

Abstract
In a continuing effort to analyze the selectivity/redundancy of the three glutaredoxin (Grx) enzymes of the model cyanobacterium Synechocystis PCC6803, we have characterized an enzyme system that plays a crucial role in protection against two toxic metal pollutants, mercury and uranium. The present data show that Grx1 (Slr1562 in CyanoBase) selectively interacts with the presumptive mercuric reductase protein (Slr1849). This MerA enzyme plays a crucial role in cell defense against both mercuric and uranyl ions, in catalyzing their NADPH-driven reduction. Like MerA, Grx1 operates in cell protection against both mercury and uranium. The Grx1-MerA interaction requires cysteine 86 (C86) of Grx1 and C78 of MerA, which is critical for its reductase activity. MerA can be inhibited by glutathionylation and subsequently reactivated by Grx1, likely through deglutathionylation. The two Grx1 residues C31, which belongs to the redox active site (CX2C), and C86, which operates in MerA interactions, are both required for reactivation of MerA. These novel findings emphasize the role of glutaredoxins in tolerance to metal stress as well as the evolutionary conservation of the glutathionylation process, so far described mostly for eukaryotes.
INTRODUCTION
Cyanobacteria, the very abundant photosynthetic prokaryotes which support much of the life on Earth in using solar energy to produce oxygen and biomass for the food chain, are continuously challenged with the toxic reactive oxygen species (ROS) generated by their respiration and photosynthesis. Among other processes, these ROS impair the redox homeostasis of cellular thiols, which can be subsequently restored by the thioredoxin and glutaredoxin systems (1). In the glutaredoxin system (2), NADPH is used to sequentially reduce (i) the glutathione reductase (GR) enzyme present in many, but not all (3), organisms; (ii) the glutathione (GSH) tripeptide; and (iii) the glutaredoxin (Grx) enzymes, which can also be reduced by the thioredoxin reductase enzymes (3–5). Grxs are widely conserved proteins that comprise two main families: the dithiol enzymes with a CX2C redox center (where C stands for cysteine and X stands for any other amino acid) and the monothiol enzymes with a CX2S redox-active center (where S stands for serine). The dithiol Grxs catalyze the reduction of disulfides (protein-S-S) or glutathione mixed disulfide (protein-S-SG) by a dithiol (CX2C-requiring) or a monothiol (CX2S-dependent) mechanism. In contrast, monothiol Grxs operate in the sensing of cellular iron and in the biogenesis of iron-sulfur clusters that play crucial roles in electron transfer processes (6–8). Some monothiol Grxs have also been reported to catalyze protein deglutathionylation (5, 9). However, the selectivity/redundancy of the Grx enzymes is poorly understood for photosynthetic organisms, even for cyanobacteria that possess a small number of Grxs (1), compared to plants (10). Whereas Arabidopsis thaliana has 31 Grxs (10), the model cyanobacterium Synechocystis PCC6803 (here Synechocystis) has only one monothiol Grx (11) and two dithiol enzymes (1). These two dithiol Grxs, Grx1 and Grx2, were shown to specifically interact with various proteins operating in tolerance to arsenate (12) and selenate (3).
In this study, we pursued the analysis of the Grx enzymes, emphasizing their possible role in protection against mercury (13) and uranium (14), which are massively used by human industries, leading to pollutions (15–17). Grx1 (Slr1562 in CyanoBase [http://genome.kazusa.or.jp/cyanobase/]), but neither Grx2 nor Grx3, appeared to interact with the Slr1849 protein, which is presented as a presumptive mercuric reductase (MerA) enzyme in CyanoBase. This MerA-like protein was found to operate in the protection against, and the NADPH-driven reduction of, not only mercuric but also uranyl ions (thereby challenging the notion of metal selectivity). Hence, cyanobacterial enzymes like MerA with the capacity to remediate both mercury and uranium might be interesting for future utilization of cyanobacteria for bioremediation (18), as most polluted sites contain cocktails of toxic metals. Consistent with the MerA-Grx1 interaction, Grx1 appeared to be crucial for protection against both mercury and uranium, like MerA. Furthermore, the activity of MerA was found to be inhibited by glutathionylation (formation of a glutathione mixed disulfide between a Cys residue of the protein and the Cys residue of glutathione) and subsequently reactivated by Grx1, likely through a deglutathionylation process. These findings emphasize the evolutionary conservation of the glutathionylation/deglutathionylation control of enzyme activity, a biological process described mostly for eukaryotes (19, 20).
MATERIALS AND METHODS
Microbial strains, growth conditions, and gene transfer procedures.
Synechocystis PCC6803 (here Synechocystis) was grown at 30°C or 39°C (depending on the strain) on mineral medium (MM) derived from BG11 (21), as described previously (22). For plate assays, 10-μl aliquots of 10-fold serial dilutions of mid-log-phase cultures (2.5 × 107 cells · ml−1) were spotted onto solid MM with or without the indicated concentration of HgCl2 and UO2(CH3COO)2, which were incubated for 5 to 7 days prior to image acquisition. Escherichia coli strains used for gene manipulations (HB101, XL1-Blue MRF′, and TOP10 [Invitrogen]), production of recombinant proteins [BL21(DE3) (Novagen) and KY2266 (23)], two-hybrid assays (DHM1 [24]), or conjugation (CM404) of Synechocystis (22) were grown on LB medium at 30°C (CM404 and KY2266) or 37°C (all other strains). Antibiotics used for selection were as follows: ampicillin (Ap) at 100 μg · ml−1, chloramphenicol (Cm) at 50 μg · ml−1, kanamycin (Km) at 50 μg · ml−1, nalidixic acid at 20 μg · ml−1, and spectinomycin (Sp) at 100 μg · ml−1 for E. coli, and Cm at 10 μg · ml−1, Km at 50 to 300 μg · ml−1, Sp at 5 μg · ml−1, and streptomycin (Sm) at 5 μg · ml−1 for Synechocystis.
Gene cloning and manipulation.
Synechocystis DNA was PCR amplified with specific primers to generate the studied genes without or with their two flanking genomic regions for homologous recombinations mediating targeted gene replacement upon transformation (25). After cloning into the appropriate plasmids (Table 1), the studied protein-coding sequences were either mutagenized (QuikChange mutagenesis kit; Stratagene) or replaced by an antibiotic-resistant marker inserted in the same orientation. These constructions were verified by PCR and DNA sequencing (BigDye kit; ABI Perkin-Elmer), before and after propagation in Synechocystis. PCR was also used to assay whether segregation between the wild-type (WT) and mutant copies of the polyploid (25) chromosome was complete (the studied gene is dispensable for cell growth) or not (the gene is essential for cell viability).
Table 1.
Characteristics of the plasmids used in this study
| Plasmid | Relevant feature(s)a | Reference or source |
|---|---|---|
| Analysis of protein-protein interactions with the bacterial two-hybrid system | ||
| pUT18 | Ampr plasmid encoding the T18 domain of the Bordetella pertussis adenylate cyclase in frame with an upstream multiple-cloning site | 24 |
| pKT25 | Kmr plasmid encoding the T25 domain of the B. pertussis adenylate cyclase in frame with a downstream multiple-cloning site | 24 |
| pUT18-zip | pUT18 with the CS for the self-interacting Zip control protein | 24 |
| pKT25-zip | pKT25 with the CS for the self-interacting Zip control protein | 24 |
| pUT18-merA | pUT18 with the full-length Synechocystis merA CS (slr1849) | This study |
| pKT25-merA | pKT25 with the full-length Synechocystis merA CS | This study |
| pUT18-arsC | pUT18 with the full-length Synechocystis arsC CS (slr0946) | This study |
| pKT25-arsC | pKT25 with the full-length Synechocystis arsC CS | This study |
| pUT18-grx1 | pUT18 with the full-length Synechocystis grx1 CS (slr1562) | 3 |
| pKT25-grx1 | pKT25 with the full-length Synechocystis grx1 CS | 3 |
| pUT18-grx2 | pUT18 with the full-length Synechocystis grx2 CS (ssr2061) | 3 |
| pKT25-grx2 | pKT25 with the full-length Synechocystis grx2 CS | 3 |
| pKT18-grx3 | pKT25 with the full-length Synechocystis grx3 CS (slr1846) | This study |
| pKT25-grx3 | pKT25 with the full-length Synechocystis grx3 CS | This study |
| Gene inactivation in Synechocystis | ||
| pGEMT | Ampr AT overhang cloning vector | Promega |
| pUC4K | Source of the Kmr marker gene | Pharmacia |
| pHP45Ω | Source of the Smr Spr marker gene | 39 |
| pΔgrx1::Kmr | Kmr cassette for deletion of the grx1 CS | 3 |
| pΔgrx2::Smr/Spr | Smr Spr for deletion of the grx2 CS | 3 |
| pmerA | pGEMT with the 534-bp merA CS preceded by its 177-bp upstream region | This study |
| pΔmerA::Kmr | pmerA with the Kmr marker in a SmaI site created at 84 bp of MerA CS | This study |
| Gene expression in Synechocystis | ||
| pFC1 | Replicating plasmid for heat-inducible gene expression in Synechocystis | 32 |
| pFmerA | pFC1 with the merA CS cloned between NdeI and EcoRI for heat-inducible production of MerA | This study |
| pSB2A | Replicating plasmid for expressing genes from their own promoter | 34 |
| pSmerA | pSB2A expressing the merA gene from its 150-bp promoter region | This study |
| pSmerAC78S | pSmerA where cysteine 78 of MerA is replaced by a serine | This study |
| pSgrx1 | pSB2A expressing the grx1 gene from its 150-bp promoter region | This study |
| pSgrx1C86S | pSgrx1 where cysteine 86 of Grx1 is replaced by a serine | This study |
| Protein production in E. coli | ||
| pET21b(+) | Plasmid for fusion of the 6×His tag at the C terminus of proteins | Novagen |
| pETM30 | Plasmid for fusion of the GST tag at the N terminus of proteins | Novagen |
| pTrc2 | Plasmid for fusion of the cMyc and 6×His tags at the C terminus of proteins | Invitrogen |
| pgrx1-6His | pET21b(+) with the grx1 CS cloned at the NdeI-EcoRI sites | This study |
| pgrx1-6HisC31S | pgrx1-6His with the C31S mutation in grx1 | This study |
| pgrx1-6HisC86S | pgrx1-6His with the C86S mutation in grx1 | This study |
| pmerA-6His | pTrc2 with the merA CS cloned at the BamHI-EcoRI sites | This study |
| pmerAC78S-6His | pmerA-6His harboring the C78S mutation in merA | This study |
| pgrx2-6His | pTRc2 with the grx2 CS cloned at the BamHI-EcoRI sites | This study |
| pGST-grx1 | pETM30 with the grx1 CS cloned at the SalI-XhoI sites | This study |
| pMaL-c2 | Plasmid for fusion of the MBP tag at the C terminus of proteins | New England BioLabs |
| pSS9 | pMaL-c2 with the merA CS cloned at the EcoRI-BamHI sites | This study |
CS, coding sequence.
Analysis of protein-protein interactions with the bacterial two-hybrid system.
Each Synechocystis full-length coding sequence was translationally fused to the intrinsically inactive adenylate cyclase (AC) domains of the replication-compatible plasmids pKT25 and pUT18, which were subsequently doubly transformed into E. coli reporter strain DHM1 to search for AC reconstitution, which turns on β-galactosidase production (24), generating the blue color of the cells grown during 2 days at 30°C on indicator plates containing 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) (40 mg · ml−1; Eurobio), isopropyl-1-thio-β-d-galactopyranoside (IPTG) (0.5 mM; Invitrogen), Ap, Km, and nalidixic acid. β-Galactosidase (β-Gal) activity was measured as described previously (3). One β-Gal unit is equal to 1 nmol o-nitrophenyl-β-d-galactopyranoside (ONPG) min−1 · mg−1 protein.
Overproduction of affinity-tagged proteins and GST pulldown.
The coding sequence of each studied protein was PCR amplified and tagged by cloning into the appropriate E. coli vectors (Table 1). The resulting plasmids were introduced into E. coli strain BL21(DE3) for the production of recombinant proteins tagged with either 6×His or glutathione S-transferase (GST) or into triple-protease-defective mutant strain KY2266 (23) for the production of the maltose-binding protein (MBP)-tagged MerA protein. Cells grown at 30°C for 2 h (up to an optical density at 600 nm [OD600] of 0.5 to 0.7) were induced with 0.5 to 1 mM IPTG for 4 to 12 h at 30°C, washed, and resuspended in 2 ml of lysis buffer (10 mM Tris-HCl [pH 7.4], 150 mM NaCl, 10% glycerol, and 1% IGEPAL; Sigma) for the purification of 6×His-tagged proteins. In contrast, 2 ml of PBS (10 mM phosphate buffer [pH 7.4], 120 mM NaCl, 2.7 mM KCl, 1 mM phenylmethylsulfonyl fluoride [PMSF], 10 mM dithiothreitol [DTT], and 0.1% Triton X-100) was used for the purification of GST-tagged proteins, and 5 ml buffer A (30 mM Tris-HCl [pH 8], 200 mM NaCl) was used for MBP-MerA purification. Cells were then rapidly frozen in an Eaton press chamber cooled in a dry ice ethanol bath, disrupted (250 MPa), and centrifuged at 14,000 × g for 30 min at 4°C (22) to collect the supernatant (about 1.5 ml) containing the soluble proteins. For the purification of the MBP-MerA fusion protein, the supernatant was loaded onto a 1-ml amylose resin column (New England BioLabs) preequilibrated and washed (10 bed volumes) with buffer A. Following elution with 10 mM maltose (Sigma) in buffer A, the purity of MBP-MerA, checked by SDS-PAGE, was >90%. The MBP tag was then cleaved (factor Xa protease; New England BioLabs) and removed by anion-exchange chromatography (Bio-Rad). The presence of MerA in the eluted fractions was confirmed by mass spectrometry analysis on a matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) apparatus after trypsin cleavage (peptide mass fingerprint), and its concentration was determined spectrophotometrically by using its calculated molar extinction coefficient at 280 nm (30,470 M−1 · cm−1). Purified MerA was concentrated in 30 mM Tris-HCl (pH 8)–200 mM NaCl with Amicon Ultra centrifugation filters (Millipore) and briefly stored at −80°C prior to analysis.
6×His-tagged proteins were purified on Ni-nitrilotriacetic acid (NTA) agarose beads (Qiagen) and eluted with lysis buffer containing 200 mM imidazole. For GST pulldown analysis, 1 μg of GST-tagged Grx1 was immobilized on glutathione-Sepharose beads (Pharmacia) and incubated with 80 μl of cell extract (10 mg of proteins) containing MerA-6×His in 900 μl of binding buffer (20 mM HEPES [pH 6.8], 100 mM KCl, 5 mM MgCl2, 2% glycerol, 1 mM DTT, 1 mM PMSF, and 1 mM EDTA) for 2 h at 4°C under continuous rotation. Beads collected by centrifugation were washed three times (Qiagen buffer), resuspended in 50 μl of binding buffer, and boiled for 5 min in sample buffer prior to SDS-PAGE (16% polyacrylamide). The purity of the studied proteins was >90%.
Mercuric and uranyl reductase activities.
Mercuric reductase activity was assayed (26) at 37°C with a solution containing 80 mM sodium phosphate (pH 7.4), 200 μM NADPH, 1 mM 2-mercaptoethanol, 2 μM flavin adenine dinucleotide (FAD), and 100 μM HgCl2 on either Synechocystis cell extracts or 0.5 μM purified MerA. The activity was monitored for 2 min as the decrease in A340 (molar extinction coefficient of 6,200 M−1 · cm−1) attributable to NADPH oxidation. As indicated, HgCl2 was replaced by UO2(CH3COO)2 (200 μM), and MerA was either reduced by a 15-min treatment at 22°C with 10 mM DTT in Tris-HCl (pH 8) or glutathionylated by a 30-min incubation with 20 mM oxidized glutathione (GSSG) followed by filtration and resuspension in phosphate buffer prior to the enzyme assay (27). When required, 0.5 μM Grx1 (WT or mutants) or Grx2 and 1 mM reduced glutathione (GSH) were added to the reaction mixture. One unit of reductase activity is defined as the amount of enzyme that catalyzes the oxidation of 1.0 μmol NADPH per min.
Production of biotinylated glutathione.
The water-soluble biotinylation reagent EZ Link Sulfo-NHS-Biotin (Perbio Science) was used to couple biotin to the primary amino group of GSSG, as described previously (28). The biotinylation reagent (40 μl; 48 mM) was incubated with GSSG (40 μl; 32 mM) in 50 mM potassium phosphate buffer (pH 7.2) for 1 h at room temperature, generating biotinylated GSSG (BioGSSG). Free biotin was then quenched by adding 28 μl of 0.6 M ammonium carbonate (NH4HCO3).
In vitro glutathionylation of MerA with BioGSSG.
MerA was reduced with 50 mM DTT, desalted in 30 mM Tris-HCl (pH 8)–200 mM NaCl with NAP-5 columns (GE Healthcare), treated or not with 100 mM iodoacetamide (IAM) and 20 mM N-ethylmaleimide (NEM) as alkylating agents for 30 min in the dark, and then incubated for 1 h with 2 mM BioGSSG in 30 mM Tris-HCl (pH 7.9). As indicated, the reversibility of the glutathionylation treatment was checked with a 30-min incubation with 50 mM DTT. All treatments were performed at room temperature. Proteins were then loaded onto nonreducing SDS-PAGE gels and analyzed by Western blotting using antibiotin antibodies, as described previously (28).
RESULTS
Characterization of a specific interaction between Grx1 and the presumptive mercuric reductase (MerA) with a bacterial two-hybrid system and identification of crucial amino acid residues in each partner.
To search for new protein targets of the three Grxs of Synechocystis PCC6803 (Synechocystis), we used the bacterial adenylate cyclase two-hybrid (BACTH) system (24), which works well in our hands (3, 29). Therefore, the full-length coding sequences of the dithiol glutaredoxins (3) Grx1 (Slr1562 in CyanoBase) and Grx2 (Ssr2061) and the monothiol enzyme (11) Grx3 (Slr1846) were cloned into the two BACTH reporter plasmids pUT18 and pKT25 (Table 1). The resulting plasmids, pUT18-Grx and pKT25-Grx, were independently used as baits to identify pairwise interactions restoring the production of the β-galactosidase reporter enzyme. Two percent of the thousand protein-protein interaction tests that were performed turned out to be positive (data not shown). As a positive-control test, we verified (Table 2) that the BACTH system was able to detect the interaction between Grx2 and ArsC (Slr0946), the arsenate reductase enzyme (12). This finding gave us confidence in the novel interaction reported here (Table 2) between Grx1 and the presumptive mercuric reductase protein MerA (Slr1849), a well-conserved protein in cyanobacteria (see Fig. S1 in the supplemental material). The MerA-Grx1 interaction appeared to be specific, as MerA interacted with neither Grx2 nor Grx3 (Table 2).
Table 2.
Identification and analysis of protein-protein interactions with the BACTH systema
| Coding sequence cloned into pUT18 | Coding sequence cloned into pKT25 | Mean β-Gal activity (U) ± SD |
|---|---|---|
| Controls | ||
| Zip | Zip | 4,236 ± 113 |
| None | None | 94 ± 11 |
| grx1 | None | 67 ± 18 |
| None | grx1 | 68 ± 15 |
| grx2 | None | 72 ± 11 |
| None | grx2 | 77 ± 08 |
| grx3 | None | 93 ± 10 |
| None | grx3 | 67 ± 08 |
| None | arsC | 89 ± 17 |
| merA | None | 74 ± 13 |
| Tests | ||
| grx1 | arsC | 97 ± 14 |
| grx2 | arsC | 1,687 ± 138 |
| merA | grx1 | 1,836 ± 97 |
| merA | grx2 | 65 ± 12 |
| merA | grx3 | 74 ± 12 |
| merA | grx1Q29L | 1,071 ± 47 |
| merA | grx1C31S* | 2,161 ± 25 |
| merA | grx1C34S* | 1,221 ± 54 |
| merA | grx1K52A | 39 ± 05 |
| merA | grx1TV72SL | 1,838 ± 87 |
| merA | grx1C86S | 292 ± 14 |
| merA | grx1Q97L | 308 ± 13 |
| merAE70A | grx1 | 64 ± 12 |
| merAC78S* | grx1 | 93 ± 16 |
| merAC83S* | grx1 | 2,096 ± 182 |
| merAC205S | grx1 | 1,411 ± 62 |
| merAD246A | grx1 | 1,974 ± 179 |
| merAC345S | grx1 | 1,689 ± 134 |
The occurrence of interactions between the proteins produced from the pUT18 and pKT25 reporter plasmids cotransformed into E. coli was ascertained by measuring the β-galactosidase activity (1 β-Gal unit corresponds to hydrolysis of 1 nmol o-nitrophenyl-β-d-galactopyranoside · min−1 · mg−1 of protein). The numbers are the mean values ± standard deviations from six assays (three measurements performed on two different cell extracts). The Zip-containing and empty plasmids served as positive and negative controls, respectively (24). The nature and position of amino acid substitutions are indicated by the subscripts. The presumed redox-active cysteines are indicated by asterisks.
To strengthen confidence in the Grx1-MerA interaction, interaction-disruptive amino acid substitutions we searched for, focusing on cysteine (C) residues because they can be critical for the activity of Grxs (30) and bacterial MerA enzymes (31). Hence, the cysteine residues of Grx1 and MerA were independently replaced by a serine (S). In addition, several charged amino acids were independently replaced by neutral ones. The results led to the identification of amino acids critical for the Grx1-MerA interaction (Table 2) in both the Grx1 (C86, K52, and Q97) and MerA (C78 and E70) protein partners.
Validation of the Grx1-MerA interaction with GST pulldown assays and confirmation of the crucial role of amino acids C86 of Grx1 and C78 of MerA.
The Grx1-MerA interaction detected with the two-hybrid test (Table 2) was confirmed with a GST pulldown assay. First, the GST and 6×His tags allowing facile protein purification were fused to the N terminus of Grx1 (GST-Grx1) and the C terminus of MerA (MerA-6×His), respectively. The pulldown assay then showed the MerA-6×His protein to be retained by the GST-Grx1 hybrid protein but not by the GST protein alone (Fig. 1). Furthermore, the same mutations, C86S in Grx1 and C78S in MerA (Fig. 1), abolished the Grx1-MerA interaction detected with both the two-hybrid test (Table 2) and the GST pulldown (Fig. 1).
Fig 1.
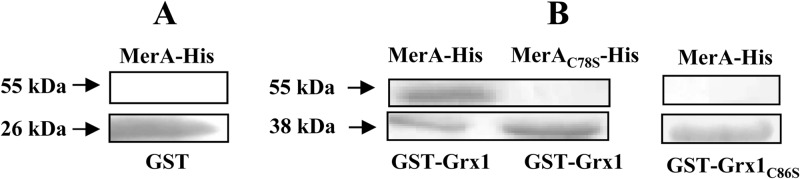
GST pulldown analysis of the MerA-Grx1 interaction. The GST (26 kDa) (A), GST-Grx1 (38 kDa) (B), or GST-Grx1C86S (38 kDa) (B) protein, independently linked to GSH-Sepharose beads, was incubated with Synechocystis cell extracts containing either the MerA-His (55 kDa) or MerAC78S-His (55 kDa) protein, and the protein complex was analyzed by SDS-PAGE and Coomassie blue staining. Arrows point to the migration of studied proteins. This experiment was performed twice.
The MerA protein operates in the protection of Synechocystis against both mercury and uranium.
To investigate the role of the MerA-like protein in the fitness of Synechocystis, a merA deletion cassette (ΔmerA::Kmr) (Table 1) was constructed and introduced by transformation into Synechocystis. The transformant clones retained no wild-type (WT) copies of the chromosome, which is polyploid (25), showing that MerA is dispensable for cell growth under standard laboratory conditions. The merA null mutant appeared to be more sensitive to mercuric ions (Hg2+) than the WT strain (Fig. 2), showing that MerA operates in protection against mercury. Having noticed the high level of sequence homology (55% identities and 71% similarities) between the MerA-like proteins from Synechocystis and the uranium-tolerant bacterium Geobacter uraniireducens (GenBank accession number ZP_01142587), we tested whether the Synechocystis MerA-like protein might operate in protection against uranyl ions (UO22+) as well. Indeed, the merA null mutant (ΔmerA::Kmr) was less tolerant to UO22+ than the WT strain (Fig. 2). These data were confirmed as follows. First, the merA protein-coding sequence (Table 1) was cloned into plasmid pFC1, which replicates autonomously in various hosts, including Synechocystis (32, 33). pFC1 harbors the λcI857 gene, encoding the temperature-sensitive repressor that tightly controls the activity of the otherwise strong λpR promoter located downstream, which is followed by the λcro ribosome-binding site (5′-AGGA-3′) and an ATG start codon (in boldface type) embedded within the unique NdeI restriction site (5′-CATATG-3′) for in-frame fusion of the studied protein-coding sequences. Hence, using pFC1, the production of the studied proteins is strongly dependent on the growth temperature (high-level production at 39°C, moderate-level of production at 34°C, and absence of production at 30°C). The results showed that a moderate heat induction of MerA production in cells incubated at 34°C increased their resistance to both mercury and uranium (Fig. 2). Second, the merA gene expressed from its own promoter (Table 1) was cloned into our pSB2A plasmid that replicates at the same 10 copies per cell (34) as the polyploid chromosome (25). The resulting plasmid, pSmerA, was introduced and stably propagated in the merA null mutant, yielding ΔmerA::Kmr/pSmerA cells, which expressed merA at a natural level and displayed the WT level of tolerance to both Hg2+ and UO22+, as expected (Fig. 2). Collectively, these data demonstrate that MerA operates in protection of Synechocystis against both mercury and uranium.
Fig 2.

Influence of mercury and uranium on growth of various Synechocystis strains. Tenfold serial dilutions of mid-log-phase cultures were spotted onto MM plates with or without Hg2+ (HgCl2) (3 μM) or UO22+ [UO2(CH3CO2)2] (500 μM) and subsequently incubated for 4 to 5 days prior to image acquisition. The strains are indicated as WT (wild type), ΔmerA (merA null mutant), Δgrx1 (grx1 null mutant), and Δgrx2 (grx2 null mutant). The complementation tests of the null mutants were performed at 30°C with the pSB2A replicating plasmid that expressed either the merA gene (pSmerA) or its C78S mutant allele (pSmerAC78S), or the grx1 gene (pSgrx1) or its mutant allele (pSgrx1C86S). The overexpression tests were done at 34°C with WT cells propagating the pFmerA replicating plasmid that expressed the merA coding sequence under the control of a temperature-regulated promoter. These experiments were repeated three times.
The conserved C78 residue of the MerA protein of Synechocystis is crucial to cell protection against mercury and uranium.
The same plasmid strategy was used to test the influence of the C78 residue of MerA, which belongs to its presumptive redox-active site (C78X4C83, conserved in cyanobacteria) (see Fig. S1 in the supplemental material), on cell tolerance to mercury and uranium. The merAC78S mutant allele was cloned into pSB2A and subsequently introduced into the merA null mutant. The resulting ΔmerA::Kmr/pSmerAC78S strain remained as sensitive to both Hg2+ and UO22+ as its parental merA null mutant (Fig. 2). This finding shows that the conserved C78 residue of MerA is crucial to MerA-driven protection of Synechocystis against both mercury and uranium.
Enzymological confirmation that the Synechocystis MerA enzyme possesses a NADPH-driven mercuric/uranyl reductase activity requiring C78 of its conserved CX4C redox-active motif.
MerA proteins with or without the C78S mutation were produced in E. coli as recombinant fusion proteins with a C-terminal 6×His tag for facile purification (Table 1). A classical test that measures the NADPH-driven reduction of mercuric ions (26) was then used to show that the Synechocystis MerA protein was able to catalyze the NADPH-dependent reduction of not only mercuric but also uranyl ions (Table 3). MerA activity appeared to be increased by the full reduction of MerA with DTT prior to the enzyme assay (Table 3). In contrast, MerA activity was abolished by the C78S mutation of its conserved redox-active cysteine (Table 3), which decreased MerA-driven cell protection against both Hg2+ and UO22+ (Fig. 2).
Table 3.
In vitro assay of MerA-like activitya
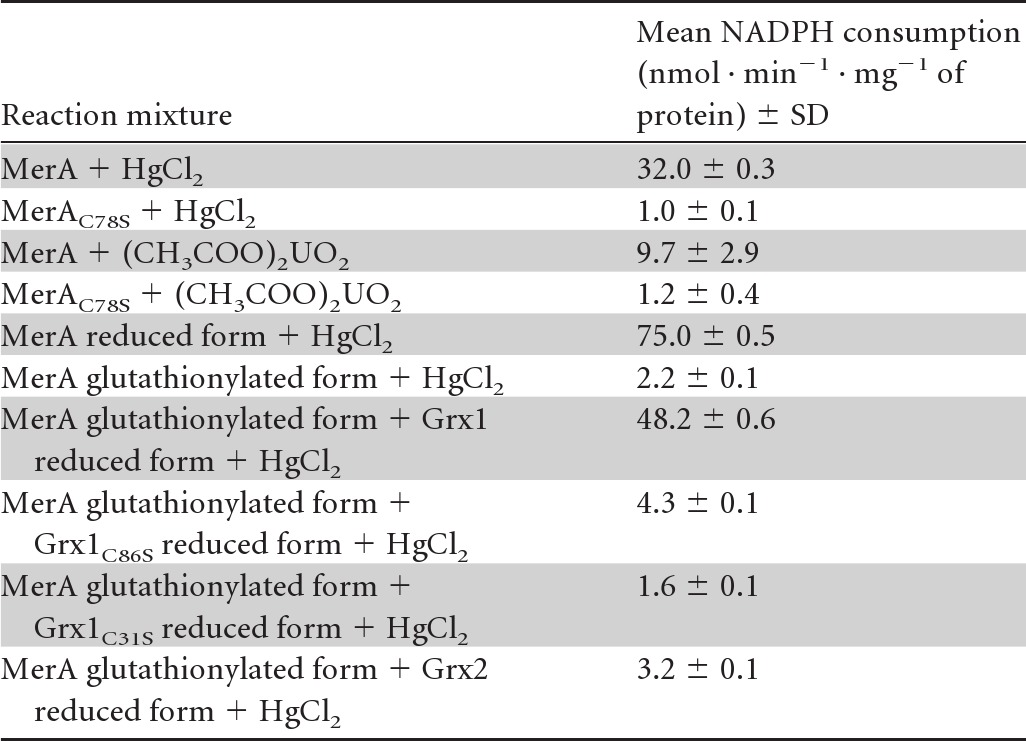
The activity of the Synechocystis MerA-like enzyme was measured as the consumption of NADPH (nmol · min−1 · mg−1 of protein) that reduces mercuric or uranyl ions. As indicated, MerA was either reduced or glutathionylated and subsequently treated or not treated with Grx1 or Grx2 before the reactions were started. The nature and position of amino acid substitutions in MerA and Grx1 are shown as subscripts. The numbers are the mean values ± standard deviations from four assays (two measurements performed on two independent enzyme preparations). The negative-control experiments performed in the absence of either NADPH, MerA, or mercuric or uranyl ions yielded only background values (1 to 3 units).
Grx1 is involved in the defense of Synechocystis against both mercury and uranium, like MerA.
The presently reported MerA-Grx1 interaction led us to speculate that Grx1 might also be involved in resistance to both mercury and uranium. The grx1 null mutant (Δgrx1::Kmr) (Table 1), which is as fit as the WT strain in the absence of stress (3), was found to be more sensitive to both Hg2+ and UO22+ than WT cells (Fig. 2). To confirm that this phenotype can be rescued by Grx1, the grx1 gene was cloned into the pSB2A vector, and the resulting pSgrx1 plasmid (Table 1) was introduced in the grx1 null mutant. The resulting Δgrx1::Kmr/pSgrx1 cells displayed WT levels of tolerance to Hg2+ and UO22+ (Fig. 2). Together, these data show that Grx1 operates in cell protection against both mercury and uranium, like MerA. In contrast, Grx2, which does not interact with MerA (Table 2), appeared to be dispensable for tolerance to Hg2+ and UO22+ (Fig. 2).
In vivo evidence that MerA needs to interact with Grx1 in order to protect cells against mercury and uranium.
The role of the MerA-Grx1 interaction in the defense against mercury and uranium was tested as follows. The grx1C86S mutant gene encoding the Grx1C86S protein, which was unable to interact with MerA (Table 2 and Fig. 2), was cloned into plasmid pSB2A. The resulting plasmid, pSgrx1C86S, was introduced into the Hg- and U-sensitive grx1 null mutant, yielding the Δgrx1::Kmr/pSgrx1C86S strain. These cells remained sensitive to both Hg2+ and UO22+, unlike the Δgrx1::Kmr/pSgrx1 strain. These findings showed that the grx1C86S mutant gene had lost the ability of the WT grx1 allele to restore tolerance to Hg2+ and UO22+ to grx1 null cells (Fig. 2). Together, these data show that the Grx1 interaction with MerA is required for tolerance to both Hg2+ and UO22+.
MerA activity is impaired by glutathionylation and subsequently restored by Grx1, which likely catalyzes MerA deglutathionylation.
The interaction between the Grx2 and ArsC enzymes (Table 2) is consistent with a previous in vitro report that ArsC can be glutathionylated upon arsenate reduction and subsequently reactivated through deglutathionylation catalyzed by the E. coli GrxA enzyme (35). By analogy with these data, the presently described MerA-Grx1 interaction led us to speculate that MerA might be glutathionylated upon Hg2+ reduction and subsequently deglutathionylated by Grx1. Hence, using standard in vitro procedures to assay glutathionylation (27, 28), MerA was incubated with biotinylated glutathione (BioGSSG). Subsequently, the presence of glutathione adducts on MerA was analyzed by Western blotting using an antibiotin antibody (Fig. 3). A clear signal was observed for the BioGSSG-treated MerA protein, which completely disappeared after subsequent reduction with DTT treatment (Fig. 3). Furthermore, MerA glutathionylation (MerASSG) was confirmed by showing that it could be prevented (Fig. 3) by MerA pretreatment with the specific cysteine-alkylating agents iodoacetamide (IAM) and N-ethylmaleimide (NEM), known to impair glutathionylation. Also, interestingly, MerA glutathionylation was shown to inhibit MerA activity, which could be restored subsequently by Grx1 (Table 3). The redox-active C31 residue of Grx1 was crucial for Grx1-mediated reactivation of MerA. Similarly, the C86 residue of Grx1, critical for the MerA-Grx1 interaction (Table 2 and Fig. 1 and 2), was also required for MerA reactivation (Table 3). To confirm the positive influence of Grx1 on MerA activity, we compared the levels observed in cell extracts prepared from the WT strain and the grx1 null mutant (Δgrx1::Kmr). As expected, WT cells displayed higher-level mercuric and uranyl reductase activities than grx1 null cells (Table 4). Collectively, these data indicate that MerA can be inactivated by glutathionylation and subsequently reactivated by Grx1, which likely catalyzes MerA deglutathionylation. In contrast, Grx2 could not reactivate glutathionylated MerA (Table 3), in agreement with the absence of a Grx2-MerA interaction (Table 2) and the dispensability of Grx2 for cell protection against Hg2+ (Fig. 2).
Fig 3.

Analysis of MerA glutathionylation with biotinylated glutathione (BioGSSG). Prereduced MerA was incubated for 1 h in the presence of BioGSSG (2 mM) with or without prior incubation with both 2-iodoacetamide (IAM) (100 mM) and N-ethylmaleimide (NEM) (20 mM). The reversibility of the glutathionylation of MerA was assessed by treatment with dithiothreitol (DTT) (50 mM). Proteins were resolved by nonreducing SDS-PAGE and transferred onto nitrocellulose for Western blotting with antibiotin antibodies. Coomassie staining of the gel shows equal loading in each lane. The results of two independent tests are shown.
Table 4.
Analysis of MerA-like activity of Synechocystis cell extracts prepared from the WT strain and the Grx1 null mutanta
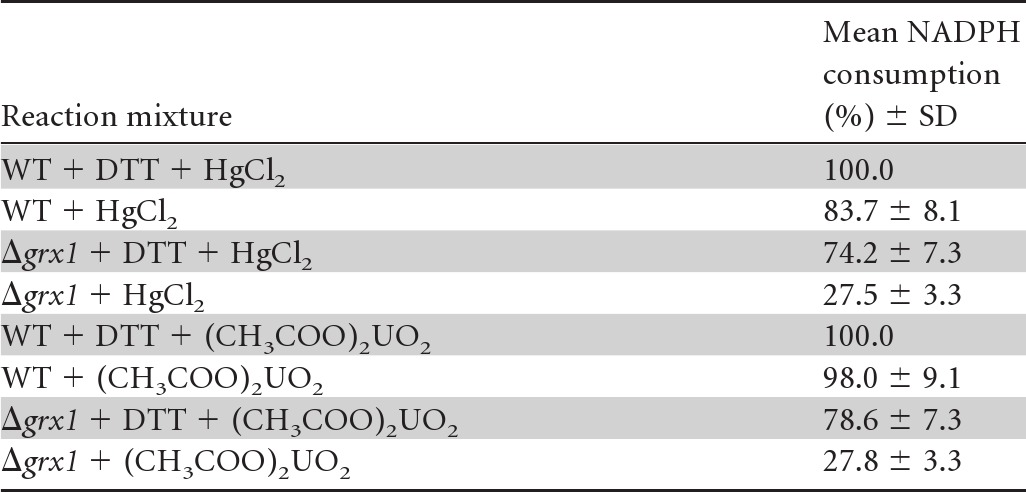
The activity of the Synechocystis MerA-like enzyme was measured as the consumption of NADPH (nmol · min−1 · mg−1 of protein) that reduces mercuric or uranyl ions. The values given are percentages of the highest activity monitored in the presence of DTT to fully reduce MerA, which was taken as 100%. The numbers are the mean values ± standard deviations from four assays (two measurements performed on two independent enzyme extracts).
DISCUSSION
We pursued the analysis of the widely conserved glutaredoxins (Grxs) in the model cyanobacterium Synechocystis PCC6803, which possesses two dithiol enzymes (Grx1 [Slr1562] and Grx2 [Ssr2061]) (3, 12) and one monothiol Grx (Grx3 [Slr1846]) (11). The bacterial two-hybrid system (24), which worked well in our hands (3, 29), was used to search for proteins selectively interacting with only one Grx. Grx1, but neither Grx2 nor Grx3, was found to interact with the Slr1849 protein (Table 2). This protein, highly conserved in cyanobacteria (see Fig. S1 in the supplemental material), with the exceptions of Prochlorococcus and marine Synechococcus species, is homologous to the bacterial mercuric reductase enzyme MerA (http://genome.kazusa.or.jp/cyanobase/). The Grx1-MerA interaction was confirmed with GST pulldown assays (Fig. 1) and the identification of critical amino acids in each partner, such as the C86 residue of Grx1 and the C78 residue of MerA (Table 2 and Fig. 1). Through gene deletion and plasmid complementation analyses (Fig. 2) with replicative vectors (32, 34), MerA was found to be (i) dispensable for cell growth in the absence of stress and (ii) crucial to cell defense against mercuric ions (HgCl2). Consistently, MerA was shown to possess genuine NADPH-driven mercuric reductase activity, which requires residue C78 residue of its invariant redox motif (CX4C) (Table 3). The high level of sequence homology between the MerA proteins from the bacterium Geobacter uraniireducens (GenBank accession number ZP_01142587) and Synechocystis suggested to us that the Synechocystis MerA enzyme might also operate in protection against uranyl ions. Indeed, MerA was found to possess a NADPH-driven uranyl reductase activity (Table 3) that is crucial to uranium tolerance (Fig. 2). This is the first report of an enzyme displaying both mercuric and uranyl reductase activities. Challenging the notion of metal selectivity, such cyanobacterial enzymes with the capacity to remediate at least mercury and uranium, two frequent pollutants (15–17), might be of value for future bioindication and/or bioremediation purposes, as most polluted sites contain cocktails of toxic metals.
Also, interestingly, Grx1 was found to be involved in the defense of Synechocystis against both Hg and U, in agreement with the Grx1-MerA interaction (Fig. 2). Furthermore, the C86 residue of Grx1, which is crucial to its interaction with MerA, was shown to be critical for cell protection against both Hg and U (Fig. 2). To our knowledge, this is the first report that a Grx enzyme operates in defense against both mercury and uranium. As a negative control, we verified that Grx2, which does not interact with MerA (Table 2), is involved in neither Hg nor U tolerance (Fig. 2). Grx2, but not Grx1, was found to interact with ArsC, the arsenate reductase enzyme (Table 2), which can be glutathionylated upon arsenate reduction and subsequently reactivated through deglutathionylation catalyzed by the heterologous E. coli GrxA enzyme (35). By analogy with these results, the presently reported MerA-Grx1 interaction led us to speculate that MerA might be glutathionylated upon Hg2+ reduction and subsequently deglutathionylated by Grx1. Indeed, MerA activity could be inhibited by glutathionylation (Table 3 and Fig. 3) and subsequently reactivated by Grx1 (Table 3). Furthermore, MerA activity appeared to be lower in the grx1 null mutant than in the WT strain (Table 4). Moreover, the C31 (redox-active site) and C86 (MerA interaction) residues of Grx1 were found to be critical for MerA reactivation (Table 3). To our knowledge, this is the first report that mercuric reductase activity can be regulated by glutathionylation, under the control of a glutaredoxin likely catalyzing deglutathionylation. Collectively, our findings shed light on the selectivity of the Grx enzymes, an as yet poorly understood phenomenon in photosynthetic organisms (36). These findings are also important for emphasizing the evolutionary conservation of the glutathionylation/deglutathionylation process, so far described mostly for eukaryotes (19, 20, 37). In prokaryotes, only few proteins are known to be regulated via glutathionylation, namely MetE (methionine biosynthesis), phosphoadenylyl-sulfate (PAPS) reductase (sulfur assimilation), ArsC (arsenate reduction), and OxyR (transcriptional regulation) (38). We propose to include MerA in this list.
Supplementary Material
ACKNOWLEDGMENTS
We thank Daniel Ladant (Pasteur Institute, Paris, France) for the gift of the bacterial two-hybrid system, Pierre Genevaux for E. coli strain KY226, and Tania Arcondéguy for plasmid pMaLC2.
This work was supported in part by the French scientific programs Toxicologie Nucléaire Environnementale, ANR Biosys06-134823: SULFIRHOM, and ANR Blanc08-0153: GLUTAPHOTO. B.M., S.C., S.S., and M.B. were recipients of fellowships from the CEA (B.M.) and the ANR (S.C., S.S., and M.B.).
Footnotes
Published ahead of print 12 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00272-13.
REFERENCES
- 1.Narainsamy K, Marteyn B, Sakr S, Cassier-Chauvat C, Chauvat F. 2013. Genomics of the pleiotropic glutathione system in cyanobacteria, p. 157–188 In Cassier-Chauvat C, Chauvat F. (ed), Genomics of cyanobacteria, vol 65 Elsevier Academic Press, New York, NY [Google Scholar]
- 2.Lillig CH, Berndt C. 21 December 2012. Glutaredoxins in thiol/disulfide exchange. Antioxid. Redox Signal. 10.1089/ars.2012.5007 [DOI] [PubMed] [Google Scholar]
- 3.Marteyn B, Domain F, Legrain P, Chauvat F, Cassier-Chauvat C. 2009. The thioredoxin reductase-glutaredoxins-ferredoxin crossroad pathway for selenate tolerance in Synechocystis PCC6803. Mol. Microbiol. 71:520–532 [DOI] [PubMed] [Google Scholar]
- 4.Johansson C, Lillig CH, Holmgren A. 2004. Human mitochondrial glutaredoxin reduces S-glutathionylated proteins with high affinity accepting electrons from either glutathione or thioredoxin reductase. J. Biol. Chem. 279:7537–7543 [DOI] [PubMed] [Google Scholar]
- 5.Zaffagnini M, Michelet L, Massot V, Trost P, Lemaire SD. 2008. Biochemical characterization of glutaredoxins from Chlamydomonas reinhardtii reveals the unique properties of a chloroplastic CGFS-type glutaredoxin. J. Biol. Chem. 283:8868–8876 [DOI] [PubMed] [Google Scholar]
- 6.Herrero E, Belli G, Casa C. 2010. Structural and functional diversity of glutaredoxins in yeast. Curr. Protein Pept. Sci. 11(8):659–668 [DOI] [PubMed] [Google Scholar]
- 7.Rouhier N, Couturier J, Johnson MK, Jacquot JP. 2010. Glutaredoxins: roles in iron homeostasis. Trends Biochem. Sci. 35:43–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lill R, Hoffmann B, Molik S, Pierik AJ, Rietzschel N, Stehling O, Uzarska MA, Webert H, Wilbrecht C, Muhlenhoff U. 2012. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim. Biophys. Acta 1823:1491–1508 [DOI] [PubMed] [Google Scholar]
- 9.Tamarit J, Belli G, Cabiscol E, Herrero E, Ros J. 2003. Biochemical characterization of yeast mitochondrial Grx5 monothiol glutaredoxin. J. Biol. Chem. 278:25745–25751 [DOI] [PubMed] [Google Scholar]
- 10.Michelet L, Zaffagnini M, Massot V, Keryer E, Vanacker H, Miginiac-Maslow M, Issakidis-Bourguet E, Lemaire SD. 2006. Thioredoxins, glutaredoxins, and glutathionylation: new crosstalks to explore. Photosynth. Res. 89:225–245 [DOI] [PubMed] [Google Scholar]
- 11.Picciocchi A, Saguez C, Boussac A, Cassier-Chauvat C, Chauvat F. 2007. CGFS-type monothiol glutaredoxins from the cyanobacterium Synechocystis PCC6803 and other evolutionary distant model organisms possess a glutathione-ligated [2Fe-2S] cluster. Biochemistry 46:15018–15026 [DOI] [PubMed] [Google Scholar]
- 12.Lopez-Maury L, Sanchez-Riego AM, Reyes JC, Florencio FJ. 2009. The glutathione/glutaredoxin system is essential for arsenate reduction in Synechocystis sp. strain PCC 6803. J. Bacteriol. 191:3534–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lefebvre DD, Kelly D, Budd K. 2007. Biotransformation of Hg(II) by cyanobacteria. Appl. Environ. Microbiol. 73:243–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lorenz MG, Krumbein WE. 1985. Influence of leaching parameters on the biological removal of uranium from coal by a filamentous cyanobacterium. Appl. Environ. Microbiol. 50:1296–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ariya PA, Dastoor AP, Amyot M, Schroeder WH, Barrie L, Anlauf K, Raofie F, Ryzhkov A, Davignon D, Lalonde J, Steffen A. 2004. The artic: a sink for mercury. Tellus B Chem. Phys. Meteorol. 56:397–403 [Google Scholar]
- 16.Clarkson TW, Magos L. 2006. The toxicology of mercury and its chemical compounds. Crit. Rev. Toxicol. 36:609–662 [DOI] [PubMed] [Google Scholar]
- 17.Wall JD, Krumholz LR. 2006. Uranium reduction. Annu. Rev. Microbiol. 60:149–166 [DOI] [PubMed] [Google Scholar]
- 18.De Philippis R, Colica G, Micheletti E. 2011. Exopolysaccharide-producing cyanobacteria in heavy metal removal from water: molecular basis and practical applicability of the biosorption process. Appl. Microbiol. Biotechnol. 92:697–708 [DOI] [PubMed] [Google Scholar]
- 19.Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. 2009. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem. Sci. 34:85–96 [DOI] [PubMed] [Google Scholar]
- 20.Zaffagnini M, Bedhomme M, Marchand CH, Morisse S, Trost P, Lemaire SD. 2012. Redox regulation in photosynthetic organisms: focus on glutathionylation. Antioxid. Redox Signal. 16(6):567–586 [DOI] [PubMed] [Google Scholar]
- 21.Rippka R, Deruelles J, Waterbury JB, Herdman M, Stanier RY. 1979. Generic assignments, strains histories and properties of pure cultures of cyanobacteria. J. Gen. Microbiol. 111:1–61 [Google Scholar]
- 22.Domain F, Houot L, Chauvat F, Cassier-Chauvat C. 2004. Function and regulation of the cyanobacterial genes lexA, recA and ruvB: LexA is critical to the survival of cells facing inorganic carbon starvation. Mol. Microbiol. 53:65–80 [DOI] [PubMed] [Google Scholar]
- 23.Kanemori M, Nishihara K, Yanagi H, Yura T. 1997. Synergistic roles of HslVU and other ATP-dependent proteases in controlling in vivo turnover of sigma32 and abnormal proteins in Escherichia coli. J. Bacteriol. 179:7219–7225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karimova G, Pidoux J, Ullmann A, Ladant D. 1998. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. U. S. A. 95:5752–5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Labarre J, Chauvat F, Thuriaux P. 1989. Insertional mutagenesis by random cloning of antibiotic resistance genes into the genome of the cyanobacterium Synechocystis PCC6803. J. Bacteriol. 171:3449–3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fox B, Walsh CT. 1982. Mercuric reductase. Purification and characterization of a transposon-encoded flavoprotein containing an oxidation-reduction-active disulfide. J. Biol. Chem. 257:2498–2503 [PubMed] [Google Scholar]
- 27.Lillig CH, Potamitou A, Schwenn JD, Vlamis-Gardikas A, Holmgren A. 2003. Redox regulation of 3′-phosphoadenylylsulfate reductase from Escherichia coli by glutathione and glutaredoxins. J. Biol. Chem. 278:22325–22330 [DOI] [PubMed] [Google Scholar]
- 28.Bedhomme M, Zaffagnini M, Marchand CH, Gao XH, Moslonka-Lefebvre M, Michelet L, Decottignies P, Lemaire SD. 2009. Regulation by glutathionylation of isocitrate lyase from Chlamydomonas reinhardtii. J. Biol. Chem. 284:36282–36291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marbouty M, Saguez C, Cassier-Chauvat C, Chauvat F. 2009. ZipN, a FtsA-like orchestrator of divisome assembly in the model cyanobacterium Synechocystis PCC6803. Mol. Microbiol. 74:409–420 [DOI] [PubMed] [Google Scholar]
- 30.Fernandes AP, Holmgren A. 2004. Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid. Redox Signal. 6(1):63–74 [DOI] [PubMed] [Google Scholar]
- 31.Silver S, Phung L. 2005. A bacterial view of the periodic table: genes and proteins for toxic inorganic ions. J. Ind. Microbiol. Biotechnol. 32:587–605 [DOI] [PubMed] [Google Scholar]
- 32.Mermet-Bouvier P, Chauvat F. 1994. A conditional expression vector for the cyanobacteria Synechocystis sp. PCC6803 and PCC6714 or Synechococcus sp. PCC7942 and PCC6301. Curr. Microbiol. 28:145–148 [DOI] [PubMed] [Google Scholar]
- 33.Poncelet M, Cassier-Chauvat C, Leschelle X, Bottin H, Chauvat F. 1998. Targeted deletion and mutational analysis of the essential (2Fe-2S) plant-like ferredoxin in Synechocystis PCC6803 by plasmid shuffling. Mol. Microbiol. 28:813–821 [DOI] [PubMed] [Google Scholar]
- 34.Marraccini P, Bulteau S, Cassier-Chauvat C, Mermet-Bouvier P, Chauvat F. 1993. A conjugative plasmid vector for promoter analysis in several cyanobacteria of the genera Synechococcus and Synechocystis. Plant Mol. Biol. 23:905–909 [DOI] [PubMed] [Google Scholar]
- 35.Li R, Haile JD, Kennelly PJ. 2003. An arsenate reductase from Synechocystis sp. strain PCC 6803 exhibits a novel combination of catalytic characteristics. J. Bacteriol. 185:6780–6789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Couturier J, Jacquot JP, Rouhier N. 2009. Evolution and diversity of glutaredoxins in photosynthetic organisms. Cell. Mol. Life Sci. 66:2539–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gallogly MM, Mieyal JJ. 2007. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 7:381–391 [DOI] [PubMed] [Google Scholar]
- 38.Masip L, Veeravalli K, Georgiou G. 2006. The many faces of glutathione in bacteria. Antioxid. Redox Signal. 8(5–6):753–762 [DOI] [PubMed] [Google Scholar]
- 39.Prentki P, Binda A, Epstein A. 1991. Plasmid vectors for selecting IS1-promoted deletions in cloned DNA: sequence analysis of the omega interposon. Gene 103:17–23 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.