

Abstract
The combination of paramagnetic tagging strategies with NMR or EPR spectroscopic techniques can revolutionize de novo structure determination of helical membrane proteins. Leveraging the full potential of this approach requires optimal labeling strategies and prediction of membrane protein topology from sparse and low-resolution distance restraints, as addressed by Chen et al. (2011).
Based on their secondary structure composition, membrane proteins can be classified into α-helical bundles, examples of which are voltage-gated ion channels and G-protein coupled receptors (GPCRs), and β-barrels such as outer membrane proteins or porins. About 30% of the sequenced genome represents integral membrane proteins, many of which are important drug targets. Determining the three dimensional structure of membrane proteins is the starting point towards an atomic-detail understanding of the mechanisms that define their biological function.
Despite their biological significance, membrane protein comprise only about 1%–2% of experimentally determined protein structures available today, mostly due to the difficulties of producing suitable amount of proteins for structural studies. Additional complication arises from the fact that membrane proteins need to be incorporated into a membrane mimetic, such as lipid bilayers, micelles, or bicelles, to retain their native fold. This leads to the third challenge of adapting techniques for protein structure determination to work with a protein in the membrane mimetic.
The use of Nuclear Magnetic Resonance (NMR) is limited by the size of the protein/membrane-mimetic complex and resulting difficulties in unambiguous signal assignment, in particular for amino acid side chains. In turn, restraints from nuclear overhauser effect (NOEs) used for structure calculations are often limited to the protein backbone. In particular for helical membrane proteins this links residues that are close in sequence but excludes distance restraints between remote amino acids. However, these high “information content” restraints define the protein fold (Alexander et al 2008). Topology determination through residual dipolar couplings (RDCs), another NMR technique that gives information on the protein fold, is hampered, as many of the traditional alignment media do not apply to membrane proteins. Nevertheless, substantial progress has been made in the development of technologies such as transverse-relaxation optimized spectroscopy (TROSY), alternative labeling strategies, and perdeuteration in conjunction with the increase in magnetic field strength (Sanders and Sonnichsen, 2006).
The introduction of paramagnetic labels is an alternative strategy to obtain distance restraints for helical membrane proteins. Typically a paramagnetic probe is attached to the protein of interest through a cysteine residue via a disulfide linkage. The cysteine is strategically introduced using site directed mutagenesis of a native residue –“site directed spin labeling (SDSL)”. The probe selectively broadens NMR signals in its proximity in a distance-dependent manner. These paramagnetic relaxation enhancements (PREs) result primarily from a dipole–dipole interaction between an unpaired electron and a nucleus. Distances of up to 25 Å can be measured through the intensities of the peaks (Battiste and Wagner 2000). This technology proved critical for the structure determination of helical membrane proteins such as DsbB (Zhou et. al. 2008) and DAGK (Van Horn et al 2009). Similarly, introduction of two spin labels allows distance measurements in the range from 5 – 80Å by measuring the dipole–dipole interaction between the unpaired electrons using EPR spectroscopy (Borbat et al., 2002).
Since a dedicated mutant protein needs to be prepared for every measurement, along with verifying structural and functional integrity of the mutant, SDSL-NMR and SDSL-EPR experiments are resource intensive. Effective labeling strategies are needed to minimize the number of experiments for unambiguous topology determination. Further, distance restraints obtained from these experiments are not only sparse but also intrinsically low in resolution. The unpaired electron resides on a flexible linker arm up to 8.5Å from the Cβ-atom of the cysteine. As a result, the distance restraints alone are insufficient to define the protein backbone at atomic detail accuracy.
This setting poses three formidable challenges for computational structural biology (Figure 1): (1) to determine optimal labeling strategies that minimizes the number of experiments needed to determine the membrane protein topology unambiguously, (2) to define the topology of the transmembrane segments from the low-resolution distance restraints and assign a confidence measure, and (3) to complete and refine these initial models to atomic detail that is invisible in the experimental data. Recently a number of isolated computational techniques have been introduced that when combined have the potential to provide an integrated approach to tackle all three challenges.
Figure 1.
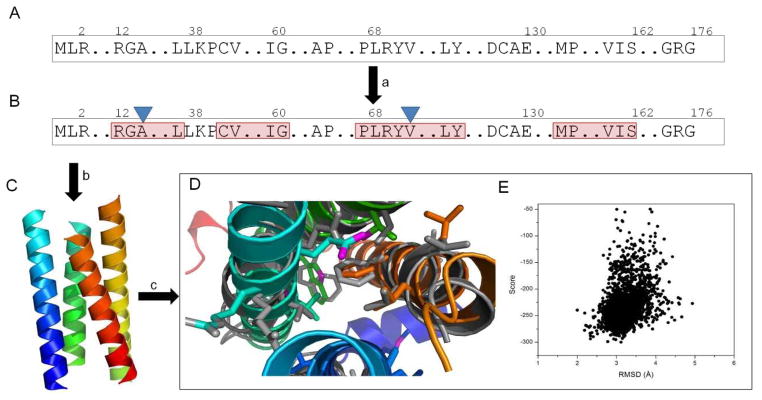
Protocol for Structure Prediction from Paramagnetic Restraints.
(A and B) Primary sequence (A) is used to predict secondary structure elements and transmembrane span region (specified by red frames) (B). (C) PRE distance restraints from two different sites (blue triangles, B) drive determination of the topology of the four transmembrane helices with minimum backbone rmsd of 3.2 Å compared with crystal structure. (D) The Rosetta refined model (rainbow) has a backbone rmsd of 2.7 Å compared with the crystal structure (gray), with similar side-chain conformation in the protein score. (E) A Rosetta score versus rmsd plot of the models generated during refinement process.
In this issue of Structure, Chen et al. (2011) discuss a computational method to determine the optimal labeling sites for collecting PRE data for helical membrane proteins from sequence information alone. The complex three dimensional packing problem was reduced to a problem of determining the two dimensional geometry of the interacting helices by assuming ideal helix geometries in the transmembrane region, parallel to each other and perpendicular to the membrane surface. Using distance geometry, the correct topology of the four-helix membrane protein DsbB was successfully determined with by considering PRE data from two tagging sites. The concept was extended to predict the optimal tagging sites of five, six and seven helix bundle proteins. The results suggest that, to correctly predict the topology, the tags should be attached to helices which are furthest apart in the structure as estimated by predicted lipophilicity.
Kazmier et al. (2011) describe a computational algorithm for selection of optimized labeling sites for de novo structure determination of helical proteins from SDSL-EPR distance restraints. The data suggest that one distance restraint between each pair of helices is needed for efficient determination of protein topology. In another study, the structure prediction program Rosetta was used in conjunction with a cone model that maps distance information from the flexible spin labels back onto the protein backbone to determinate the protein topology (Alexander et al. 2008, Hirst et al 2011). The same authors demonstrate refinement of initial topology models to atomic detail accuracy using RosettaEPR. It is expected that this approach can be extended to membrane proteins as Barth et al. (Barth et al 2009) predicted membrane protein structures with complex topologies using limited constraints in conjunction with Rosetta and refined some of these models to high resolution.
In the coming years, substantial progress in membrane protein structure determination from spectroscopic techniques is expected through development and integration of these and similar computational approaches. Figure 1 Illustrates a possible protocol for this combined approach by generating a high-resolution model of DsbB topology that has been determined using the PRE distance restraints from labeling sites A14C and V72C (Chen et al., 2011). Transmembrane helices have been assembled using BCL::Fold (Lindert et al 2009). The initial topology model has been refined using Rosetta to an accuracy of 2.7 Å and can be identified by superior agreement with experimental data as well as Rosetta energy.
Acknowledgments
This work was supported by grant R01-GM080403 from the National Institute of General Medical Sciences.
References
- Alexander N, Al-Mestarihi A, Bortolus M, Mchaourab H, Meiler J. De Novo High-Resolution Protein Structure Determination from Sparse Spin-Labeling EPR Data. Structure. 2008;16:181–95. doi: 10.1016/j.str.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth P, Wallner B, Baker D. Prediction of membrane protein structures with complex topologies using limited constraints. Proc Natl Acad Sci U S A. 2009;106:1409–1414. doi: 10.1073/pnas.0808323106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- Borbat PP, Mchaourab HS, Freed JH. Protein structure determination using long-distance constraints from double-quantum coherence ESR: study of T4 lysozyme. J Am Chem Soc. 2002;124:5304–14. doi: 10.1021/ja020040y. [DOI] [PubMed] [Google Scholar]
- De Angelis AA, Jones DH, Grant CV, Park SH, Mesleh MF, Opella SJ. NMR experiments on aligned samples of membrane proteins. Methods Enzymol. 2005;394:350–82. doi: 10.1016/S0076-6879(05)94014-7. [DOI] [PubMed] [Google Scholar]
- Hirst SJ, Alexander N, McHaourab HS, Meiler J. RosettaEPR: An integrated tool for protein structure determination from sparse EPR data. Journal of Structural Biology. 2011;173:506–514. doi: 10.1016/j.jsb.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K, Murakami S, Suzuki M, Nakagawa A, Yamashita E, Okada K, Ito K. Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell. 2006;127:789–801. doi: 10.1016/j.cell.2006.10.034. [DOI] [PubMed] [Google Scholar]
- Kazmier K, Alexander NS, Meiler J, McHaourab HS. Algorithm for selection of optimized EPR distance restraints for de novo protein structure determination. Journal of Structural Biology. 2011;173:549–557. doi: 10.1016/j.jsb.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindert S, Staritzbichler R, Wötzel N, Karakas M, Stewart PL, Meiler J. EM-Fold: De Novo Folding of [alpha]-Helical Proteins Guided by Intermediate-Resolution Electron Microscopy Density Maps. Structure. 2009;17:990–1003. doi: 10.1016/j.str.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders CR, Sonnichsen F. Solution NMR of membrane proteins: practice and challenges. Magn Reson Chem. 2006;44(Spec No):S24–40. doi: 10.1002/mrc.1816. [DOI] [PubMed] [Google Scholar]
- Van Horn WD, Kim HJ, Ellis CD, Hadziselimovic A, Sulistijo ES, Karra MD, Tian C, Sonnichsen FD, Sanders CR. Solution nuclear magnetic resonance structure of membrane-integral diacylglycerol kinase. Science. 2009;324:1726–9. doi: 10.1126/science.1171716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Cierpicki T, Jimenez RH, Lukasik SM, Ellena JF, Cafiso DS, Kadokura H, Beckwith J, Bushweller JH. NMR solution structure of the integral membrane enzyme DsbB: functional insights into DsbB-catalyzed disulfide bond formation. Mol Cell. 2008;31:896–908. doi: 10.1016/j.molcel.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]