and John Maurer for helpful discussions and Kyle Friez for his help with enzyme assays. We thank Jean Guard and Todd Stewart for the use of equipment and assistance with the Biolog assays, Diana Downs and Mary Anderson for use of equipment and advice on growth assays, Anthony Maurelli and Erick Denamur for providing E. coli IAI1, and Diana Downs and Jorge Escalante-Semerena for providing S. Typhimurium strains.
This research was supported by grant MCB-1051175 from the National Science Foundation to T.R.H. and by Agriculture and Food Research Initiative competitive grant 2012-67011-19934 from the USDA National Institute of Food and Agriculture to K.A.M.
Footnotes
Published ahead of print 8 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00290-13.
REFERENCES
- 1.Jones TF, Ingram LA, Cieslak PR, Vugia DJ, Tobin-D'Angelo M, Hurd S, Medus C, Cronquist A, Angulo FJ. 2008. Salmonellosis outcomes differ substantially by serotype. J. Infect. Dis. 198:109–114 [DOI] [PubMed] [Google Scholar]
- 2.Figueira R, Holden DW. 2012. Functions of the Salmonella pathogenicity island 2 (SPI-2) type III secretion system effectors. Microbiology 158:1147–1161 [DOI] [PubMed] [Google Scholar]
- 3.Marcus SL, Brumell JH, Pfeifer CG, Finlay BB. 2000. Salmonella pathogenicity islands: big virulence in small packages. Microbes Infect. 2:145–156 [DOI] [PubMed] [Google Scholar]
- 4.Que F, Wu S, Huang R. 2013. Salmonella pathogenicity island 1 (SPI-1) at work. Curr. Microbiol. 66:582–587. 10.1007/s00284-013-0307-8 [DOI] [PubMed] [Google Scholar]
- 5.Wagner C, Hensel M. 2011. Adhesive mechanisms of Salmonella enterica. Adv. Exp. Med. Biol. 715:17–34 [DOI] [PubMed] [Google Scholar]
- 6.van Asten AJ, van Dijk JE. 2005. Distribution of “classic” virulence factors among Salmonella spp. FEMS Immunol. Med. Microbiol. 44:251–259 [DOI] [PubMed] [Google Scholar]
- 7.Bumann D. 2009. System-level analysis of Salmonella metabolism during infection. Curr. Opin. Microbiol. 12:559–567 [DOI] [PubMed] [Google Scholar]
- 8.Poncet S, Milohanic E, Maze A, Nait Abdallah J, Ake F, Larribe M, Deghmane AE, Taha MK, Dozot M, De Bolle X, Letesson JJ, Deutscher J. 2009. Correlations between carbon metabolism and virulence in bacteria. Contrib. Microbiol. 16:88–102 [DOI] [PubMed] [Google Scholar]
- 9.AbuOun M, Suthers PF, Jones GI, Carter BR, Saunders MP, Maranas CD, Woodward MJ, Anjum MF. 2009. Genome scale reconstruction of a Salmonella metabolic model: comparison of similarity and differences with a commensal Escherichia coli strain. J. Biol. Chem. 284:29480–29488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adkins JN, Mottaz HM, Norbeck AD, Gustin JK, Rue J, Clauss TR, Purvine SO, Rodland KD, Heffron F, Smith RD. 2006. Analysis of the Salmonella typhimurium proteome through environmental response toward infectious conditions. Mol. Cell. Proteomics 5:1450–1461 [DOI] [PubMed] [Google Scholar]
- 11.Holzer SU, Hensel M. 2012. Divergent roles of Salmonella pathogenicity island 2 and metabolic traits during interaction of S. enterica serovar Typhimurium with host cells. PLoS One 7:e33220. 10.1371/journal.pone.0033220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim YM, Schmidt BJ, Kidwai AS, Jones MB, Deatherage Kaiser BL, Brewer HM, Mitchell HD, Palsson BO, McDermott JE, Heffron F, Smith RD, Peterson SN, Ansong C, Hyduke DR, Metz TO, Adkins JN. 2013. Salmonella modulates metabolism during growth under conditions that induce expression of virulence genes. Mol. Biosyst. 9:1522–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raghunathan A, Reed J, Shin S, Palsson B, Daefler S. 2009. Constraint-based analysis of metabolic capacity of Salmonella typhimurium during host-pathogen interaction. BMC Syst. Biol. 3:38. 10.1186/1752-0509-3-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gutnick D, Calvo JM, Klopotowski T, Ames BN. 1969. Compounds which serve as the sole source of carbon or nitrogen for Salmonella typhimurium LT-2. J. Bacteriol. 100:215–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jarvik T, Smillie C, Groisman EA, Ochman H. 2010. Short-term signatures of evolutionary change in the Salmonella enterica serovar Typhimurium 14028 genome. J. Bacteriol. 192:560–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shakeri-Garakani A, Brinkkotter A, Schmid K, Turgut S, Lengeler JW. 2004. The genes and enzymes for the catabolism of galactitol, d-tagatose, and related carbohydrates in Klebsiella oxytoca M5a1 and other enteric bacteria display convergent evolution. Mol. Genet. Genomics 271:717–728 [DOI] [PubMed] [Google Scholar]
- 17.Deutscher J, Francke C, Postma PW. 2006. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol. Mol. Biol. Rev. 70:939–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erni B, Zanolari B, Graff P, Kocher HP. 1989. Mannose permease of Escherichia coli. Domain structure and function of the phosphorylating subunit. J. Biol. Chem. 264:18733–18741 [PubMed] [Google Scholar]
- 19.Erni B, Zanolari B, Kocher HP. 1987. The mannose permease of Escherichia coli consists of three different proteins. Amino acid sequence and function in sugar transport, sugar phosphorylation, and penetration of phage lambda DNA. J. Biol. Chem. 262:5238–5247 [PubMed] [Google Scholar]
- 20.Postma PW, Lengeler JW, Jacobson GR. 1993. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 57:543–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erni B, Zanolari B. 1985. The mannose-permease of the bacterial phosphotransferase system. Gene cloning and purification of the enzyme IIMan/IIIMan complex of Escherichia coli. J. Biol. Chem. 260:15495–15503 [PubMed] [Google Scholar]
- 22.Studholme DJ. 2002. Enhancer-dependent transcription in Salmonella enterica Typhimurium: new members of the sigmaN regulon inferred from protein sequence homology and predicted promoter sites. J. Mol. Microbiol. Biotechnol. 4:367–374 [PubMed] [Google Scholar]
- 23.Xu H, Hoover TR. 2001. Transcriptional regulation at a distance in bacteria. Curr. Opin. Microbiol. 4:138–144 [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, Chaney M, Wigneshweraraj SR, Schumacher J, Bordes P, Cannon W, Buck M. 2002. Mechanochemical ATPases and transcriptional activation. Mol. Microbiol. 45:895–903 [DOI] [PubMed] [Google Scholar]
- 25.Neidhardt FC, Bloch PL, Smith DF. 1974. Culture medium for enterobacteria. J. Bacteriol. 119:736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maloy SR, Stewart VJ, Taylor RK. 1996. Genetic analysis of pathogenic bacteria: a laboratory manual. Cold Spring Harbor Laboratory Press, Plainview, NY [Google Scholar]
- 27.Dalcanale E, Montanari F. 1986. Selective oxidation of aldehydes to carboxylic acids with sodium chlorite-hydrogen peroxide. J. Org. Chem. 51:567–569 [Google Scholar]
- 28.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warren JW, Walker JR, Roth JR, Altman E. 2000. Construction and characterization of a highly regulable expression vector, pLAC11, and its multipurpose derivatives, pLAC22 and pLAC33. Plasmid 44:138–151 [DOI] [PubMed] [Google Scholar]
- 30.Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. 1990. Use of T7 RNA polymerase to direct the expression of cloned genes. Methods Enzymol. 185:60–89 [DOI] [PubMed] [Google Scholar]
- 31.Studier FW. 2005. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41:207–234 [DOI] [PubMed] [Google Scholar]
- 32.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3:1101–1108 [DOI] [PubMed] [Google Scholar]
- 33.Karlin S, Campbell AM, Mrazek J. 1998. Comparative DNA analysis across diverse genomes. Annu. Rev. Genet. 32:185–225 [DOI] [PubMed] [Google Scholar]
- 34.Karlin S, Mrazek J, Campbell AM. 1997. Compositional biases of bacterial genomes and evolutionary implications. J. Bacteriol. 179:3899–3913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karlin S, Ladunga I, Blaisdell BE. 1994. Heterogeneity of genomes: measures and values. Proc. Natl. Acad. Sci. U. S. A. 91:12837–12841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campbell AM, Mrazek J, Karlin S. 1999. Genome signature comparisons among prokaryote, plasmid, and mitochondrial DNA. Proc. Natl. Acad. Sci. U. S. A. 96:9184–9189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karlin S, Burge C. 1995. Dinucleotide relative abundance extremes: a genomic signature. Trends Genet. 11:283–290 [DOI] [PubMed] [Google Scholar]
- 38.Garcia E, Bancroft S, Rhee SG, Kustu S. 1977. The product of a newly identified gene, glnF, is required for synthesis of glutamine synthetase in Salmonella. Proc. Natl. Acad. Sci. U. S. A. 74:1662–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chin AM, Feldheim DA, Saier MH., Jr 1989. Altered transcriptional patterns affecting several metabolic pathways in strains of Salmonella typhimurium which overexpress the fructose regulon. J. Bacteriol. 171:2424–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sutrina SL, Chin AM, Esch F, Saier MH., Jr 1988. Purification and characterization of the fructose-inducible HPr-like protein, FPr, and the fructose-specific enzyme III of the phosphoenolpyruvate:sugar phosphotransferase system of Salmonella typhimurium. J. Biol. Chem. 263:5061–5069 [PubMed] [Google Scholar]
- 41.Schneider G, Kack H, Lindqvist Y. 2000. The manifold of vitamin B6 dependent enzymes. Structure 8:R1–R6 [DOI] [PubMed] [Google Scholar]
- 42.Deutscher J. 2008. The mechanisms of carbon catabolite repression in bacteria. Curr. Opin. Microbiol. 11:87–93 [DOI] [PubMed] [Google Scholar]
- 43.Debarbouille M, Martin-Verstraete I, Klier A, Rapoport G. 1991. The transcriptional regulator LevR of Bacillus subtilis has domains homologous to both σ54- and phosphotransferase system-dependent regulators. Proc. Natl. Acad. Sci. U. S. A. 88:2212–2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin-Verstraete I, Charrier V, Stulke J, Galinier A, Erni B, Rapoport G, Deutscher J. 1998. Antagonistic effects of dual PTS-catalysed phosphorylation on the Bacillus subtilis transcriptional activator LevR. Mol. Microbiol. 28:293–303 [DOI] [PubMed] [Google Scholar]
- 45.Curtis SJ, Epstein W. 1975. Phosphorylation of d-glucose in Escherichia coli mutants defective in glucosephosphotransferase, mannosephosphotransferase, and glucokinase. J. Bacteriol. 122:1189–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kornberg HL, Jones-Mortimer MC. 1975. PtsX: a gene involved in the uptake of glucose and fructose by Escherichia coli. FEBS Lett. 51:1–4 [DOI] [PubMed] [Google Scholar]
- 47.Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Anderson I, Lykidis A, Mavromatis K, Ivanova NN, Kyrpides NC. 2010. The integrated microbial genomes system: an expanding comparative analysis resource. Nucleic Acids Res. 38:D382–D390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barrios H, Valderrama B, Morett E. 1999. Compilation and analysis of σ54-dependent promoter sequences. Nucleic Acids Res. 27:4305–4313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iwamoto R, Imanaga Y, Sawada S, Soda K. 1983. Stereochemistry of an alpha, beta-elimination reaction by d-glucosaminate dehydratase. FEBS Lett. 156:33–36 [DOI] [PubMed] [Google Scholar]
- 50.Iwamoto R, Imanaga Y, Soda K. 1979. Purification and properties of d-glucosaminate dehydratase from Agrobacterium radiobacter. FEBS Lett. 104:131–134 [DOI] [PubMed] [Google Scholar]
- 51.Iwamoto R, Imanaga Y, Soda K. 1982. d-Glucosaminate dehydratase from Agrobacterium radiobacter. Physicochemical and enzymological properties. J. Biochem. 91:283–289 [DOI] [PubMed] [Google Scholar]
- 52.Iwamoto R, Imanaga Y, Soda K. 1984. d-Glucosaminate dehydratase: spectrometric properties of the enzyme-bound pyridoxal 5′-phosphate. J. Biochem. 95:13–18 [DOI] [PubMed] [Google Scholar]
- 53.Arrach N, Zhao M, Porwollik S, Hoffman RM, McClelland M. 2008. Salmonella promoters preferentially activated inside tumors. Cancer Res. 68:4827–4832 [DOI] [PubMed] [Google Scholar]
- 54.Bourret TJ, Porwollik S, McClelland M, Zhao R, Greco T, Ischiropoulos H, Vazquez-Torres A. 2008. Nitric oxide antagonizes the acid tolerance response that protects Salmonella against innate gastric defenses. PLoS One 3:e1833. 10.1371/journal.pone.0001833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eriksson S, Lucchini S, Thompson A, Rhen M, Hinton JC. 2003. Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Mol. Microbiol. 47:103–118 [DOI] [PubMed] [Google Scholar]
- 56.Ge S, Danino V, He Q, Hinton JC, Granfors K. 2010. Microarray analysis of response of Salmonella during infection of HLA-B27-transfected human macrophage-like U937 cells. BMC Genomics 11:456. 10.1186/1471-2164-11-456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harvey PC, Watson M, Hulme S, Jones MA, Lovell M, Berchieri A, Jr, Young J, Bumstead N, Barrow P. 2011. Salmonella enterica serovar Typhimurium colonizing the lumen of the chicken intestine grows slowly and upregulates a unique set of virulence and metabolism genes. Infect. Immun. 79:4105–4121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hautefort I, Thompson A, Eriksson-Ygberg S, Parker ML, Lucchini S, Danino V, Bongaerts RJ, Ahmad N, Rhen M, Hinton JC. 2008. During infection of epithelial cells Salmonella enterica serovar Typhimurium undergoes a time-dependent transcriptional adaptation that results in simultaneous expression of three type 3 secretion systems. Cell. Microbiol. 10:958–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lamichhane-Khadka R, Frye JG, Porwollik S, McClelland M, Maier RJ. 2011. Hydrogen-stimulated carbon acquisition and conservation in Salmonella enterica serovar Typhimurium. J. Bacteriol. 193:5824–5832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prouty AM, Brodsky IE, Manos J, Belas R, Falkow S, Gunn JS. 2004. Transcriptional regulation of Salmonella enterica serovar Typhimurium genes by bile. FEMS Immunol. Med. Microbiol. 41:177–185 [DOI] [PubMed] [Google Scholar]
- 61.Yoon H, McDermott JE, Porwollik S, McClelland M, Heffron F. 2009. Coordinated regulation of virulence during systemic infection of Salmonella enterica serovar Typhimurium. PLoS Pathog. 5:e1000306. 10.1371/journal.ppat.1000306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fink RC, Evans MR, Porwollik S, Vazquez-Torres A, Jones-Carson J, Troxell B, Libby SJ, McClelland M, Hassan HM. 2007. FNR is a global regulator of virulence and anaerobic metabolism in Salmonella enterica serovar Typhimurium (ATCC 14028s). J. Bacteriol. 189:2262–2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lawhon SD, Frye JG, Suyemoto M, Porwollik S, McClelland M, Altier C. 2003. Global regulation by CsrA in Salmonella typhimurium. Mol. Microbiol. 48:1633–1645 [DOI] [PubMed] [Google Scholar]
- 64.Troxell B, Fink RC, Porwollik S, McClelland M, Hassan HM. 2011. The Fur regulon in anaerobically grown Salmonella enterica sv. Typhimurium: identification of new Fur targets. BMC Microbiol. 11:236. 10.1186/1471-2180-11-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Steeb B, Claudi B, Burton NA, Tienz P, Schmidt A, Farhan H, Maze A, Bumann D. 2013. Parallel exploitation of diverse host nutrients enhances Salmonella virulence. PLoS Pathog. 9:e1003301. 10.1371/journal.ppat.1003301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bhat UR, Forsberg LS, Carlson RW. 1994. Structure of lipid A component of Rhizobium leguminosarum bv. phaseoli lipopolysaccharide. Unique nonphosphorylated lipid A containing 2-amino-2-deoxygluconate, galacturonate, and glucosamine. J. Biol. Chem. 269:14402–14410 [PubMed] [Google Scholar]
- 67.Cozier GE, Salleh RA, Anthony C. 1999. Characterization of the membrane quinoprotein glucose dehydrogenase from Escherichia coli and characterization of a site-directed mutant in which histidine-262 has been changed to tyrosine. Biochem. J. 340:639–647 [PMC free article] [PubMed] [Google Scholar]
- 68.Erni B, Zanolari B. 1986. Glucose-permease of the bacterial phosphotransferase system. Gene cloning, overproduction, and amino acid sequence of enzyme IIGlc. J. Biol. Chem. 261:16398–16403 [PubMed] [Google Scholar]
- 69.Matsushita K, Arents JC, Bader R, Yamada M, Adachi O, Postma PW. 1997. Escherichia coli is unable to produce pyrroloquinoline quinone (PQQ). Microbiology 143:3149–3156 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.