

Abstract
Nε-lysine acetylation was recently discovered on many bacterial proteins that function in diverse cellular processes. Thus, many questions remain unanswered. For example, what mechanisms regulate lysine acetylation? Does acetylation affect physiology? To help answer these questions, we studied the Escherichia coli response regulator and transcription factor RcsB, which is reported to be acetylated in vitro. To characterize RcsB acetylation, we monitored transcription from the rprA promoter, which requires RcsB. The conventional view is that RcsB is activated by phosphorylation through either the Rcs phosphorelay or acetyl phosphate. We affirmed that rprA transcription requires phosphorylated RcsB and showed that acetyl-phosphate (AcP) is a phosphoryl group donor to RcsB. However, a mutant that accumulates AcP (ackA) exhibited a reduction in rprA transcription instead of the predicted increase. rprA transcription also diminished in the cobB mutant, which lacks the only known E. coli protein deacetylase. This suggests the existence of an inhibitory mechanism that involves lysine acetylation, a supposition supported by the observation that RcsB isolated from the ackA or cobB mutant was hyperacetylated. Finally, we used a genetic approach to identify an AckA- and CobB-sensitive lysine (Lys-154) that controls RcsB activity. We propose that acetylation inhibits RcsB activity and that some of this inhibition acts through the acetylation of Lys-154.
INTRODUCTION
More than 300 posttranslational modifications (PTMs) have been identified (1, 2). By decorating proteins in various combinations, PTMs can alter charge, size, hydrophobicity, or conformation and thus a protein's activity, stability, or cellular location and/or its affinity for its binding partners (3). Phosphorylation is one of the most common PTMs. In bacteria and lower eukaryotes, phosphorylation of histidinyl and aspartyl residues allows two-component signal transduction systems to sense environmental change and, in response, to appropriately alter cellular behavior. The Rcs phosphorelay is a complex two-component signal transduction pathway composed of three proteins, the hybrid sensor RcsC, the histidine phosphotransferase RcsD, and the response regulator (RR) RcsB (Fig. 1A). In response to certain extracellular stimuli, the hybrid sensor RcsC autophosphorylates its conserved histidinyl residue and donates that phosphoryl group to a conserved aspartyl residue within its receiver (REC) domain. Next, the phosphoryl group transfers to a conserved histidinyl residue on RcsD, which passes the phosphoryl group to aspartyl residue 56 (Asp-56) within the REC domain of the RR RcsB. Once phosphorylated, RcsB is reported either to form an RcsB homodimer or to form heterodimers with its coactivator RcsA (4) or the LuxR-type transcription factors BglJ (5) and GadE (6). The result is regulated transcription of more than 150 genes in Escherichia coli (7), including the small nonencoding RNA rprA, which activates the translation of stationary-phase sigma factor RpoS (8).
Fig 1.
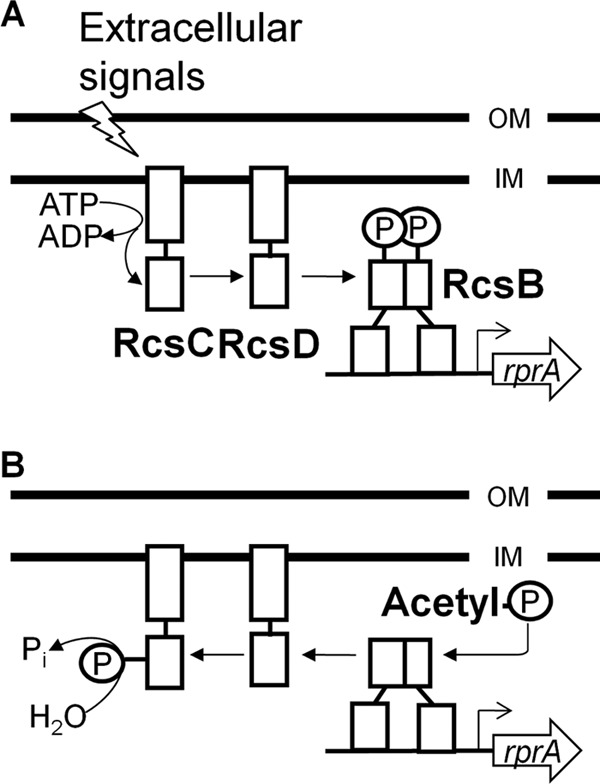
Schematic diagrams of the Rcs phosphorelay. (A) Model of Rcs kinase signaling. In the presence of an activating environmental stimulus, RcsC acts as a net kinase, using ATP as a phosphoryl group donor, this phosphoryl group is transferred from RcsC to the Hpt RcsD, which donates the phosphoryl group and activates RcsB. (B) Model of Rcs phosphatase signaling. Under conditions that stimulate RcsC net phosphatase activity, AcP is an alternative phosphoryl group donor for RcsB. The RcsCD pathway can remove phosphoryl groups from phosphorylated RcsB to generate inorganic phosphate. OM, outer membrane; IM inner membrane.
In the absence of an extracytoplasmic stimulus, the Rcs phosphorelay works in reverse, with RcsD and RcsC functioning as a net phosphatase, dephosphorylating phosphorylated RcsB (Fig. 1B). Under such conditions, it is proposed that the phosphoryl group donor for RcsB is acetyl-phosphate (AcP), the intermediate of the Pta-AckA pathway (9). Disruptions of this central metabolic pathway alter intracellular AcP concentrations (10) and result in correlative changes in in vivo RcsB activities (11). While AcP has been shown to function as the phosphoryl donor to various RRs in vitro (12, 13), its ability to act in this capacity for RcsB has not been demonstrated.
Another PTM is Nε-lysine acetylation, which is thought to use acetyl coenzyme A (acetyl-CoA) as the acetyl donor (3). It was recently reported that RcsB is one of several E. coli transcription factors acetylated in vitro by Pat, a Salmonella enterica Gcn5-like acetyltransferase (14). Both Pat and its E. coli homolog YfiQ (15, 16) (also called Pka [17] or PatZ [18]) acetylated RcsB lysine residue 180 (Lys-180) (14). Deacetylation of Lys-180Ac was catalyzed by CobB (14), a member of the sirtuin family of NAD+-dependent deacetylases (19). Since Lys-180 is located in the DNA-binding helix-turn-helix motif of RcsB, Lys-180 acetylation reduced RcsB's affinity for its DNA site at the flhDC promoter (14). Whether RcsB is acetylated in vivo and whether acetylated RcsB has physiological relevance remained unknown (14, 20).
Here, we provide evidence that the RcsB-dependent rprA promoter is regulated by both phosphorylation and acetylation of RcsB. First, we identify a novel RcsB-dependent behavior and demonstrate that it depends on RcsB phosphorylation. We then show that this behavior is inhibited by loss of CobB's deacetylase activity or by deletion of ackA, which causes AcP to accumulate. The relevant target of CobB and AckA appears to be RcsB, as mass spectrometry (MS) detected several acetylated RcsB lysines, some of which were sensitive to both CobB and AckA. One of those acetylated lysines (Lys-154) appears to play an essential role in RcsB's ability to activate rprA transcription but not in its ability to inhibit migration through semisolid agar. To the best of our knowledge, this is the first report that acetylation of a bacterial transcription factor could have an effect in vivo.
MATERIALS AND METHODS
Bacterial strains, plasmids, and bacteriophage.
All of the bacterial strains used in this study are listed in Table 1. Derivatives were constructed by generalized transduction with P1kc as described previously (21). The transcriptional fusion ϕ(PrprA-lacZ), carried by λPrprA as described previously (8), was a generous gift from Susan Gottesman (National Institutes of Health, Bethesda, MD). Construction of monolysogens was performed and verified as described previously (22, 23).
Table 1.
Bacterial strains, bacteriophage, plasmids, and primers used in this study
| Strain, phage, plasmid, or primer | Relevant characteristic(s) | Source and/or reference(s) |
|---|---|---|
| E. coli strains | ||
| BW25113 | F− λ− Δ(araD-araB)567 Δ(rhaD-rhaB)568 ΔlacZ4787 rrnB3 rph-1 hsdR514 | 69 |
| AJW678 | thi-1 thr-1(Am) leuB6 metF159(Am) rpsL136 ΔlacX74 | 70 |
| AJW3759 | BW25113 λΦ(PrprA142-lacZ) | λ: λrprA142 → BW25113 |
| AJW3331 | AJW678 λΦ(PrprA142-lacZ) | λ: λrprA142 → AJW678 |
| AJW4884 | AJW3759 ΔrcsB::FRT | P1: JW2205 (71) → AJW3759, then antibiotic marker removed |
| AJW3976 | AJW3759 ΔrcsC::tet | P1: MZ63 (72) → AJW3759 |
| AJW3981 | AJW3759 ΔackA::FRT-kan-FRT | P1: JW2293 (71) → AJW3759 |
| EPB6 | Δ(ackA pta)::cat | Mark Goulian (University of Pennsylvania) |
| AJW4030 | AJW3759 Δ(ackA pta)::cat | P1: EPB6 → AJW3759 |
| AJW5187 | AJW3759 Δ(ackA pta)::cat ΔrcsC::tet | P1: MZ63 (72) → AJW4030 |
| JE8659 | ΔcobB::cat | J. Escalante-Semerena (University of Georgia) |
| AJW5011 | AJW3759 ΔcobB::cat | P1: JE8659 → AJW3759 |
| AJW3797 | AJW3759 ΔyfiQ::FRT-kan-FRT | P1: JW2568 (71) → AJW3759 |
| EPI300TM-T1R | F− mcrA Δ(mrr-hsdRMS-mcrBC) φ80dlacZΔM15 lacX74 recA1 endA1 araD139 (ara-leu)7697 galU galK rpsL nupG trfA tonA dhfR | Epicentre Biotechnologies |
| BL21(DE3) Magic | Derivative of BL21 cells carrying a plasmid encoding rare tRNAs | 29 |
| Phage λrprA142 | rprA142-lacZ | 8 |
| Plasmids | ||
| pVEC | Single-copy, lacZ-null derivative of pCC1 (Epicentre), control for pLIH002 and pLIH003, Cmr | 27 |
| pLIH001 | pVEC derivative with kanamycin cassette removed | This study |
| pLIH002 | pLIH001 carrying rcsB(WT), Cmr | This study |
| pLIH003 | pLIH001 carrying rcsBD56A allele, Cmr | This study |
| pLIH004 | pLIH001 carrying rcsBK154Q allele, Cmr | This study |
| pLIH005 | pLIH001 carrying rcsBK154R allele, Cmr | This study |
| pLIH006 | pLIH001 carrying rcsBK154A allele, Cmr | This study |
| pTrc99a | Vector control for pPSG980, Apr | 73 |
| pPSG980 | pTrc99a carrying Φ(PlacUV5-rcsC), Apr | 7, 74 |
| pMH300 | pTrc99a carrying Φ(PlacUV5-rcsF), Apr | 37 |
| pCA24n | Control plasmid, Cmr | 28 |
| pCA24n-rcsB | pCA24n expressing His6-RcsB from an IPTG-inducible promoter, Cmr | 28 |
| pHis6rcsB | pIM10 vector expressing His6-RcsB from an IPTG-inducible promoter, Knr Spr | 31 |
| Primers (5′-3′) | ||
| rcsBFBamHI | GGATCCAGGAAGGTAGCCTATTACATGAAC | This study |
| rcsBRBamHI | GGATCCTTAGTCTTTATCTGCCGGACTTAAGG | This study |
| pCC1RBZ | GTAAAACGACGGCCAGTGAATTG | This study |
| rcsBK154Q | ACAAGCGTCTCTCGCCACAGGAGAGTGAAGTTCTGCG, AAA→CAG | This study |
| rcsBK154A | TGACAAGCGTCTCTCGCCAGCAGAGAGTGAAGTTCTGC, AAA→GCA | This study |
| rcsBK154R | ACAAGCGTCTCTCGCCAAGAGAGAGTGAAGTTCTG, AAA→AGA | This study |
| rcsBD56A | CATGTGTTGATTACCGCTCTCTCCATGCCTGGC, GAT→GCT | This study |
Culture conditions.
For strain construction, cells were grown in LB containing 1% (wt/vol) tryptone, 0.5% (wt/vol) yeast extract, and 0.5% (wt/vol) sodium chloride; LB plates also contained 1.5% agar. For promoter activity assays, cells were grown in TB7, which contains 1% (wt/vol) tryptone buffered at pH 7.0 with potassium phosphate (100 mM). Transformations were performed by electroporation or through the use of either transformation buffers 1 and 2 (24) or transformation-and-storage solution (25). Cell growth was monitored spectrophotometrically (DU640; Beckman Instruments, Fullerton, CA) by determining the absorbance at 600 nm (A600). Spectinomycin (100 μg/ml), ampicillin (100 μg/ml), and chloramphenicol (25 μg/ml for experiments with pCA24n-yfiQ or 17.5 μg/ml for experiments with pCC1-derived plasmids) were added to growth media when needed. To induce the expression of genes carried from various plasmids, isopropyl-β-d-thiogalactopyranoside (IPTG) was added at the concentrations indicated.
Promoter activity assays.
To monitor the promoter activity of ϕ(PrprA-lacZ), cells were grown aerobically at 37°C in TB7 overnight. The overnight cultures were diluted in fresh TB7 and grown aerobically at 37°C until early stationary phase. At regular intervals, 50-μl aliquots were harvested and added to 50 μl of All-in-One β-galactosidase reagent (Pierce Biochemical). β-Galactosidase activity was determined quantitatively by using a microtiter format as described previously (26). As a blank, 50 μl of sterile TB7 was used. Promoter activity was always determined across the entire growth curve and was plotted versus either time or A600; for some experiments, however, only peak activity during exponential phase is shown. Each experiment included three biological replicates unless otherwise mentioned. The values represent the means with standard deviations. All experiments were performed at least twice.
RcsB cloning into a single-copy vector.
To construct a single-copy plasmid that permits the expression of moderate amounts of wild-type (WT) RcsB, we cloned the WT rcsB allele into a derivative of CopyControl pCC1 (EpiCentre). We began with pVEC, a derivative of pCC1 in which plasmid-encoded lacZα is disrupted by a kanamycin resistance cassette (Table 1) (27). To replace the kanamycin resistance cassette with the WT rcsB allele, we first constructed a derivative of pVEC that lacked the kanamycin resistance cassette. pVEC was BamHI digested, ligated, transformed into TransforMax EPI300-T1 chemically competent E. coli (EpiCentre), and plated onto an LB plate containing chloramphenicol (17 μg/ml) and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal). A blue transformant was isolated and grown overnight at 37°C in LB supplemented with chloramphenicol (17 μg/ml) to maintain the plasmid and with CopyControl BAC Autoinduction Solution (EpiCentre) to amplify the plasmid. The plasmid DNA was purified, and this pCC1 derivative was designated pLIH001 (Table 1). We then amplified the WT rcsB allele from the chromosome of strain AJW3759 (Table 1) by using primers rcsBFBamHI and rcsBRBamHI (Table 1). The resulting amplicon was purified with the CloneJET PCR purification kit (Thermo Scientific) and cloned into pJET1.2 with the CloneJET PCR cloning kit (Thermo Scientific). The ligated product was transformed into competent DH5α and plated onto LB ampicillin plates. The WT rcsB allele was excised from pJET1.2 with BamHI, gel extracted, subcloned into BamHI-digested pLIH001, transformed into EPI300 cells, and plated onto LB plates containing chloramphenicol and X-Gal. White colonies were used to inoculate LB supplemented with chloramphenicol and CopyControl BAC Autoinduction Solution (EpiCentre) and shaken overnight at 37°C. Plasmid DNA was purified and sequenced with primer pCC1RBZ (Table 1). The resulting plasmid was named pLIH002 (Table 1). For experiments that involved pLIH002 or derivatives, pVEC functioned as the negative control.
Site-directed mutagenesis.
Site-directed mutagenesis of RcsB in pLIH002 was conducted with the QuikChange Lightning Multi site-directed mutagenesis kit (Agilent Technologies) in accordance with the manufacturer's instructions by using the mutagenic primers listed in Table 1.
Cloning, recombinant expression, and purification.
To purify proteins used for in vitro studies, we transformed the chloramphenicol resistance ASKA plasmids that encode YfiQ or RcsB with a noncleavable N-terminal polyhistidine (His6) tag (28) into kanamycin-resistant BL21 Magic cells (29). The ASKA-encoded, His6-tagged protein was expressed in the presence of 34 μg/ml chloramphenicol and 35 μg/ml kanamycin and purified by immobilized metal affinity chromatography, followed by size exclusion chromatography, as described previously (30, 35).
Dynamic light scattering.
For characterization of molecular size in solution, dynamic light scattering (DLS) provides a measure of the translational diffusion coefficient of a protein in solution. All DLS measurements were performed on a Zetasizer Nano-S Zen 1600 DLS instrument with a 633-nm wavelength laser. The fluctuations in light intensity due to the Brownian motion of the molecules were measured by a photodiode at a 90° angle. Photons were counted, and the time dependence of the light intensity fluctuations was analyzed by autocorrelation. Assumptions include a solution viscosity equal to 1.019 and that the proteins are spherical in nature. The proteins were centrifuged for 5 min with 14,000 × g prior to the collection of scattering data. Experiments were run at 25°C, and at least six measurements were taken under each condition. Regularization histogram analyses of samples were carried out with the Zetasizer Nano software.
LTQ-Orbitrap Velos liquid chromatography (LC)-tandem MS (MS/MS) analysis.
For identification of in vivo acetylation sites, cells transformed with pHis6rcsB (which expresses His6-tagged RcsB [31]) were grown aerobically at 37°C at 250 rpm in TB7 supplemented with 50 μM IPTG and spectinomycin until entry into stationary phase, and cell lysates were prepared. For the identification of in vivo acetylation of cells inhibited for sirtuin activity, 20 mM nicotinamide (NAM) was added to the growth medium. For identification of in vitro acetylation sites, His6-tagged RcsB and His6-tagged YfiQ proteins were purified from E. coli cells (see below). The proteins in the in vivo and in vitro samples were separated by SDS-PAGE. To normalize the number of cells to lyse per milliliter, the inverse of the culture A600 was used (32). To avoid cross-contamination, we electrophoresed the samples from each strain and/or condition on separate SDS-polyacrylamide gels with bioreplicates separated by an empty lane(s). Electrophoresis was terminated before proteins could run off the bottom of the gel. The gel was cut down the middle of the empty lane to separate the bioreplicates. Each half of the gel was stained with SimplyBlue SafeStain (Invitrogen) and destained with water. The bands were then excised and subjected to tryptic digestion as described previously (33). Peptides eluted from tryptic digests of gel pieces were subjected to reversed-phase column chromatography (self-packed C18 column, 100-μm diameter by 200-mm length) operated on an Easy-nLC II (Thermo Fisher Scientific, Waltham, MA). Elution was performed with a binary gradient of buffer A (0.1% [vol/vol] acetic acid) and buffer B (99.9% [vol/vol] acetonitrile; 0.1% [vol/vol] acetic acid) over a period of 100 min with a flow rate of 300 nl/min. MS and MS/MS data were acquired with the LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) equipped with a nanoelectrospray ion source. The Orbitrap Velos was operated in data-dependent MS/MS mode by using the lock-mass option for real-time recalibration. After a survey scan in the Orbitrap (resolution of 30,000), MS/MS data were recorded for the 20 most intensive precursor ions in the linear ion trap. For MS/MS analysis, singly charged ions were not taken into account.
Identification of lysine acetylation sites.
Posttranslational lysine acetylation sites on proteins were identified by searching all MS/MS spectra in .dta format against an E. coli database (extracted from the Uniprot-KB database [http://www.uniprot.org/uniprot/?query=Escherichia+coli+K12&sort=score]), with Sorcerer-SEQUEST (Sequest v. 2.7 rev. 11 Thermo Electron including Scaffold_3_00_05; Proteome Software Inc., Portland, OR). The Sequest search was carried out within the parameters of a parent ion mass tolerance of 10 ppm and a fragment ion mass tolerance of 1.00 Da. Up to two tryptic miscleavages were allowed. Methionine oxidation (+15.99492 Da), cysteine carbamidomethylation (+57.021465 Da), and lysine acetylation (+42.010571 Da) were set as variable modifications. Proteins were identified by at least two peptides by applying a stringent SEQUEST filter. Sequest identifications required deltaCn scores of greater than 0.10 and XCorr scores of greater than 2.2, 3.3, and 3.8 for doubly, triply, and quadruply charged peptides. Acetylated peptides that met these filter criteria were examined manually and accepted only when b or y ions confirmed the acetylation site.
Quantification of acetylated peptides.
To compare the abundance of acetylated peptides in different biological samples, we used Scaffold (Sequest v. 2.7 rev. 11 Thermo Electron including Scaffold_3_00_05; Proteome Software Inc., Portland, OR) to perform spectral count (SC) analysis. After fragmentation of a specific peptide, Scaffold can quantify the protein amounts in different samples according to the number of MS/MS fragment ion spectra. If a peptide was abundant in the overview MS scan, then many MS/MS fragment ion spectra were generated for this peptide. The number of these spectra is considered to be the SC for that peptide. To compare the abundances of any given acetylated peptide between samples, we calculated the ratio of the SC of one sample to that of the other. In-gel tryptic digests for MS analysis are not suited to added peptide standards; therefore, we used a semiquantitative approach. We processed and measured all of the samples to be compared by the same methods, at the same time, in parallel, and loaded equal amounts of tryptic digest onto the LC column. Each comparison was performed with multiple replicates.
In vitro acetylation reactions.
Two different methods were used to detect in vitro acetylated proteins using acetyl-CoA as the acetyl group donor: (i) detection by anti-acetylated lysine Western immunoblot assay and (ii) detection of acetyl-CoA hydrolysis.
(i) In vitro acetylation followed by anti-acetyllysine Western immunoblot analysis.
A combination of 20 μM RcsB, 0.4 mM acetyl-CoA, and 2 μM YfiQ was incubated at 37°C for 1 h in 50 mM Tris HCl (pH 8)–10% glycerol–1 mM dithiothreitol (DTT)–10 mM sodium butyrate–0.1 mM EDTA as described previously (34). (For the Western immunoblot analysis, see below.)
(ii) In vitro acetylation, followed by measurement of acetyl-CoA hydrolysis.
In vitro acetylation, followed by measurement of acetyl-CoA hydrolysis, detects acetylation by measuring the hydrolysis of acetyl-CoA. Biochemical assays were performed as described previously (35), with some modifications. First, reactions were performed in the presence of 150 mM NaCl at 25°C for 1 h to prevent protein precipitation during the reaction. Additionally, after stopping the reactions with guanidine HCl, samples were transferred to Nanosep centrifugation devices with a molecular weight cutoff of 10,000 (Pall Life Sciences, VWR) and centrifuged at 17,000 × g for 10 min to separate reduced CoA (CoASH) and the protein. Ellman's reagent was then added to the flowthrough and monitored as described previously (35). This biochemical in vitro acetylation assay was performed in the absence of DTT because this reducing agent generates false-positive signals. Reactions reached completion within 1 h, and a saturating concentration of RcsB was determined to be 0.15 mM. A large amount of YfiQ (100 μg) was necessary to detect activity, possibly because of competition for binding between acetyl-CoA and the His6 tag (35).
In vitro phosphorylation reactions.
In vitro phosphorylation was carried out by incubating lithium potassium AcP (Sigma) with purified, His-tagged RcsB at 30°C for 30 min in buffer (40 mM Tris-HCl [pH 8.0], 10 mM MgCl2, 40 mM KCl, 1 mM DTT) as described previously (36).
Detection of phosphorylated RcsB.
Similar to a previously reported procedure (36), phosphorylated RcsB from in vitro phosphorylation was separated from nonphosphorylated RcsB by zinc(II) Phos-Tag SDS-PAGE [10% acrylamide (37.5:1), 350 mM Tris (pH 6.8), 0.1% SDS, 75 μM Phos-Tag (NARD Institute Ltd.), 150 μM Zn(NO3)2]. Purified protein was detected by staining the gel with SimplyBlue (Invitrogen).
Western immunoblot analysis.
Purified proteins were separated by SDS-PAGE. Proteins were transferred onto a methanol-treated polyvinylidene difluoride membrane at 25 to 30 V at 4°C for 16 h. The blot was blocked with 5% (wt/vol) milk prepared in phosphate-buffered saline (PBS) for 1 h at room temperature. The blot was washed with PBS–Triton X-100 (PBST) for 5 min. A rabbit polyclonal antibody raised against an acetylated lysine-containing peptide (Cell Signaling 9441) was used at a 1:500 dilution in 5% milk in PBS at 4°C overnight. The blot was washed three times for 5 min each with PBST and then incubated with a horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (Cell Signaling 7074S) at a 1:1,000 dilution in 5% milk in PBS for 1 h at room temperature. The blot was washed three times for 5 min each with PBST and exposed with 20× LumiGLO Reagent and Peroxide (Cell Signaling 7003).
Motility assays.
Three independent colonies representing three biological replicates were used to inoculate 5 ml of tryptone broth (TB), which contains 1% (wt/vol) tryptone and 0.5% (wt/vol) sodium chloride, and grown aerobically with agitation at room temperature. These overnight cultures were diluted to a final optical density at 600 nm of 0.1 in TB. Five microliters was inoculated onto the surface of tryptone motility plates (TB supplemented with 0.25% [wt/vol] agar) and incubated at 28°C, and the diameter was measured over time. The values represent the means with standard deviations. The experiment included at least three replicates and was repeated at least once.
RESULTS
A novel RcsB-dependent behavior requires the phosphoryl acceptor site Asp-56.
To determine how acetylation might regulate RcsB, we monitored RcsB activity by using the canonical RcsB-dependent rprA promoter (PrprA) fused transcriptionally to lacZ. This PrprA-lacZ fusion was carried on prophage λ (8), which we inserted into its attachment site in the E. coli K-12 BW25113 chromosome. We grew the resultant λPrprA-lacZ lysogen (strain AJW3759, Table 1) at 37°C in buffered tryptone broth (TB7), monitored β-galactosidase activity over time, and observed that rprA transcription peaked during exponential phase (Fig. 2A and B), a behavior that required the RR RcsB (Fig. 2B).
Fig 2.
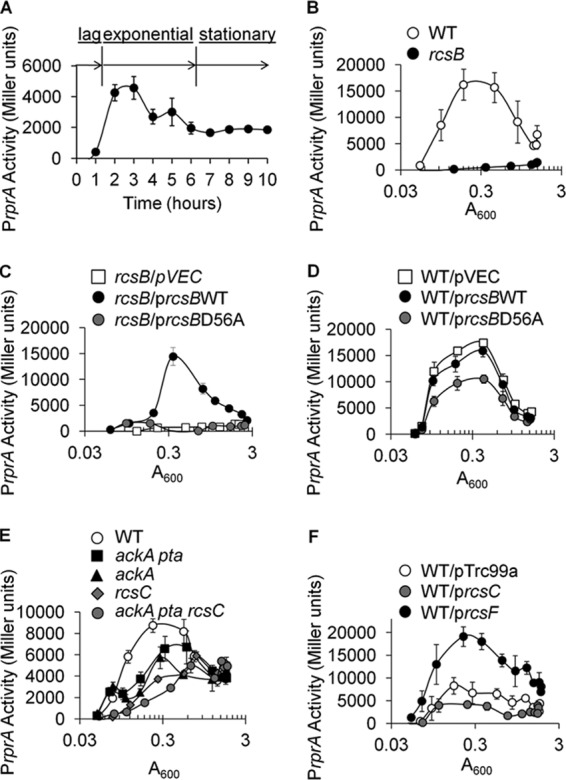
rprA transcription in exponential phase requires conserved aspartyl residue 56 of RcsB and is regulated by RcsC and AcP. (A) PrprA activity of WT BW25113 cells lysogenized with a λPrprA-lacZ transcriptional fusion (strain AJW3759) and aerated at 37°C in TB7. Cells were harvested at multiple points throughout growth to measure A600 and β-galactosidase activity. The values represent the means of triplicate independent cultures with standard deviations. (B) PrprA activity of WT cells (AJW3759) and the isogenic rcsB (AJW4884) mutant. The values represent the means of triplicate independent cultures with standard deviations. (C) PrprA activity of an rcsB null mutant (AJW4884) transformed with a lacZ null derivative of the single-copy expression plasmid pVEC or with a variant of that vector that encodes the WT rcsB allele (pLIH002) or the D56A mutant rcsB allele (pLIH003) aerated at 37°C in TB7 supplemented with 50 μM IPTG. The values represent the means of triplicate independent cultures with standard deviations. (D) PrprA activity of WT cells (AJW3759) transformed with pVEC or pVEC encoding WT rcsB (pLIH002) or the D56A mutant rcsB allele (pLIH003) aerated at 37°C in TB7 supplemented with 50 μM IPTG. The values represent the means of triplicate independent cultures with standard deviations. (E) PrprA activity of WT (AJW3759) cells and ackA (AJW3981), ackA pta (AJW4030), rcsC (AJW3976), and ackA pta rcsC (AJW5187) mutant cells aerated at 37°C in TB7. The values represent the means of triplicate independent cultures with standard deviations. (F) PrprA activity of WT cells transformed with the RcsC expression plasmid prcsC (pPSG980), the RcsF expression plasmid (pMH300), or the vector control (pTrc99a) and aerated at 37°C in TB7 supplemented with 50 μM IPTG to induce rcsC or rcsF expression. The values represent the means of triplicate independent cultures with standard deviations.
To further characterize this novel RcsB-dependent behavior, we asked whether PrprA activity, like other RcsB-regulated behaviors, depends on the phosphoryl acceptor residue (Asp-56) in the REC domain of RcsB. From a single-copy plasmid, we expressed WT rcsB (pLIH002, Table 1) or the D56A mutant allele of rcsB (pLIH003, Table 1) in an rcsB null mutant of the λPrprA-lacZ lysogen (strain AJW4884) and found that WT rcsB complemented exponential-phase PrprA activity while the rcsB-D56A allele did not (Fig. 2C). Since the rcsB-D56A allele carried by pLIH003 inhibited the activity of endogenous WT RcsB (Fig. 2D), we conclude that RcsB-D56A is expressed and that exponential-phase rprA transcription requires Asp-56 of RcsB and thus active, phosphorylated RcsB. Since both the Rcs phosphorelay and AcP have been reported to activate RcsB (11, 37, 38), we tested if these phosphoryl sources could activate PrprA activity. Because the sensor protein RcsC has been reported to function as either a net kinase or a net phosphatase (11, 37, 38), we assessed the influence of RcsC on rprA promoter activity. First, we deleted rcsC and found that PrprA activity diminished relative to that of the WT parent (Fig. 2E), indicating that RcsC activates PrprA transcription. We then overexpressed RcsC, which is reported to decrease phospho-RcsB-dependent mucoidy (11; see Fig. S1 in the supplemental material), and observed inhibition. In contrast, RcsF overexpression, which has been reported to increase mucoidy (11; see Fig. S1), activated rprA transcription (Fig. 2F). We conclude that endogenous RcsC functions as a net kinase in this genetic background grown under these conditions. We further propose that the equilibrium between RcsC kinase and phosphatase activities is sensitive to the RcsC concentration.
AckA influences exponential-phase rprA transcription.
AcP has been shown genetically to activate the Asp-56-dependent RcsB activity required for mucoidy and for inhibition of flagellar biogenesis (11). AcP also has been shown both biochemically and genetically to donate phosphoryl groups to RRs (38). We therefore assessed the role of AcP in activating RcsB at the rprA promoter. To test whether AcP can act as a phosphoryl group donor for RcsB, we used Phos-Tag, a dinuclear metal complex {i.e., 1,3-bis[bis(pyridin-2-ylmethyl)amino]propan-2-olato dizinc(II)} that has affinity for phosphomonoester dianions such as the one found in a phosphorylated RR. When included in SDS-PAGE, Phos-Tag slows the migration of the phosphorylated protein, allowing it to be distinguished from the nonphosphorylated protein by mobility shift. When purified RcsB was incubated with AcP, we observed a shifted band that was sensitive to heat (Fig. 3), indicative of a heat-labile aspartyl phosphate. We conclude that this shifted band is composed of phosphorylated RcsB and that RcsB can catalyze its own phosphorylation by using AcP as the phosphodonor.
Fig 3.

AcP can act as a phosphoryl group donor to RcsB. Purified His6-RcsB peak 2 was incubated in the absence (lane 1) or presence (lanes 2 to 4) of 5 mM AcP for 30 min at 30°C. A fraction of the reaction mixture was heated at 95°C for 15 min to hydrolyze the phosphoryl group (lane 4). The reaction products were separated in a Phos-Tag SDS-polyacrylamide gel, and the gel was stained with Coomassie brilliant blue.
Since AcP can donate its phosphoryl group to RcsB in vitro, we asked whether AcP activates rprA transcription in vivo. The enzyme phosphotransacetylase (Pta) converts acetyl-CoA to AcP, while acetate kinase (AckA) converts AcP to acetate (Fig. 4A). Deletion of both ackA and pta results in a lack of AcP (10). In contrast, deletion of ackA alone causes accumulation of AcP (10). If AcP activated the observed Asp-56-dependent rprA transcription, then PrprA activity would be expected to decrease in the ackA pta double mutant and increase in the ackA null mutant. As predicted, the ackA pta double mutant displayed a small but reproducible reduction in rprA transcription whether or not rcsC was intact (Fig. 2E). These results support the hypothesis that both AcP and RcsC contribute to RcsB phosphorylation. Contrary to expectations, however, rprA transcription was reduced in the ackA mutant rather than increased (Fig. 2E and 4B). The latter result suggests that the accumulation of some metabolic intermediate in the ackA mutant inhibits RcsB activity. We reasoned that this inhibitory mechanism might involve protein acetylation because we have observed that a deletion of ackA changes the acetylation profile of RNA polymerase (36) and enhances global E. coli protein acetylation (B. Zemaitaitis and A. J. Wolfe, unpublished results). We thus considered the possibility that protein acetylation was involved in the reduced rprA transcription exhibited by the ackA mutant.
Fig 4.
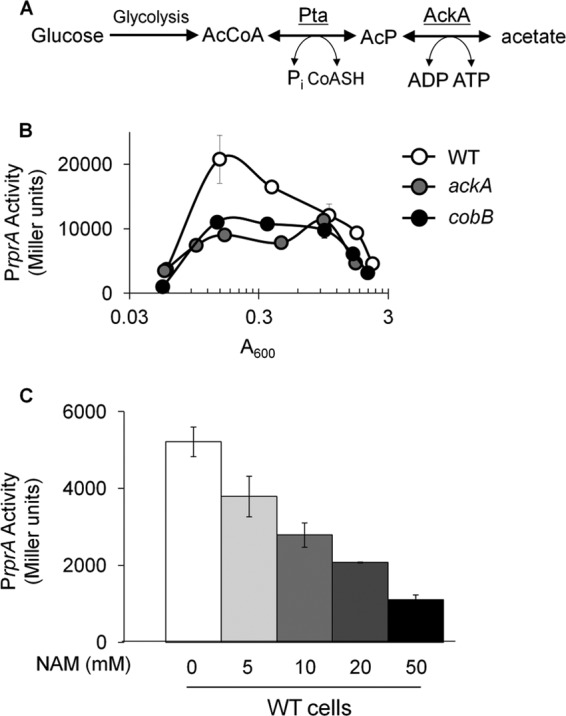
AckA and CobB activate rprA transcription in BW25113. (A) The Pta-AckA pathway. Glucose is metabolized to acetyl-CoA (AcCoA). Pta reversibly converts Pi and acetyl-CoA to generate AcP and reduced CoA (CoASH). AckA reversibly converts AcP and ADP to ATP and excreted acetate. (B) WT cells lysogenized with λPrprA-lacZ (AJW3759) and the isogenic ackA (AJW3981) and cobB (AJW5011) mutants were aerated at 37°C in TB7. Cells were harvested at multiple points throughout growth to measure A600 and β-galactosidase activity. The values represent the means of triplicate independent cultures with standard deviations. (C) Peak β-galactosidase activities between A600s of 0.6 and 0.8 of WT (AJW3759) cells aerated at 37°C in TB7 in the presence or absence of increasing concentrations of NAM. The values indicate the average of triplicate independent cultures with standard deviations.
The deacetylase CobB activates PrprA activity and regulates RcsB acetylation in vivo.
Consistent with the hypothesis that acetylation may regulate rprA transcription, others have reported that RcsB could be acetylated in vitro, that this acetylation reduces RcsB DNA binding affinity, and that this acetylation can be reversed by CobB (14). We therefore tested if protein acetylation inhibits rprA transcription. In the absence of cobB, which encodes the only known E. coli deacetylase, there was a strong reduction in rprA transcription compared to that of its WT parent, similar to that of the ackA mutant (Fig. 4B). Similar results were obtained when WT cells were grown in the presence of NAM, an inhibitor of CobB (39) (Fig. 4C). We conclude that CobB enhances rprA transcription, likely by deacetylating some protein that inhibits rprA transcription in its acetylated form.
Because deletion or inhibition of CobB reduced PrprA activity (Fig. 4B and C) and since CobB deacetylates Lys-180Ac of RcsB in vitro (14), we investigated whether RcsB was acetylated in vivo and whether CobB regulates RcsB deacetylation in vivo. We first attempted to detect acetylated endogenous RcsB but failed because the concentration of RcsB was too low. We therefore used pHis6rcsB (31) to overexpress RcsB in WT cells and in isogenic cobB mutant cells and grew the resulting transformants in TB7 supplemented with and without NAM. For each strain and condition, we performed four biological replicates in parallel and prepared cell lysates. We excised RcsB protein from each SDS-polyacrylamide gel and evaluated the acetylation status by Orbitrap Velos LC-MS/MS analysis. To ensure comparable protein abundances for tryptic digestion and MS analysis, the RcsB proteins obtained from all of the strains were processed in parallel and similar protein concentrations were loaded onto an SDS-polyacrylamide gel. This allowed us to use the Scaffold software to perform a semiquantitative comparison of abundances (given as SCs [40]) for each detected peptide that contained an acetylated lysine (Table 2; see Tables S1 to S3 in the supplemental material).
Table 2.
SCs of lysine-acetylated RcsB peptides isolated from WT cells, cobB mutants, and WT cells exposed to NAMa
| Acetylated lysine | Avg SCb |
SC ratiob |
|||
|---|---|---|---|---|---|
| WT | cobB mutant | WT + NAM | cobB/WT | NAM/WT | |
| 72 | 1.0 | 0.8 | 1.0 | 0.8 | 1.0 |
| 118 | 0.0 | 3.0 | 2.3 | >3.0c | >2.3 |
| 125 | 1.5 | 2.0 | 1.5 | 1.3 | 1.0 |
| 128 | 0.5 | 0.8 | 1.0 | 1.5 | 2.0 |
| 140 | 0.0 | 2.0 | 0.8 | >2.0 | >0.8 |
| 149 | 0.0 | 1.5 | 1.5 | >1.5 | >1.5 |
| 154 | 1.0 | 2.0 | 2.0 | 2.0 | 2.0 |
| 173 | 0.0 | 0.0 | 0.5 | NA | >0.5 |
| 180 | 0.0 | 0.5 | 0.0 | >0.5 | NA |
| Total | 4.0 | 12.5 | 10.5 | 3.1 | 2.6 |
Average SCs of detected lysine acetylation sites in the WT, the isogenic cobB mutant, and the WT treated with NAM are shown. For the SCs of the detected lysine acetylation sites in each replicate and the peptide scores and mass deviations, as well as the complete MS/MS spectra and fragment ion series, see Table S1A to D, S2A to D, and S3A to D in the supplemental material, where A to D denote the four bioreplicates.
Using data obtained from Scaffold 3 proteome software, the SCs of four biological replicates of each strain and condition were determined manually, averaged over the four replicates, and compared as ratios of the mutant or the treated WT relative to the untreated WT.
When the SC of a peptide with an acetylated lysine in the cobB mutant or in the NAM-treated WT is greater than the SC of the same peptide in the untreated WT control, the ratio is underlined. A ratio of >n refers to any acetylated peptide that was not detected in the WT (SC = 0) but was detected in the cobB mutant or the NAM-treated WT (SC = n).
RcsB is acetylated in vivo. In four parallel bioreplicates of the untreated WT strain, Orbitrap Velos MS reproducibly detected the acetylation of four lysine residues, Lys-72, Lys-125, Lys-128, and Lys-154. Each acetylated lysine was identified by at least one SC in at least two replicates, with an average total SC of 4.0 over all four replicates (Table 2; see Table S1A to D in the supplemental material).
CobB affects RcsB acetylation in vivo. In the cobB mutant, MS reproducibly detected the acetylation of eight lysine residues, the four listed above plus Lys-118, Lys-140, Lys-149, and Lys-180 (Table 2; see Table S2A to D in the supplemental material). In the parent strain treated with NAM, MS also reproducibly detected the acetylation of eight lysine residues, the first seven identified in the cobB mutant and Lys-173 instead of Lys-180 (Table 2; see Table S3A to D). Furthermore, the acetylated peptides were more abundant in the cobB mutant (average total SC of 12.5) and in the NAM-treated parent strain (average total SC of 10.5) than in the parent control (average total SC of 4.0). However, these SC increases were not distributed evenly over all of the acetylated lysines, suggesting that CobB does not regulate all of the acetylated RcsB lysines similarly. For example, the average SC for peptides containing acetylated Lys-72 or Lys-125 did not differ significantly between strains or conditions; in contrast, we reproducibly detected the acetylation of three residues (Lys-118, Lys-140, and Lys-149) in the cobB mutant and the NAM-treated parent that was not detected in the untreated parent (Table 2). We conclude that RcsB is acetylated in vivo and that CobB may promote the deacetylation of certain RcsB lysines in vivo.
AckA impacts the acetylation status of RcsB in vivo.
If RcsB acetylation is responsible for the weak rprA transcription in the ackA mutant, then the RcsB acetylation pattern of the ackA mutant should differ from that of WT cells and perhaps resemble that of the cobB mutant. We therefore overexpressed RcsB in the WT strain and its isogenic ackA and cobB mutants, separated the proteins by SDS-PAGE, and subjected the tryptic digests to MS as described above. Once again, we performed replicate experiments in parallel and processed the same amount of protein from all of the samples so that we could compare the SCs for each acetylated peptide in each mutant with those of the WT. When comparing the results of this experiment (Table 3) to those of the previously described experiment (Table 2), we noticed a general increase in the numbers of detected acetylated lysine residues and their SCs. We attribute this difference primarily to the larger amounts of RcsB analyzed in the second experiment, which was performed a year later, and to the use of a different LC column prior to Orbitrap Velos MS.
Table 3.
SCs of lysine acetylated RcsB peptides isolated from WT cells or yfiQ, cobB, or ackA mutant cellsa
| Acetylated lysine | Avg SCb |
SC ratiob |
|||||
|---|---|---|---|---|---|---|---|
| WT | cobB mutant | ackA mutant | yfiQ mutant | cobB/WT | ackA/WT | yfiQ/WT | |
| 63 | 1.0 | 1.0 | 1.5 | 0.0 | 1.0 | 1.5 | 0.0 |
| 72 | 2.0 | 0.5 | 4.5 | 2.0 | 0.3c | 2.3c | 1.0 |
| 118 | 1.5 | 3.5 | 4.5 | 2.5 | 2.4 | 3.0 | 1.7 |
| 125 | 1.5 | 1.5 | 2.0 | 1.5 | 1.0 | 1.4 | 1.0 |
| 128 | 0.5 | 2.0 | 1.0 | 0.0 | 4.0 | 2.0 | 0.0 |
| 140 | 0.0 | 1.5 | 0.5 | 0.0 | >1.5 | >0.5 | 0.0 |
| 149 | 0.0 | 0.5 | 0.0 | 0.0 | >0.5 | 0.0 | 0.0 |
| 154 | 0.5 | 3.0 | 2.0 | 1.0 | 6.0 | 4.0 | 2.0 |
| 173 | 1.5 | 0.0 | 0.0 | 1.5 | 0.0 | 0.0 | 1.0 |
| 180 | 0.0 | 1.0 | 1.0 | 0.0 | >1.0 | >1.0 | 0.0 |
| Total | 8.5 | 14.5 | 17.0 | 8.5 | 1.7 | 2.0 | 1.0 |
Average SCs of detected lysine acetylation sites in the WT and isogenic cobB, ackA, and yfiQ mutants. For the SCs of the detected lysine acetylation sites in each replicate and the peptide scores and mass deviations, as well as the complete MS/MS spectra and fragment ion series, see Tables S4A and B, S5A and B, S6A and B, and S7A and B in the supplemental material, where A and B denote the two bioreplicates.
Using data obtained from Scaffold 3 proteome software, the SCs of two biological replicates of each strain were determined manually, averaged over the two replicates, and compared as ratios of the mutants relative to the untreated WT.
When the SC of a peptide with an acetylated lysine in a mutant is less than the SC of the same peptide in the WT control, the ratio is italicized. When the SC of a peptide with an acetylated lysine in a mutant is greater than the SC of the same peptide in the WT control, the ratio is underlined. A ratio of >n refers to any acetylated peptide that was not detected in WT (SC = 0) but was detected in a mutant (SC = n).
The general pattern, however, was reproducible; acetylated peptides were more abundant in the cobB mutant (average total SC of 14.5) than in the parent control (average total SC of 8.5) (Table 3), supporting the conclusion that CobB may deacetylate RcsB in vivo. For the parent, we reproducibly detected the acetylation of five lysine residues, each identified by an SC of 1 to 2 per replicate (Table 3; see Table S4A and B in the supplemental material). For the cobB mutant, we observed reproducible acetylation of seven lysines, each identified by an SC of 1 to 4 per replicate (Table 3; see Table S5A and B). Moreover, some acetylated peptides were reproducibly more abundant than the same peptides from the parent, suggesting that CobB deacetylates these sites in vivo.
As predicted, the acetylation profile of RcsB isolated from the ackA mutant (Table 3; see Table S6A and B in the supplemental material) generally resembled that of the cobB mutant (Table 3; see Table S5A and B). Of the seven RcsB lysines acetylated in the cobB mutant, six were reproducibly acetylated in the ackA mutant. Several acetylated lysines (Lys-118, Lys-128, and Lys-154) were detected more often in both mutants than in their WT parent. Like RcsB from the cobB mutant (average total SC of 14.5), acetylated peptides from the ackA mutant were 2-fold more abundant (average total SC of 17) than the same peptides from WT cells (average total SC of 8.5). We conclude that disruption of ackA promotes the acetylation of some RcsB lysines in vivo. We further propose that the reduction in rprA transcription in the ackA and cobB mutants results from the same mechanism, an increase in RcsB acetylation.
YfiQ regulates PrprA activity independently of RcsB acetylation.
Because S. enterica Pat and its E. coli homolog YfiQ were reported to acetylate E. coli RcsB in vitro and inhibit its ability to bind DNA (14), we asked if the acetyltransferase YfiQ affects PrprA activity and found that it does; deletion of yfiQ inhibited rprA transcription (Fig. 5). We therefore investigated whether YfiQ affects RcsB acetylation in vivo. However, the acetylation profile of RcsB isolated from the yfiQ mutant did not differ much from that of its parent (Table 3; see Table S7A and B in the supplemental material). While not identical, the lysine acetylation sites were quite similar. Most notably, the overall SCs of the yfiQ mutant and its parent were identical (average total SC of 8.5) and there was no substantial change in the SC for individual acetylated lysines. Thus, the acetyltransferase YfiQ appears to have no effect on the acetylation status of RcsB in vivo under this growth condition. We conclude that YfiQ enhances rprA transcription by acetylating a protein other than RcsB.
Fig 5.
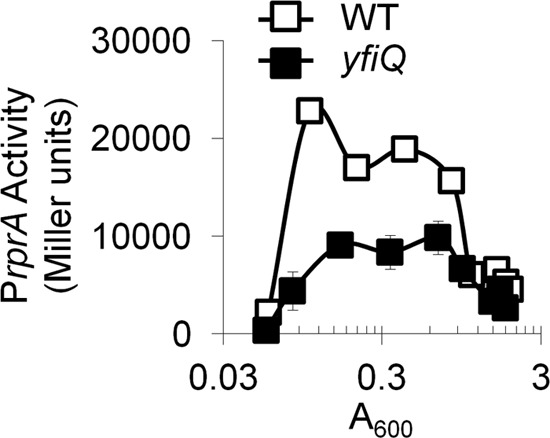
Analysis of the effect of YfiQ at the rprA promoter. λrprA of WT cells (AJW3759) and the isogenic yfiQ null mutant (AJW3797) were grown at 37°C in TB7 with shaking. Samples were harvested for A600 and β-galactosidase activity measurements. The values represent the average of triplicate independent cultures, and error bars indicate standard deviations.
In vitro acetylation of RcsB.
Since our in vivo observations argue that YfiQ does not regulate the acetylation status of RcsB (Table 3; see Table S7A and B in the supplemental material) but the published in vitro results show that YfiQ can acetylate RcsB (14), we investigated the effect of purified YfiQ on the acetylation status of purified RcsB. We first purified RcsB by gel filtration chromatography, finding that the 23.7-kDa RcsB protein purified from WT E. coli cells resolved into two peaks with estimated molecular masses of 100 kDa (high molecular weight or peak 1 [RcsB-1]) and 60 kDa (low molecular weight or peak 2 [RcsB-2]) (see Fig. S2A). Dynamic light scattering revealed that monodisperse solutions of RcsB-1 and RcsB-2 have globular species with exclusion radii of 8.9 and 7.2 nm, respectively (see Fig. S2B and C). Taken together, these results are consistent with the formation in solution of both tetramers (RcsB-1) and dimers (RcsB-2).
Anti-acetyllysine immunoblot analysis detected the acetylation of purified YfiQ (Fig. 6A). It also detected the acetylation of RcsB-1 and RcsB-2, but this acetylation was visible only after a long exposure (see Fig. S3 in the supplemental material). This suggests that YfiQ and a small fraction of both RcsB oligomers were acetylated under the conditions used for purification. Indeed, Orbitrap Velos mass spectrometric analysis (33, 41) detected 10 acetylated lysines in RcsB-1 (Table 4; see Table S8A), 9 acetylated lysines in RcsB-2 (Table 4; see Table S8D), and 8 acetylated lysines in YfiQ (see Table S9A to C). The average total SCs of acetylated peptides for both RcsB-1 (SC of 22) and RcsB-2 (SC of 20) were similar. The distributions of acetylated RcsB peptides also were similar but not identical.
Fig 6.
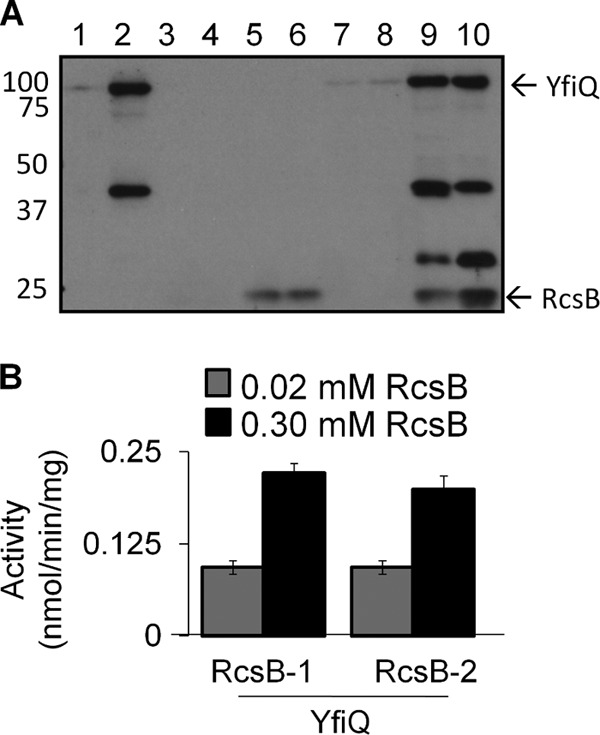
In vitro acetylation of RcsB. (A) In vitro acetylation reactions with RcsB, YfiQ, and acetyl-CoA. Fifty-microliter reaction mixtures containing 21 μM RcsB-1 or -2, 2 μM YfiQ, and 0.4 mM acetyl-CoA were incubated at 37°C for 2 h. The reactions were quenched with 50 μl of 2× SDS-PAGE loading buffer, and the mixtures were heated at 95°C for 5 min. Fifteen microliters was resolved by 13% SDS-PAGE, proteins were transferred, and acetylated proteins were detected by Western immunoblotting with anti-acetyl-lysine antibody. Lane 1, YfiQ alone; lane 2, YfiQ plus acetyl-CoA; lane 3, RcsB-1 alone; lane 4, RcsB-2 alone; lane 5, RcsB-1 plus acetyl-CoA; lane 6, RcsB-2 plus acetyl-CoA; lane 7, YfiQ plus RcsB-1; lane 8, YfiQ plus RcsB-2; lane 9, YfiQ plus RcsB-1 plus acetyl-CoA; lane 10, YfiQ plus RcsB-2 plus acetyl-CoA. These results are representative of three independent experiments. The high-molecular-weight signal in lanes 1, 2, 9, and 10 corresponds to acetylated YfiQ. The acetylated sites were determined by semiquantitative MS (see Table S9A to E in the supplemental material). MS also identified the signal visible between the 37- and 50-kDa markers in lanes 2, 9, and 10 as the 43.3-kDa EF-Tu protein and determined the acetylated sites (see Table S10). MS identified the signal between the 25- and 37-kDa markers in lanes 9 and 10 as the 29.9-kDa RplB protein and determined its acetylated sites (see Table S11). The values to the left are molecular sizes in kilodaltons. (B) Activity of YfiQ with different concentrations of RcsB1 and RcsB2. Fifty-microliter reaction mixtures containing 0.02 mM YfiQ were incubated at 25°C for 1 h. To initiate the reaction, 0.5 mM acetyl-CoA was used. Activity (nmol/min/mg) is shown as the average of three separate trials. Black bars correspond to 0.02 mM RcsB, and red bars correspond to 0.3 mM RcsB.
Table 4.
SCs of acetylated peptides of RcsBa
| Acetylated lysine | RcsB-1 |
RcsB-2 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Avg SCb |
SC ratiob |
Avg SC |
SC ratio |
|||||||
| Control | Acetyl-CoA | Acetyl-CoA + YfiQ | Acetyl-CoA/control | Acetyl-CoA + YfiQ/acetyl-CoA | Control | Acetyl-CoA | Acetyl-CoA + YfiQ | Acetyl-CoA/control | Acetyl-CoA + YfiQ/acetyl-CoA | |
| 63 | 2.0 | 1.0 | 0.0 | 0.5c | 0.0 | 0.0 | 0.0 | 0.0 | NA | NA |
| 72 | 2.0 | 2.0 | 1.5 | 1.0 | 0.8 | 0.5 | 2.5 | 1.5 | 5.0 | 0.6 |
| 118 | 5.5 | 7.0 | 10.0 | 1.3 | 1.4 | 6.0 | 8.5 | 10.5 | 1.4 | 1.2 |
| 125 | 2.0 | 4.0 | 13.5 | 2.0c | 3.4 | 3.0 | 4.0 | 9.0 | 1.3 | 2.3 |
| 127 | 0.0 | 1.0 | 2.0 | >1.0 | 2.0 | 0.0 | 1.0 | 2.0 | >1.0 | 2.0 |
| 128 | 3.0 | 4.0 | 6.0 | 1.3 | 1.5 | 1.5 | 4.5 | 7.0 | 3.0 | 1.6 |
| 140 | 0.5 | 19.0 | 3.0 | 38.0 | 0.2 | 0.0 | 3.0 | 3.5 | >3.0 | 1.2 |
| 149 | 1.0 | 2.0 | 5.0 | 2.0 | 2.5 | 2.5 | 5.0 | 5.0 | 2.0 | 1.0 |
| 154 | 3.5 | 8.0 | 4.5 | 2.3 | 0.6 | 3.0 | 6.5 | 5.0 | 2.2 | 0.9 |
| 173 | 2.0 | 2.0 | 2.5 | 1.0 | 1.3 | 1.0 | 2.5 | 2.0 | 2.5 | 0.8 |
| 180 | 0.5 | 1.0 | 6.0 | 2.0 | 6.0 | 1.5 | 2.5 | 6.5 | 1.6 | 2.3 |
| 186 | 0.0 | 1.0 | 3.0 | >1.0 | 3.0 | 1.0 | 0.5 | 2.5 | 0.5 | 5.0 |
| Total | 22 | 52 | 57 | 2.4 | 1.1 | 20 | 40.5 | 54.5 | 2.0 | 1.3 |
Average SCs of detected lysine acetylation sites in purified RcsB (control), purified RcsB incubated with acetyl-CoA, or purified RcsB incubated with acetyl-CoA plus YfiQ are shown. For the SCs of the detected lysine acetylation sites in each replicate and the peptide scores and mass deviations, as well as the complete MS/MS spectra and fragment ion series, see Table S10A to F in the supplemental material, where A to C, respectively, denote RcsB-1 alone, RcsB-1 incubated with acetyl-CoA, and RcsB-1 incubated with both acetyl-CoA and YfiQ and D to F, respectively, denote RcsB-2 alone, RcsB-2 incubated with acetyl-CoA, and RcsB-2 incubated with both acetyl-CoA and YfiQ.
Using data obtained from Scaffold 3 proteome software, the SCs of two replicates of each condition were determined manually, averaged over the two replicates, and compared as the ratio of the SC detected for RcsB incubated with acetyl-CoA or with acetyl-CoA and YfiQ to the SC detected with purified RcsB alone.
When the SC of a peptide with an acetylated lysine following acetyl-CoA incubation is substantially more than the SC of the same peptide in the absence of acetyl-CoA or if the SC of a peptide with an acetylated lysine following incubation with both acetyl-CoA and YfiQ is substantially more than the SC of the same peptide in the presence of acetyl-CoA alone, the ratio is underlined. If the ratio is substantially less, then it is italicized. A ratio of >n refers to any acetylated peptide that was not detected in the absence of acetyl-CoA (SC = 0) but was detected in the presence of acetyl-CoA (SC = n).
For both RcsB oligomers, incubation with acetyl-CoA dramatically increased acetylation (Fig. 6A, compare lanes 5 and 6 to lanes 3 and 4) and increased the abundance of acetylated RcsB peptides by about 2-fold (Table 4; see Table S8B and E in the supplemental material). The distribution of acetylated lysines differed somewhat between the two oligomers. This difference suggests the possibility that the oligomeric structure could influence the acetylation pattern. For YfiQ, incubation with acetyl-CoA also detected increased acetylation of YfiQ (Fig. 6A, compare lane 2 to lane 1; see Table S9D and E), suggesting that YfiQ might autoacetylate.
To determine whether RcsB can be a substrate of YfiQ, we used a biochemical assay that detects acetyl-CoA hydrolysis (35). When RcsB and acetyl-CoA were present at saturating concentrations, the activity of YfiQ toward either RcsB-1 or RcsB-2 was nearly the same (Fig. 6B). For both oligomers, acetyl-CoA hydrolysis increased as the concentration of RcsB was increased; incubation of 0.02 mM YfiQ with 15 times the concentration of RcsB (0.30 mM) hydrolyzed more acetyl-CoA than incubation with an equimolar concentration of RcsB (0.02 mM). This behavior indicates that RcsB acts as a substrate of YfiQ. We also found that YfiQ hydrolyzed acetyl-CoA (see Fig. S4 in the supplemental material), adding support to the supposition that YfiQ might autoacetylate.
To identify the RcsB lysines acetylated in the presence of YfiQ, we used mass spectrometric analysis (33, 41). Following the incubation of either RcsB oligomer with YfiQ and acetyl-CoA, we observed acetylation of Lys-180, consistent with the previous report that YfiQ acetylates Lys-180 in vitro (14), but we also observed YfiQ-dependent acetylation of several other RcsB lysines, Lys-125, Lys-127, Lys-149, and Lys-186 (Table 4; see Table S8C and F in the supplemental material). Surprisingly, incubation with both acetyl-CoA and YfiQ did not dramatically change the abundance of acetylated lysines relative to that seen upon incubation with acetyl-CoA alone (Table 4; see Table S8B and E). Anti-acetyl-lysine immunoblot analysis yielded a similar result (Fig. 6A, compare lanes 9 and 10 to lanes 5 and 6). Instead, the pattern of acetylation changed. For some residues, the level of acetylation increased relative to their status after incubation with acetyl-CoA alone. For other residues, the acetylation decreased (Table 4). For example, both RcsB-1 and RcsB-2 exhibited increased SCs for acetylated Lys-125, Lys-180, and Lys-186. In contrast, the SC for acetylated Lys-140 of RcsB-1 decreased by more than 5-fold (Table 4). While these in vitro results show that YfiQ can affect the acetylation status of multiple RcsB lysines, our in vivo results showing that YfiQ does not affect RcsB acetylation (Table 3; see Table S7A and B) argue against a direct role for YfiQ in RcsB acetylation, at least under the conditions tested.
Analysis of the function of acetylated RcsB lysines in rprA transcription.
In the absence of ackA or cobB, rprA transcription was inhibited and RcsB acetylation increased. One RcsB lysine (Lys-154) that was reproducibly more acetylated in the mutants than in the WT parent (Tables 2 and 3) was found to be critical for RcsB activity. To assess the effect of acetylation on RcsB function, we applied an approach commonly used in studies of acetylation in eukaryotes (42), converting this AckA- and/or CobB-sensitive lysine to a glutamine (which mimics a neutral, acetylated lysine) or to an arginine (which mimics a positive, nonacetylatable lysine). We introduced this rcsB variant (carried by pVEC) into the rcsB null strain (AJW4884) and monitored PrprA activity. The acetylated and nonacetylated mimics of Lys-154 exhibited opposite behaviors. The mimic of acetylated Lys-154 (RcsB-K154Q) exhibited a strong reduction in PrprA activity, a behavior that was indistinguishable from that of the vector control (Fig. 7A), the nonphosphorylatable mutant RcsB-D56A, and the RcsB-K154A mutant (data not shown). This contrasted with the behavior of the nonacetylated mimic (RcsB-K154R), which more closely resembled that of the WT protein (Fig. 7A). We next tested whether the inability of RcsB-K154Q to support PrprA activity was due to a lack of expression or to a loss of function. To do so, we took advantage of the report that RcsB inhibits migration through semisolid agar by repressing the transcription of flhDC, which encodes the master regulator of flagellar biosynthesis (11, 43). We investigated if the expression of the mutant rcsB alleles could inhibit the migration of highly motile strain AJW3331. Indeed, each mutant rcsB allele (D56A, K154A, K154Q, and K154R) inhibited migration to a level similar to that of the WT allele (Fig. 7B). We conclude that the mutants are expressed and that they retain function. We therefore propose that the positive charge of Lys-154 is critical to RcsB-dependent activation of rprA transcription and that acetylation of Lys-154 may regulate RcsB activity.
Fig 7.
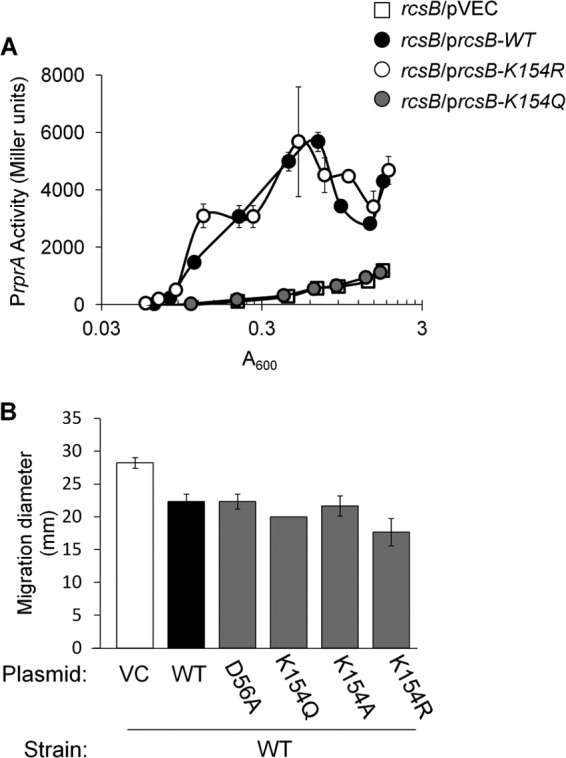
Analysis of the role of Lys-154 in rprA transcription and migration. (A) PrprA activity of rcsB mutant (AJW4884) cells transformed with the single-copy plasmid pVEC or pVEC carrying the WT and K154 mutant rcsB alleles. Cells were aerated at 37°C in TB7 supplemented with 50 μM IPTG. (B) Migration analysis of the highly motile WT strain (AJW3331) transformed with pVEC or pVEC carrying the WT and mutant rcsB alleles. The histogram shows the last measurement of an experiment with an 8.5-h time course. These results are representative of the whole experiment.
DISCUSSION
Given the diversity of the bacterial proteins that have been detected to be acetylated (44–46), there is great potential for this PTM to impact many facets of bacterial physiology. However, the study of bacterial protein acetylation is young and many fundamental questions remain unanswered. In this report, we describe genetic and biochemical analyses of RcsB acetylation that have led to four key findings. First, we provided the first evidence that in vivo acetylation of a bacterial transcription factor could regulate its activity. Second, we verified that RcsB is a substrate of CobB (14) and showed that it regulates the acetylation status of RcsB in vivo. Third, we showed that rprA transcription is activated by YfiQ but likely not by direct acetylation of RcsB. Since YfiQ is the only known protein acetyltransferase in E. coli, this result suggests that other mechanisms of acetylation might exist in E. coli. Fourth, we showed that the Pta-AckA pathway influences protein acetylation.
Both phosphorylation and acetylation regulate rprA transcription.
Majdalani et al. reported that PrprA activity and rprA RNA levels depend on RcsB. As cultures transition into stationary phase, rprA RNA levels increase 8-fold, while PrprA activity increases only 2.5-fold. Thus, these authors proposed that RprA is stabilized during the transition, allowing accumulation in stationary phase. The net result is increased expression of the alternative sigma factor σS (8, 47). We have extended this model, showing that PrprA activity, and thus RprA synthesis, can peak during exponential growth in buffered tryptone broth (Fig. 2A and B). While the physiological impact of this novel behavior remains obscure, it has given us a unique opportunity to investigate the effect that Nε-lysine acetylation exerts on the function of a phosphorylated transcription factor.
Like other reported RcsB-activated behaviors, this exponential burst of rprA transcription requires the phosphoacceptor site (Asp-56) of RcsB (Fig. 2C). Like many of these behaviors, this novel exponential-phase rprA transcription required the kinase activity of bifunctional RcsC (Fig. 2E). It also involved AcP, as reported previously for two other RcsB-regulated promoters, those of cps and flhD (11). In this study, we showed that purified RcsB can become phosphorylated when incubated with AcP (Fig. 3).
If AcP donates its phosphoryl group to RcsB, then why is phospho-RcsB-dependent rprA transcription inhibited in the ackA mutant? The answer appears to be acetylation. Escalante-Semerena and colleagues previously reported that RcsB could become acetylated on Lys-180 in vitro by YfiQ or Pat, the S. enterica YfiQ homolog, and deacetylated by CobB (14). We have confirmed this report and extended its significance. We confirmed that YfiQ acetylates RcsB in vitro (Table 4 and Fig. 6). We further showed that RcsB acetylation occurs in vivo on multiple lysines (Tables 2 and 3) and that both CobB and AckA affect RcsB acetylation in vivo (Fig. 4). We also provided genetic evidence that acetylation of a specific lysine may exert an effect on RcsB activity (Fig. 7).
What is the effect of acetylation on RcsB?
We propose that the positive charge at position 154 is required for RcsB-dependent rprA transcription and that acetylation of Lys-154 would inhibit RcsB activity, possibly by interfering with the binding of RcsB to its DNA site. In the absence of ackA or cobB, rprA transcription diminished (Fig. 4B) and acetylation of Lys-154 increased substantially (Table 3). Furthermore, a genetic mimic of acetylated Lys-154 (RcsB-K154Q) appeared inactive at the rprA promoter, while a genetic mimic of nonacetylated Lys-154 (RcsB-K154R) remained active (Fig. 7A). Our proposal is supported by a previous report on the structural analysis of the Erwinia amylovora RcsB DNA-binding domain (48). The authors reported that Lys-153 of E. amylovora RcsB (homologous to Lys-154 in E. coli RcsB), positioned in supporting helix a7 of the DNA-binding helix-turn-helix motif, is capable of interacting with DNA. Because the RcsB-K154Q and RcsB-K154R mutants retained the ability to inhibit migration on semisolid agar plates (Fig. 7B), which requires the binding of RcsB to the flhDC promoter, acetylation could impact some later event in the initiation of rprA transcription, such as the recruitment of RNA polymerase. However, while the inhibition of migration provides evidence that the mutant proteins are functional and that RcsB does not require Lys-154 to bind at the flhDC promoter, it does not directly address the role of Lys-154 at the rprA promoter. This is because the flhDC promoter is quite complex and the binding of the mutant RcsB proteins could be aided by one of the many proteins that also regulate flhDC.
CobB regulates RcsB acetylation.
CobB enhanced PrprA activity (Fig. 4B and C), suggesting that CobB activates by deacetylating some acetylated protein. Given that we found that RcsB is hyperacetylated in the cobB mutant (Tables 2 and 3), we propose that acetylation of RcsB is responsible for reducing PrprA activity and that CobB regulates the deacetylation of certain RcsB lysines in vivo. While certain RcsB lysines (e.g., Lys-118 and Lys-154) appeared to be sensitive to CobB (i.e., they exhibited increased acetylation in the absence of CobB activity (Tables 2 and 3), others (e.g., Lys-72 and Lys-125) appeared to be insensitive to CobB, suggesting that some acetylated lysines are not deacetylated or that some as-yet-unknown deacetylase or other deacetylation mechanism exists.
Does AckA influence protein acetylation?
We hypothesize that AckA is a regulator of protein acetylation. We base this hypothesis on the following observations. First, we showed that RcsB is hyperacetylated when isolated from the ackA mutant and that its acetylation profile was similar to that of RcsB isolated from the cobB mutant (Table 3). Second, we previously reported that the acetylation pattern of RNA polymerase isolated from an ackA mutant grown in the presence of glucose is substantially different from the pattern obtained with the WT parent (36). Indeed, preliminary analyses show that ackA mutants exhibit a dramatic increase in global protein acetylation that is eliminated when pta is also deleted (Zemaitaitis and Wolfe, unpublished). Efforts to understand the underlying mechanism are under way.
Does a lysine acetyltransferase regulate RcsB?
The mechanism by which RcsB becomes acetylated in vivo remains unknown. Deletion of yfiQ diminished rprA transcription (Fig. 5), suggesting that YfiQ is an activator of rprA transcription. However, YfiQ is unlikely to act directly upon RcsB, as deletion of yfiQ did not exert a major effect on the acetylation status of RcsB (Table 3). Further support for the proposition that YfiQ is not the physiological acetyltransferase for RcsB acetylation comes from the observation that deletion of yfiQ enhances migration in semisolid agar rather than inhibiting it as predicted if the main role of YfiQ were to acetylate Lys-180 and thus inhibit the binding of RcsB to its DNA site (J. Escalante-Semerena, personal communication).
If YfiQ does not acetylate RcsB in vivo, two scenarios are possible, the existence of another protein acetyltransferase or a mechanism that does not involve an acetyltransferase. The ability of purified RcsB to become acetylated when incubated with acetyl-CoA alone is suggestive of an alternative mechanism of acetylation (Fig. 6 and Table 4). It is unlikely that purified RcsB was contaminated by an acetyltransferase, as in-solution mass spectrometric analysis did not detect a contaminating acetyltransferase (data not shown). An alternative is that RcsB autoacetylates in vivo with acetyl-CoA as the acetyl donor. This mechanism is not unprecedented, as others have reported the autoacetylation of several other proteins (49–52), including proteins in the mitochondrion, where little is known about a potential mitochondrial lysine acetyltransferase(s) and the conditions are favorable for acetyl-CoA-dependent lysine acetylation, i.e., an alkaline pH (∼8.0) and a high acetyl-CoA concentration (∼5 mM) (53–55).
Why is RcsB regulated by both aspartyl phosphorylation and Nε-lysine acetylation?
Since RcsB regulates over 100 different genes that are involved in many aspects of bacterial physiology, it is likely that acetylation affects other RcsB-regulated promoters. Regulation of RcsB with a simple phosphorylation-dependent “on-off” switch may not provide enough sophisticated control of gene expression. Indeed, this study shows that RcsB can be posttranslationally modified in vivo on eight different residues (i.e., phosphorylated on Asp-56 and acetylated on at least seven lysines), raising the opportunity for 256 different isoforms. The existence of multiple RcsB isoforms could increase functional diversity, which could dictate promoter specificity or affinity for the diverse RcsB-interacting proteins (5). Furthermore, multiple and different modifications could interact (56). In bacteria, such an interaction is not unprecedented. In Mycoplasma pneumoniae, serine/threonine/tyrosine phosphorylation and Nε-lysine acetylation are common comodifiers of the same proteins and phosphorylation and acetylation are proposed to regulate each other (57). Like RcsB, CheY is an RR that can be coregulated by phosphorylation and acetylation and these modifications of CheY inhibit each other (58–65). Additionally, we show that RcsB can form dimers or tetramers (see Fig. S2 in the supplemental material), a behavior that has been described for some other RRs (66–68).
Concluding remarks.
In summary, we propose that RcsB can become acetylated on multiple lysines in vivo. We further propose that one of these acetylations (Lys-154) inhibits the ability of RcsB to activate rprA transcription in vivo. Much remains to be learned, including the mechanism(s) by which RcsB becomes acetylated, how acetylation inhibits RcsB function, whether acetylation interacts with phosphorylation, and how the Pta-AckA pathway regulates protein acetylation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Sandy Thao and Jorge Escalante-Semerena for allowing us access to unpublished data. We thank Susan Gottesman, Robert Blumenthal, Kaymeuang Cam, and Jorge Escalante-Semerena for their generous donations of phage, plasmids, or strains. We thank Dhaval Nanavati for his MS identification of acetylated RplB. We thank all of the members of both the Wolfe and Visick labs for critical and open discussions. We specifically thank Sylvia A. Reimann for her help and support, Bruno P. Lima for his help with Phos-Tag, Bozena Zemaitaitis for her analyses of global acetylation in E. coli, and Richard M. Schultz for his modeling of RcsB structure.
This work was supported by NIH grant GM066130 and Loyola University Chicago Potts Foundation award LU11200 awarded to A.J.W. and by Deutsche Forschungsgemeinschaft project AN746/2-1 awarded to H.A. It also was supported with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contracts HHSN272200700058C and HHSN272201200026C awarded to W.F.A.
Footnotes
Published ahead of print 12 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00383-13.
REFERENCES
- 1.Witze ES, Old WM, Resing KA, Ahn NG. 2007. Mapping protein post-translational modifications with mass spectrometry. Nat. Methods 4:798–806 [DOI] [PubMed] [Google Scholar]
- 2.Gupta N, Tanner S, Jaitly N, Adkins JN, Lipton M, Edwards R, Romine M, Osterman A, Bafna V, Smith RD, Pevzner PA. 2007. Whole proteome analysis of post-translational modifications: applications of mass-spectrometry for proteogenomic annotation. Genome Res. 17:1362–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu LI, Lima BP, Wolfe AJ. 2010. Bacterial protein acetylation: the dawning of a new age. Mol. Microbiol. 77:15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majdalani N, Gottesman S. 2005. The Rcs phosphorelay: a complex signal transduction system. Annu. Rev. Microbiol. 59:379–405 [DOI] [PubMed] [Google Scholar]
- 5.Venkatesh GR, Kembou Koungni FC, Paukner A, Stratmann T, Blissenbach B, Schnetz K. 2010. BglJ-RcsB heterodimers relieve repression of the Escherichia coli bgl operon by H-NS. J. Bacteriol. 192:6456–6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castanié-Cornet MP, Cam K, Bastiat B, Cros A, Bordes P, Gutierrez C. 2010. Acid stress response in Escherichia coli: mechanism of regulation of gadA transcription by RcsB and GadE. Nucleic Acids Res. 38:3546–3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrières L, Clarke DJ. 2003. The RcsC sensor kinase is required for normal biofilm formation in Escherichia coli K-12 and controls the expression of a regulon in response to growth on a solid surface. Mol. Microbiol. 50:1665–1682 [DOI] [PubMed] [Google Scholar]
- 8.Majdalani N, Hernandez D, Gottesman S. 2002. Regulation and mode of action of the second small RNA activator of RpoS translation, RprA. Mol. Microbiol. 46:813–826 [DOI] [PubMed] [Google Scholar]
- 9.Wolfe AJ. 2005. The acetate switch. Microbiol. Mol. Biol. Rev. 69:12–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klein AH, Shulla A, Reimann SA, Keating DH, Wolfe AJ. 2007. The intracellular concentration of acetyl phosphate in Escherichia coli is sufficient for direct phosphorylation of two-component response regulators. J. Bacteriol. 189:5574–5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fredericks CE, Shibata S, Aizawa S, Reimann SA, Wolfe AJ. 2006. Acetyl phosphate-sensitive regulation of flagellar biogenesis and capsular biosynthesis depends on the Rcs phosphorelay. Mol. Microbiol. 61:734–747 [DOI] [PubMed] [Google Scholar]
- 12.Lukat GS, McCleary WR, Stock AM, Stock JB. 1992. Phosphorylation of bacterial response regulator proteins by low molecular weight phospho-donors. Proc. Natl. Acad. Sci. U. S. A. 89:718–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng J, Atkinson MR, McCleary W, Stock JB, Wanner BL, Ninfa AJ. 1992. Role of phosphorylated metabolic intermediates in the regulation of glutamine synthetase synthesis in Escherichia coli. J. Bacteriol. 174:6061–6070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thao S, Chen CS, Zhu H, Escalante-Semerena JC. 2010. Nepsilon-lysine acetylation of a bacterial transcription factor inhibits its DNA-binding activity. PLoS One 5:e15123. 10.1371/journal.pone.0015123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lima BP, Antelmann H, Gronau K, Chi BK, Becher D, Brinsmade SR, Wolfe AJ. 2011. Involvement of protein acetylation in glucose-induced transcription of a stress-responsive promoter. Mol. Microbiol. 81:1190–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma Q, Wood TK. 2011. Protein acetylation in prokaryotes increases stress resistance. Biochem. Biophys. Res. Commun. 410:846–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang W, Deutscher MP. 2012. Post-translational modification of RNase R is regulated by stress-dependent reduction in the acetylating enzyme Pka (YfiQ). RNA 18:37–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castaño-Cerezo S, Bernal V, Blanco-Catala J, Iborra JL, Canovas M. 2011. cAMP-CRP co-ordinates the expression of the protein acetylation pathway with central metabolism in Escherichia coli. Mol. Microbiol. 82:1110–1128 [DOI] [PubMed] [Google Scholar]
- 19.Blander G, Guarente L. 2004. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 73:417–435 [DOI] [PubMed] [Google Scholar]
- 20.Thao S, Escalante-Semerena JC. 2011. Control of protein function by reversible Nepsilon-lysine acetylation in bacteria. Curr. Opin. Microbiol. 14:200–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Silhavy TJ, Berman ML, Enquist LW. 1984. Experiments with gene fusions. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 22.Powell BS, Rivas MP, Court DL, Nakamura Y, Turnbough CL., Jr 1994. Rapid confirmation of single copy lambda prophage integration by PCR. Nucleic Acids Res. 22:5765–5766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simons RW, Houman F, Kleckner N. 1987. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene 53:85–96 [DOI] [PubMed] [Google Scholar]
- 24.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580 [DOI] [PubMed] [Google Scholar]
- 25.Chung CT, Niemela SL, Miller RH. 1989. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc. Natl. Acad. Sci. U. S. A. 86:2172–2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beatty CM, Browning DF, Busby SJ, Wolfe AJ. 2003. Cyclic AMP receptor protein-dependent activation of the Escherichia coli acsP2 promoter by a synergistic class III mechanism. J. Bacteriol. 185:5148–5157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lintner RE, Mishra PK, Srivastava P, Martinez-Vaz BM, Khodursky AB, Blumenthal RM. 2008. Limited functional conservation of a global regulator among related bacterial genera: Lrp in Escherichia, Proteus and Vibrio. BMC Microbiol. 8:60. 10.1186/1471-2180-8-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. 2005. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12:291–299 [DOI] [PubMed] [Google Scholar]
- 29.Dieckman L, Gu M, Stols L, Donnelly MI, Collart FR. 2002. High throughput methods for gene cloning and expression. Protein Expr. Purif. 25:1–7 [DOI] [PubMed] [Google Scholar]
- 30.Millard CS, Stols L, Quartey P, Kim Y, Dementieva I, Donnelly MI. 2003. A less laborious approach to the high-throughput production of recombinant proteins in Escherichia coli using 2-liter plastic bottles. Protein Expr. Purif. 29:311–320 [DOI] [PubMed] [Google Scholar]
- 31.Carballès F, Bertrand C, Bouche JP, Cam K. 1999. Regulation of Escherichia coli cell division genes ftsA and ftsZ by the two-component system rcsC-rcsB. Mol. Microbiol. 34:442–450 [DOI] [PubMed] [Google Scholar]
- 32.DiGiuseppe PA, Silhavy TJ. 2003. Signal detection and target gene induction by the CpxRA two-component system. J. Bacteriol. 185:2432–2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chi BK, Gronau K, Mader U, Hessling B, Becher D, Antelmann H. 2011. S-bacillithiolation protects against hypochlorite stress in Bacillus subtilis as revealed by transcriptomics and redox proteomics. Mol. Cell. Proteomics 10:M111.009506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP. 2010. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327:1004–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuhn ML, Majorek KA, Minor W, Anderson WF. 2013. Broad-substrate screen as a tool to identify substrates for bacterial Gcn5-related N-acetyltransferases with unknown substrate specificity. Protein Sci. 22:222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lima BP, Huyen TT, Bäsell K, Becher D, Antelmann H, Wolfe AJ. 2012. Inhibition of acetyl phosphate-dependent transcription by an acetylatable lysine on RNA polymerase. J. Biol. Chem. 287:32147–32160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Majdalani N, Heck M, Stout V, Gottesman S. 2005. Role of RcsF in signaling to the Rcs phosphorelay pathway in Escherichia coli. J. Bacteriol. 187:6770–6778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolfe AJ. 2010. Physiologically relevant small phosphodonors link metabolism to signal transduction. Curr. Opin. Microbiol. 13:204–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Denu JM. 2005. Vitamin B3 and sirtuin function. Trends Biochem. Sci. 30:479–483 [DOI] [PubMed] [Google Scholar]
- 40.Lundgren DH, Hwang SI, Wu L, Han DK. 2010. Role of spectral counting in quantitative proteomics. Expert Rev. Proteomics 7:39–53 [DOI] [PubMed] [Google Scholar]
- 41.Chi BK, Gronau K, Mäder U, Hessling B, Becher D, Antelmann H. 2011. S-bacillithiolation protects against hypochlorite stress in Bacillus subtilis as revealed by transcriptomics and redox proteomics. Mol. Cell. Proteomics 10:M111.009506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, Kaeberlein M, Kennedy BK, Berger SL. 2009. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459:802–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Francez-Charlot A, Laugel B, Van Gemert A, Dubarry N, Wiorowski F, Castanié-Cornet MP, Gutierrez C, Cam K. 2003. RcsCDB His-Asp phosphorelay system negatively regulates the flhDC operon in Escherichia coli. Mol. Microbiol. 49:823–832 [DOI] [PubMed] [Google Scholar]
- 44.Yu BJ, Kim JA, Moon JH, Ryu SE, Pan JG. 2008. The diversity of lysine-acetylated proteins in Escherichia coli. J. Microbiol. Biotechnol. 18:1529–1536 [PubMed] [Google Scholar]
- 45.Zhang J, Sprung R, Pei J, Tan X, Kim S, Zhu H, Liu CF, Grishin NV, Zhao Y. 2009. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol. Cell. Proteomics 8:215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL. 2010. Regulation of cellular metabolism by protein lysine acetylation. Science 327:1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Majdalani N, Chen S, Murrow J, St John K, Gottesman S. 2001. Regulation of RpoS by a novel small RNA: the characterization of RprA. Mol. Microbiol. 39:1382–1394 [DOI] [PubMed] [Google Scholar]
- 48.Pristovsek P, Sengupta K, Lohr F, Schafer B, von Trebra MW, Ruterjans H, Bernhard F. 2003. Structural analysis of the DNA-binding domain of the Erwinia amylovora RcsB protein and its interaction with the RcsAB box. J. Biol. Chem. 278:17752–17759 [DOI] [PubMed] [Google Scholar]
- 49.Barak R, Yan J, Shainskaya A, Eisenbach M. 2006. The chemotaxis response regulator CheY can catalyze its own acetylation. J. Mol. Biol. 359:251–265 [DOI] [PubMed] [Google Scholar]
- 50.Karanam B, Jiang L, Wang L, Kelleher NL, Cole PA. 2006. Kinetic and mass spectrometric analysis of p300 histone acetyltransferase domain autoacetylation. J. Biol. Chem. 281:40292–40301 [DOI] [PubMed] [Google Scholar]
- 51.Santos-Rosa H, Valls E, Kouzarides T, Martinez-Balbas M. 2003. Mechanisms of P/CAF auto-acetylation. Nucleic Acids Res. 31:4285–4292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang C, Wu J, Zheng YG. 2012. Function of the active site lysine autoacetylation in Tip60 catalysis. PLoS One 7:e32886. 10.1371/journal.pone.0032886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, Schilling B, Mooney SD, Kahn CR, Verdin E, Gibson BW. 2013. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci. U. S. A. 110:6601–6606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wagner GR, Payne RM. 2011. Mitochondrial acetylation and diseases of aging. J. Aging Res. 2011:234875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scott I, Webster BR, Li JH, Sack MN. 2012. Identification of a molecular component of the mitochondrial acetyltransferase programme: a novel role for GCN5L1. Biochem. J. 443:655–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang XJ, Seto E. 2008. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol. Cell 31:449–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Noort V, Seebacher J, Bader S, Mohammed S, Vonkova I, Betts MJ, Kuhner S, Kumar R, Maier T, O'Flaherty M, Rybin V, Schmeisky A, Yus E, Stulke J, Serrano L, Russell RB, Heck AJ, Bork P, Gavin AC. 2012. Cross-talk between phosphorylation and lysine acetylation in a genome-reduced bacterium. Mol. Syst. Biol. 8:571. 10.1038/msb.2012.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barak R, Welch M, Yanovsky A, Oosawa K, Eisenbach M. 1992. Acetyladenylate or its derivative acetylates the chemotaxis protein CheY in vitro and increases its activity at the flagellar switch. Biochemistry 31:10099–10107 [DOI] [PubMed] [Google Scholar]
- 59.Barak R, Abouhamad WN, Eisenbach M. 1998. Both acetate kinase and acetyl coenzyme A synthetase are involved in acetate-stimulated change in the direction of flagellar rotation in Escherichia coli. J. Bacteriol. 180:985–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramakrishnan R, Schuster M, Bourret RB. 1998. Acetylation at Lys-92 enhances signaling by the chemotaxis response regulator protein CheY. Proc. Natl. Acad. Sci. U. S. A. 95:4918–4923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barak R, Eisenbach M. 1992. Correlation between phosphorylation of the chemotaxis protein CheY and its activity at the flagellar motor. Biochemistry 31:1821–1826 [DOI] [PubMed] [Google Scholar]
- 62.Yan J, Barak R, Liarzi O, Shainskaya A, Eisenbach M. 2008. In vivo acetylation of CheY, a response regulator in chemotaxis of Escherichia coli. J. Mol. Biol. 376:1260–1271 [DOI] [PubMed] [Google Scholar]
- 63.Barak R, Eisenbach M. 2004. Co-regulation of acetylation and phosphorylation of CheY, a response regulator in chemotaxis of Escherichia coli. J. Mol. Biol. 342:375–381 [DOI] [PubMed] [Google Scholar]
- 64.Barak R, Prasad K, Shainskaya A, Wolfe AJ, Eisenbach M. 2004. Acetylation of the chemotaxis response regulator CheY by acetyl-CoA synthetase purified from Escherichia coli. J. Mol. Biol. 342:383–401 [DOI] [PubMed] [Google Scholar]
- 65.Liarzi O, Barak R, Bronner V, Dines M, Sagi Y, Shainskaya A, Eisenbach M. 2010. Acetylation represses the binding of CheY to its target proteins. Mol. Microbiol. 76:932–943 [DOI] [PubMed] [Google Scholar]
- 66.Chen MW, Kotaka M, Vonrhein C, Bricogne G, Rao F, Chuah ML, Svergun D, Schneider G, Liang ZX, Lescar J. 2012. Structural insights into the regulatory mechanism of the response regulator RocR from Pseudomonas aeruginosa in cyclic di-GMP signaling. J. Bacteriol. 194:4837–4846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.De N, Pirruccello M, Krasteva PV, Bae N, Raghavan RV, Sondermann H. 2008. Phosphorylation-independent regulation of the diguanylate cyclase WspR. PLoS Biol. 6:e67. 10.1371/journal.pbio.0060067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Najle SR, Inda ME, de Mendoza D, Cybulski LE. 2009. Oligomerization of Bacillus subtilis DesR is required for fine tuning regulation of membrane fluidity. Biochim. Biophys. Acta 1790:1238–1243 [DOI] [PubMed] [Google Scholar]
- 69.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kumari S, Beatty CM, Browning DF, Busby SJ, Simel EJ, Hovel-Miner G, Wolfe AJ. 2000. Regulation of acetyl coenzyme A synthetase in Escherichia coli. J. Bacteriol. 182:4173–4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zuber M, Hoover TA, Court DL. 1995. Analysis of a Coxiella burnetti gene product that activates capsule synthesis in Escherichia coli: requirement for the heat shock chaperone DnaK and the two-component regulator RcsC. J. Bacteriol. 177:4238–4244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Amann E, Ochs B, Abel KJ. 1988. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene 69:301–315 [DOI] [PubMed] [Google Scholar]
- 74.Clarke DJ, Joyce SA, Toutain CM, Jacq A, Holland IB. 2002. Genetic analysis of the RcsC sensor kinase from Escherichia coli K-12. J. Bacteriol. 184:1204–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.