

Abstract
We previously reported that maternal separation (MS) sensitizes adult rats to angiotensin II (AngII)-induced hypertension. The aim of this study was to investigate the vascular reactivity to AngII and the role of renin-angiotensin system components, reactive oxygen species production and nitric oxide synthase (NOS) buffering capacity mediating the exacerbated AngII-induced responses. MS rats were separated from their mothers for 3 hours/day from day 2-14 of life. Controls (C) were non-handled littermates. At 12 weeks of age, aortic AngII-induced constriction was greater from MS rats compared to C (p<0.05), moreover, endothelial denudation abolished this difference. The response to other constrictors was unchanged. Angiotensin type 2 receptor (AT2R) function was reduced in aortic AngII-induced constriction from MS rats compared to C. Angiotensin type 1 receptor (AT1R) function was similarly abolished in both groups. However, protein expression of AT1 and AT2 receptors were similar in aortic rings from MS and C rats. Pre-incubation with superoxide inhibitor or scavenger attenuated the AngII-induced vasoconstriction in C, but not in MS rats. However, acute pre-incubation with a NOS inhibitor enhanced aortic AngII-induced constriction in aorta from C rats, but this effect was significantly reduced in MS rats compared to C. Accordingly, a further increase in AngII-induced hypertension due to chronic NOS inhibition (days 10-13) was blunted in MS rats compared to C. Similar NOS expression and activity was observed in C and MS rats. In conclusion, MS induces a phenotype with reduced endothelial NOS buffering capacity leading to dysfunctional endothelial AngII-mediated signaling and sensitization to AngII-induced vasoconstriction.
Keywords: maternal separation, vascular reactivity, AT2 receptor, NOS buffering, angiotensin II
Within the last decade, a growing body of evidence links adverse behavioral or environmental stress during early life with chronic diseases in adulthood. The quality of early life, prenatal and/or postnatal, can influence mechanism(s) of metabolic and mental disorders as well as cardiovascular disease in both humans 1-3 and animal models4-6. Significant alteration in maternal care is considered a stressful experience associated with modifications in the endocrine response7, 8 leading to enhanced acute stress-related responses in the offspring later in life. Maternal separation (MS) is a widely used model of early life behavioral stress in rodents (for review see 9). Several studies reported that exposure to MS permanently alters the “programming” of the CNS, endocrine and immune systems10-12.
Recently, we reported that MS sensitizes adult rats to angiotensin II (AngII)-induced hypertension and vascular inflammation13. These compelling experimental data led us to reason that early life stress, specifically MS, may induce defects in the renin-angiotensin system (RAS).
The RAS is comprised of classical and non-classical pathways. Classically, angiotensin-converting enzyme (ACE1) metabolizes AngI to AngII, while ACE2 and neprilysin, components of the non-classical pathway, metabolize various other angiotensin peptides14. AngII type 1 (AT1; AT1a and AT1b in the rat) receptor activation mediates vasoconstriction, vascular hypertrophy and remodeling, promotes inflammatory cell activation, as well as sodium and water retention15, 16. It is well-established that activation of NADPH oxidase and production of reactive oxygen species (ROS) is a major AT1-mediated pathway in many cell types 17. AngII type 2 (AT2) receptor activation is generally considered to be a counter-regulatory receptor to AT1 receptor activation 18. Both AT1 19, 20 and AT2 21 receptors stimulate NO synthase (NOS) activity and NO production, which blunts the vasoconstrictor actions of the AT1 receptor 19.
We hypothesized that MS induces a dysfunctional vascular phenotype, specifically an imbalance in AT1 and AT2 receptor activation that may exacerbate ROS production and/or reduce the NOS buffering capacity ultimately leading to vascular dysfunction and promoting cardiovascular disease. ROS are linked to the mechanism underlying reduced NOS buffering capacity, given that superoxide can react with NO and diminish NO bioavailability22, 23. Thus, the aim of this study is to investigate AngII-mediated vascular reactivity and the role of the endothelium, RAS components, ROS production and NOS buffering capacity mediating the exacerbated AngII-induced responses. Receptor mediated mechanism(s) as well as expression and localization of aortic RAS pathway components in MS and control rats will also be determined. In addition, we designed studies to elucidate the role of NOS buffering capacity ex vivo and in vivo mediating AngII-induced vasoconstrictor responses by using a NOS non-selective inhibitor, L-NAME.
Methods
Detailed methods are provided in the online Data Supplement (http://hyper.ahajournals.org).
Animal model
All animal use protocols received prior approval by the institutional animal care and use committee at the Georgia Health Sciences University. Using Wistar Kyoto (WKY) rats, maternal separation protocol was performed as previously described13. Briefly, approximately half of the male pups were separated from their mothers (MS) and littermates by transferring the pups to a clean cage in an incubator (30±1° C) for 3 hours from day 2 to day 14 of life. Normally-reared non-handled littermates served as the control group. Each experimental group of rats originated from a minimum of 3 different litters.
Aortic vascular reactivity
Thoracic aortic tissue was isolated as previously described24. Cumulative concentration-response curves (CRC) were generated for AngII, endothelin (ET-1, from 1×10-12 to 3×10-6 M), phenylephrine (PE, from 1×10-9 to 3×10-5 M), and KCl (4.7 mM to 100 mM). The following compounds were utilized to pre-incubate (30 min) aortic rings prior to AngII-induced constriction: candesartan (AT1 receptor antagonist; 10-5 M) and PD123,319 (AT2 receptor antagonist; 10-6 M), L-NAME (NOS inhibitor; 10-4 M), apocynin (NADPH oxidase inhibitor; 3×10-4 M) and PEG-SOD (cell permeable superoxide dismutase; 1000 U/ml).
Protein expression
Whole tissue homogenates (NOS isoforms) and membrane preparations (AT receptors, see supplement for detailed protocol) were obtained from aortic tissue. Equal protein amounts were loaded and run in SDS-PAGE analysis followed by immunoblotting on PVDF blots. Two-color immunoblots were analyzed using polyclonal primary antibodies to AT1, AT2, NOS1, NOS2 and NOS3 and detected by immunofluorescence. Densitometric results were reported normalized to β-actin.
NOS enzymatic activity
NOS enzymatic activity and expression were performed as previously described25. NOS activity was normalized to total protein concentration (Bradford method, BioRad) and expressed as pmoles citrulline/mg protein/30 min.
Immunohistochemical analysis of aortic tissue
Rats were anesthetized with pentobarbital and aortic rings were removed, rinsed in saline, and immersed in 10% buffered formalin overnight at room temperature and paraffin-embedded as previously described13. Following staining, sections were viewed with an Olympus BX40 microscope (Olympus America, Melville, NY) on brightfield setting fitted with a digital camera (Olympus DP12, Olympus America, Melville, NY).
Chronic inhibition of NOS
L-NAME was administered in the drinking water from week 14-15 (10 mg/kg/day), week 16 (50 mg/kg/day) and week 17 of life (100 mg/kg/day). MAP was monitored by telemetry as previously described13. Blood pressure was expressed as a 24hr average at baseline, day 7, 14 and 28. A separate group of telemetry-implanted rats were infused with AngII. On days 10-13 of AngII infusion, rats were treated with L-NAME in drinking water (100 mg/kg/day).
Statistical analysis
Linear and logarithmic data are presented as means ± SE. Basal parameters in table S1 were compared using unpaired Student’s t-test. Vascular reactivity data were compared between concentrations by unpaired Student’s t-test or one-way ANOVA with Bonferroni’s post hoc performed using GraphPad Prism version 5.01 for Windows (GraphPad Software, San Diego, CA). A value of p < 0.05 was considered statistically significant.
Results
Aortic vasoconstriction
Maximal AngII-induced vasoconstriction was significantly greater in aortic rings from MS than control rats (figure 1A; table S1). No differences in EC50 were observed between MS and control rats (table S1). Absolute tension in response to AngII (table S2) was also significantly higher in aortic tissue of MS rats (44.5±2.4 mN) compared to control rats (34.3±1.8 mN, p<0.05). Denudation of the endothelium revealed that AngII-induced a similar response in aortic rings from MS and control rats (figure 1B).
Figure 1.
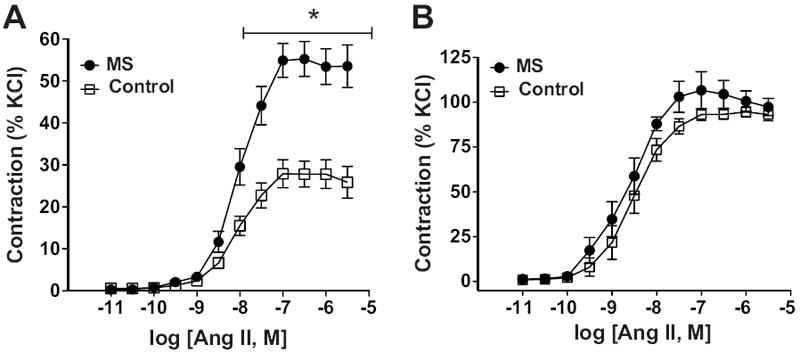
Vascular reactivity in intact thoracic aortic rings. A) Cumulative concentration-response curves of AngII from MS (n=17) and control (n=17) rats. B) AngII-induced constriction in endothelium denuded rings of MS (n=6) and control (n=6) rats. * p< 0.05.
Pre-incubation with an AT1 receptor blocker (candesartan; 10 μM) abolished the AngII-induced constriction in endothelium-intact aorta from both MS and control rats (figure 2A). Selective AT2 receptor blockade (PD123,319; 1 μM) significantly increased the AngII-induced constriction in aortic rings from control rats (figure 2B), while pre-incubation with the AT2 receptor blocker did not display a functional change in vasoconstriction in aortic rings of MS rats. EC50 in the presence and absence of receptor blockers was similar in MS and control rats (table S1).
Figure 2.
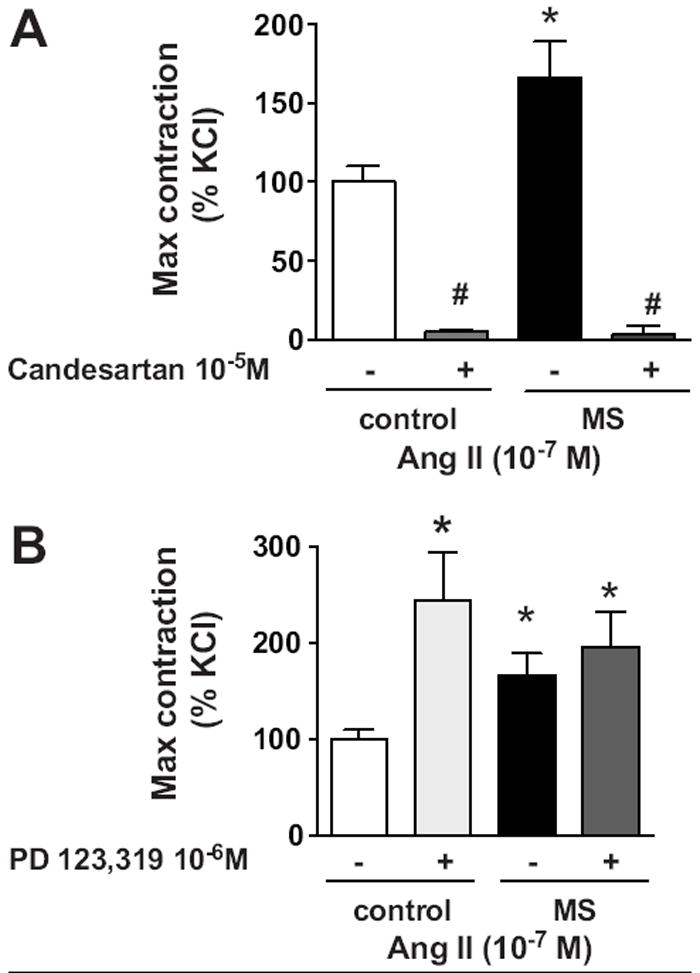
AngII receptor pharmacological blockade. A) Pre-incubation with candesartan (specific AT1 receptor blocker, 10-5 M) blunted AngII-induced constriction in MS (n=4) and control (n=4) rats. B) Pre-incubation with PD123,319 (specific AT2 receptor blocker, 10-6 M) increased the response to AngII in control rats (n=7), but had no effect in MS rats (n= 7). Data are expressed in percentage of increase from the maximal response to AngII 10-7 M. Values from the control group represent the baseline (100%). * p<0.05 vs. control.
To elucidate whether the enhanced AngII-mediated constriction is specific for AngII, we analyzed the vasoconstrictor responses to phenylephrine (PE), potassium chloride (KCl) and endothelin (ET-1) in aortic rings from MS and control rats (figure S1). No significant differences between groups were found in % max contraction or −log EC50 (table S1 and S2).
Aortic AT1 and AT2 receptor expression and immunolocalization
Protein expression of AT1 (43 kDa) and AT2 (band at 50 kDa) receptors were similar in membrane preparations of aortic tissue of control and MS rats (figure 3A). Immunolocalization of AT1 (figure 3B and 3C) and AT2 (figure 3E and 3F) receptors are similar in the endothelium and smooth muscle from control and MS rats, respectively. Specificity of the AT1 and AT2 receptor immunoreactivity was verified with antigen blockade for immunohistological analysis (figure 3D and 3G). Aortic mRNA expression of RAS components was assessed by quantitative real-time PCR from MS and control rats (figure S2). AT1 receptor, mas receptor, ACE1, ACE2, and neprilysin mRNA expression were similar between groups. AT2 receptor mRNA expression was significantly reduced in aorta from MS rats compared to control rats (p<0.05).
Figure 3.
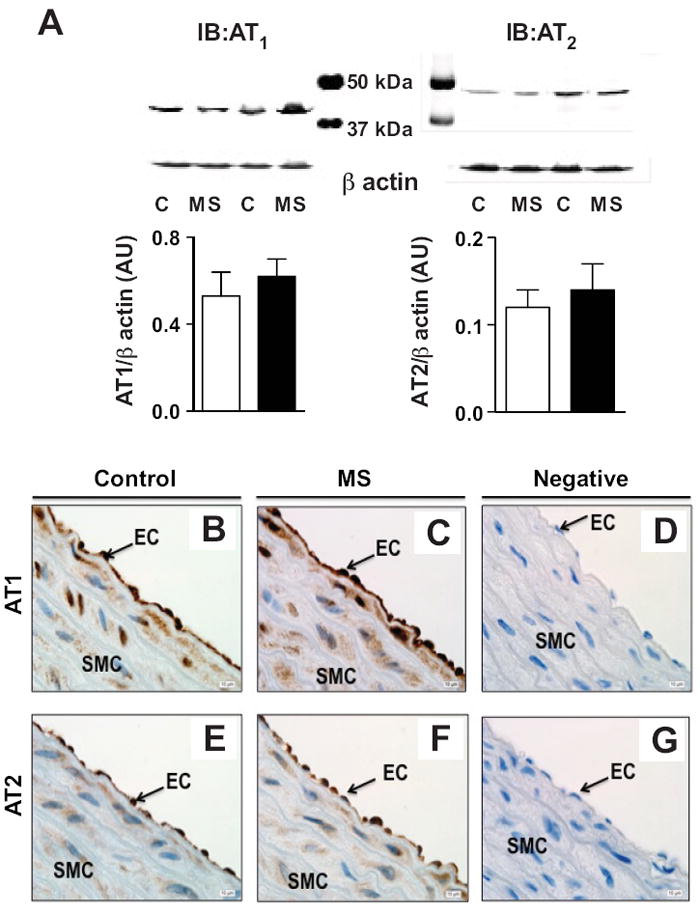
Expression and localization of AT1 and AT2 receptor. A) Representative Western blot of AT1 and AT2 protein expression in aortic tissue of control and MS rats with densitometric analysis (n=5-6). Representative immunolocalization of AT1 receptor (B and C) and AT2 receptor (E and F) in control and MS aortae. Negative control was assessed by antigen blockade (D and F). Magnification=100X.
Aortic AngII-mediated ROS and NOS signaling during vasoconstriction
Pre-incubation with apocynin (300 μM; NADPH oxidase inhibitor) or PEG-SOD (1000 Units/ml; superoxide scavenger) significantly blunted the AngII-induced vasoconstriction in intact aortic rings from control rats (figure 4A and 4C). Surprisingly, these compounds did not modify the AngII-induced response in aortic rings from MS rats (figure 4B and 4D).
Figure 4.
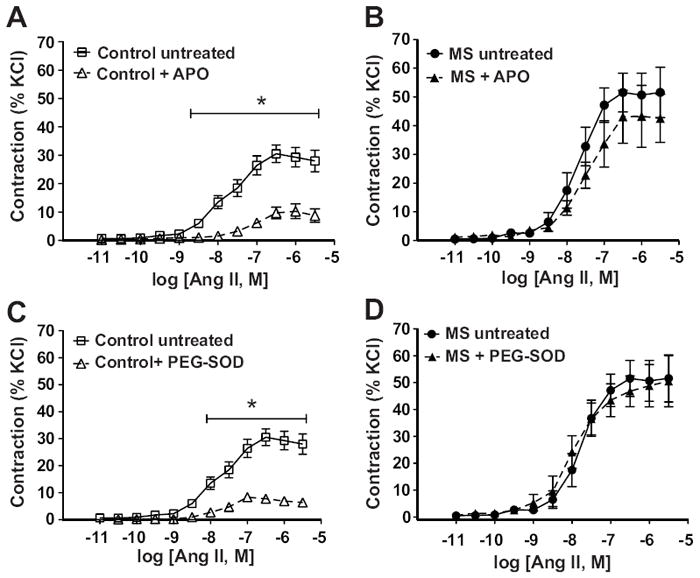
Participation of ROS in AngII-induced aortic constriction. A) AngII-induced aortic constriction in control rats (n=12) was reduced after pre-incubation with apocynin (300uM, n=7), B) MS rats showed similar AngII-induced constriction in the presence (n=5) or absence (n=12) of apocynin. C) AngII-induced aortic constriction in control rats (n=12) was reduced after pre-incubation with PEG-SOD (1000 Units/mL, n=6), D) MS rats (n=12) showed similar AngII-induced constriction using PEG-SOD. * p<0.05 vs. untreated.
Pre-incubation with the NOS inhibitor (L-NAME; 100 μM) elicited a significantly greater AngII-induced constriction in intact aortic rings compared to untreated aortic rings from control rats (figure 5A). L-NAME treatment also produced an increased AngII-induced constriction in intact aorta from MS rats (figure 5B), although the response was significantly less than the response in control rats (figure 5C). Maximal vasoconstriction in the presence of L-NAME was increased in intact aortic tissue from MS rats compared to control rats (figure S3). The AngII-induced constriction in the presence of L-NAME was similar in endothelium-denuded aortas from control and MS rats (figure S4).
Figure 5.
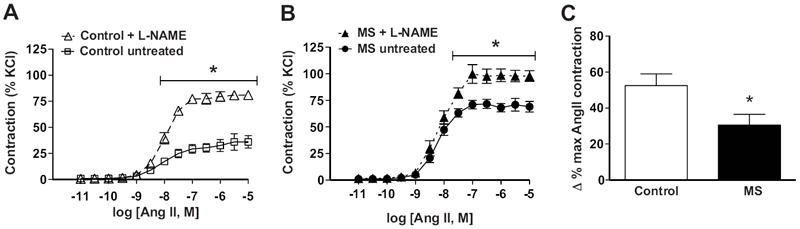
AngII-induced aortic constriction during NOS blockade. Pre-incubation of intact aortic rings with a NOS inhibitor (L-NAME, 100uM) enhanced AngII-induced constriction in both A) control (n=7) and B) MS (n=6) rats. C) The increase in the percentage of contraction in the L-NAME-treated rings was significantly less in MS compared to control rats. * p<0.05 vs. control.
Blood pressure during chronic in vivo NOS inhibition
MAP at day 10 of AngII infusion, prior to NOS blockade, was significantly greater in MS rats (142.0±0.9 mmHg) compared to control rats (132.1±2.8 mmHg, p<0.05; figure 6A) confirming our previously published results13. L-NAME treatment, during AngII infusion, significantly increased MAP within 24 hours in control rats, while MS rats displayed a blunted response (figure 6B). The total pressor response in AngII-infused rats treated with L-NAME for 3 days (days 10-13 during AngII infusion) was significantly greater in control rats (158.3±2.5 mmHg) compared to MS rats (146.4±2.9 mmHg, p<0.005) (figure S5). The heart rate increase in response to chronic NOS inhibition during AngII infusion was severely attenuated in MS rats (figure S5). Chronic in vivo administration of L-NAME (in the absence of AngII infusion) induced a dose-dependent increase in blood pressure in control and MS rats (figure S6). Analysis of the slope revealed significantly attenuated effects of L-NAME on blood pressure in MS rats (figure S6; p=0.028). Conversely, chronic L-NAME administration, in the absence of AngII infusion, induced similar decreases in heart rate in control and MS rats (figure S6).
Figure 6.
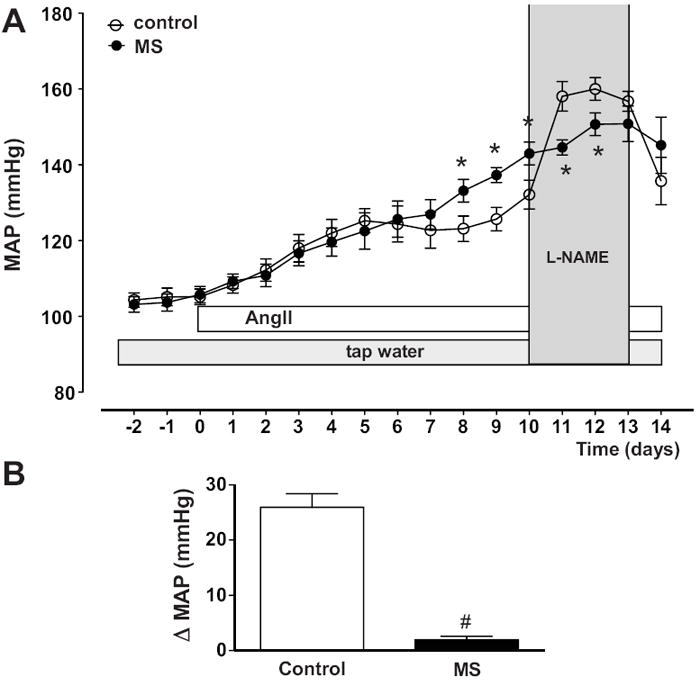
MAP responses to chronic L-NAME treatment during AngII infusion. MS rats (n=6) have exaggerated MAP to chronic AngII infusion and a blunted pressor response to chronic L-NAME compared to control rats (n=6). A) MAP expressed as 24 hr average. B) Change in the MAP in response to L-NAME treatment after 24 hours. * p<0.05 vs. AngII-infused control rats, # p<0.05 vs. AngII-infused+L-NAME control rats.
Vasorelaxation, NOS activity, and NOS expression
Endothelium-dependent and -independent vasorelaxation in aortic rings from MS and control rats were determined by analyzing responses to acetylcholine and sodium nitroprusside. Responses were similar from both groups (figure 7A, 7B and table S1). Total and isoform-specific NOS activity was determined in homogenates of aortic tissue from MS and control rats. No significant differences were observed in total or isoform-specific NOS activity (figure 7C). Aortic NOS1 (155 kDa) and NOS3 (135 kDa) expression by Western blot were found to be similar in both groups of rats (figure 7D). NOS2 expression was below detection (data not shown). In addition, NOS3 expression was determined by immunohistochemical analysis. NOS3 was highly expressed in the aortic endothelium in both MS and control rats (figure S7). Additional staining in the absence of primary antibody served as a negative control and confirmed the absence of immunoreactivity (figure S7).
Figure 7.
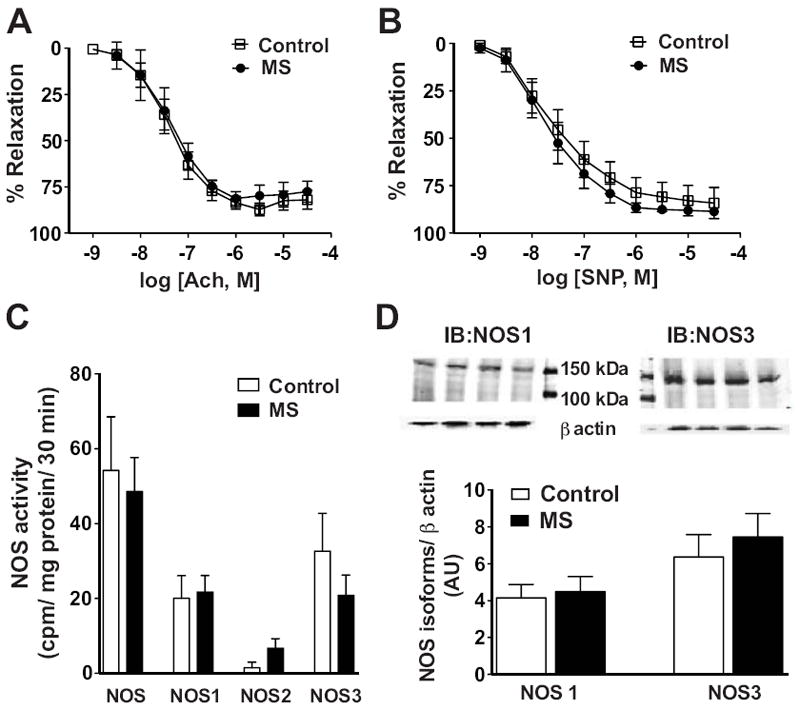
Endothelial-dependent and –independent vasorelaxation of aortic rings. A) ACh and B) SNP-induced relaxation were similar in control (n=5) and MS (n=9) rats. C) Total NOS and NOS isoform enzymatic activity were similar in MS (n=7) and control (n=7) rats. D) Representative Western blots for NOS1 and NOS3 from aortic tissue of MS and control rats (top) and densitometric analysis of NOS1 and NOS3 immunoreactivity revealed similar expression in aortic tissue between MS and control rats (bottom, n=4).
Discussion
This study revealed that maternal separation, a model of early life stress, induces a dysfunction in endothelial AngII receptor signaling and NOS buffering. Specifically, aortic tissue from MS rats demonstrated a significantly increased AngII-induced constriction compared to control rats that results from a loss in endothelial- and AngII-mediated NO production. In addition, aortic tissue from MS rats showed a loss of functional AT2 receptor activation, which appears to account for the increased AngII-induced constriction. Furthermore, the blood pressure response to chronic in vivo NOS inhibition was significantly attenuated in MS rats indicating that early life stress induces a phenotype with reduced NOS buffering capacity.
Maternal separation is considered a chronic behavioral stress model replicating the adverse environment during childhood in humans26. Therefore, maternal separation has been used to study behavioral consequences in stress-related responses, however, very few of these investigations have probed cardiovascular outcomes. Studies employing animal models using glucocorticoid exposure27, 28 or low protein diet 29 during the prenatal period, demonstrate that these fetal programming paradigms affect the sensitivity of the RAS pathway directly29, 30. We recently reported that control and MS rats have similar circulating levels of AngII, Ang1-7, aldosterone and plasma renin activity in baseline conditions, yet MS rats were found to be more sensitive to chronic AngII-infused hypertension and vascular inflammation13. The current results, taken together with the previous findings, support our hypothesis that early life stress induces a vascular phenotype that predisposes the adult rat to endothelial dysfunction, a hallmark of cardiovascular disease. Furthermore, this study extends our knowledge of potential RAS-dependent mechanisms focused on the role of the vascular endothelium as a target affected by early life stress.
Both AT1 and AT2 receptor activation includes stimulation of the NO pathway, however, specific intracellular signaling pathways and subcellular localizations are yet to be clarified. Endothelial AT1 receptor activates NO production via stimulation of protein kinase B (Akt) and phosphorylating NOS331. Siragy et al.32 have reported that a chronic infusion of AngII failed to increase NO release in AT2 null mice. In addition, Tsutsumi et al.33 have shown that the over-expression of AT2 receptors in the vasculature attenuates AngII-induced hypertension. Alterations in the AT1/AT2 ratio has been suggested as a possible mechanism leading to increased blood pressure34, 35. Our study did not observe any differences in immunolocalization of the AT1 and AT2 receptors within the vasculature nor any difference in protein expression of the receptors with a vascular membrane preparation, however, AT2 mRNA expression was reduced in MS rats as well as reduced AT2 receptor function. Given that we did not observe differences in the wall thickness of control and MS aortae 13 and that the dysfunction is localized to the endothelium, we have reasoned that the increased AngII reactivity may be intrinsic to angiotensin receptor signaling rather than expression. Further studies with isolated endothelial cells are necessary to elucidate the specific nature of the dysfunction in the AngII signaling pathway.
The role of reactive oxygen species (ROS) mediating AngII actions in the vasculature has been extensively reported to be via NADPH oxidase activation of superoxide36, 37. Accordingly, we probed this pathway and observed that a cell-permeable superoxide scavenger and an NADPH oxidase inhibitor significantly reduced the aortic AngII-induced constriction in control rats. To our surprise, neither PEG-SOD nor apocynin modulated the aortic AngII response in MS rats. Schuijt et al. reported that acute AngII stimulation in human and porcine arteries is not mediated via superoxide generated through NADPH oxidase and/or xanthine oxidase activation, but rather results in the generation of the vasodilator H2O238. At this point, it is unclear whether AngII activates other ROS dependent pathways or not and further experiments are planned to determine the status of these pathways. We propose that maternal separation induces a vascular phenotype that uncouples AngII-mediated activation of NADPH oxidase-mediated superoxide.
In vivo, NOS inhibition during chronic AngII infusion and in the absence of AngII infusion revealed a reduced sensitivity to increase blood pressure in MS rats compared to control rats. L-NAME induces a dramatic increase in blood pressure in control rats especially during chronic AngII infusion, but elicited a minimal change in MS rats. In addition, we observed that control rats displayed tachycardia as a consequence of NOS inhibition, whereas the heart rate was blunted in the MS rats in response to L-NAME. These data further support reduced NOS buffering capacity and suggests differences in the NOS pathway in cardiac function in MS rats. Others have reported that rats treated chronically with L-NAME develop hypertension 39, 40. Noteworthy, most of the reports suggest that NOS inhibition in the central nervous system may increase the sympathetic activity and vagal suppression as well41, which could be responsible, at least in part, for the increase in arterial pressure occurring in the L-NAME hypertensive model 40, 41. Thus, further investigation is necessary to determine the role that early life stress exerts on the brain, kidneys, and heart in the regulation of long-term blood pressure.
Classically, endothelial dysfunction has been defined as blunted acetylcholine-induced vasorelaxation. Surprisingly, maternal separation did not influence endothelium-dependent or -independent vasorelaxation pathways, NOS activity, NOS expression, or NOS immunolocalization. Interestingly, we compared AngII-induced maximal constriction in the presence of L-NAME and found that the vasoconstriction is higher in MS than in control rats. Therefore, MS rats display an increased AngII reactivity regardless of the status of NOS. These data suggest that early life stress mediates a dysfunctional vasoconstriction rather than vasorelaxation. Thus, the endothelial AT1 and AT2 intracellular signaling pathways require further investigation, specifically, regarding the role of NOS. Taken together, our findings point to dysfunctional AngII signaling to NOS during the contractile response as a mechanism by which early life stress induces permanent changes in the adult phenotype increasing the risk to develop cardiovascular disease. Future research will test the hypothesis that increased AngII vasoconstriction in MS rats is mediated through reduced endothelial AT/NOS molecular signaling.
Perspectives
There is a consensus that adverse environment during early life programs biological consequences associated with stress and inflammatory response mechanisms in adulthood 42. Indeed, this study distinguishes the impact of adverse early life environmental stressors on vascular function. New mechanistic strategies for the study of long-term consequences of early life stress in the development of cardiovascular disease are required.
Supplementary Material
Acknowledgments
We gratefully acknowledge the outstanding technical support from Hiram Ocasio, Janet Hobbs, Melissa Durley and Jacqueline Musall.
Funding sources This study was supported by grants from the NIH National Heart, Lung, and Blood Institute (J.S. Pollock and D.M. Pollock: P01 HL69999, R01 HL60653) and from the American Heart Association (A. S. Loria Postdoctoral Fellowship: AHASE00027).
Footnotes
Disclosures None
References
- 1.Kelishadi R, Mirghaffari N, Poursafa P, Gidding SS. Lifestyle and environmental factors associated with inflammation, oxidative stress and insulin resistance in children. Atherosclerosis. 2009;203:311–319. doi: 10.1016/j.atherosclerosis.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 2.Holness MJ, Langdown ML, Sugden MC. Early-life programming of susceptibility to dysregulation of glucose metabolism and the development of Type 2 diabetes mellitus. Biochem J. 2000;349(Pt 3):657–665. doi: 10.1042/bj3490657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caldji C, Diorio J, Meaney MJ. Variations in maternal care in infancy regulate the development of stress reactivity. Biol Psychiatry. 2000;48:1164–1174. doi: 10.1016/s0006-3223(00)01084-2. [DOI] [PubMed] [Google Scholar]
- 4.Samuelsson AM, Matthews PA, Argenton M, Christie MR, McConnell JM, Jansen EH, Piersma AH, Ozanne SE, Twinn DF, Remacle C, Rowlerson A, Poston L, Taylor PD. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension. 2008;51:383–392. doi: 10.1161/HYPERTENSIONAHA.107.101477. [DOI] [PubMed] [Google Scholar]
- 5.Enthoven L, de Kloet ER, Oitzl MS. Differential development of stress system (re)activity at weaning dependent on time of disruption of maternal care. Brain Res. 2008;1217:62–69. doi: 10.1016/j.brainres.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 6.Tucker DC, Johnson AK. Influence of neonatal handling on blood pressure, locomotor activity, and preweanling heart rate in spontaneously hypertensive and Wistar Kyoto rats. Dev Psychobiol. 1984;17:587–600. doi: 10.1002/dev.420170603. [DOI] [PubMed] [Google Scholar]
- 7.Lehmann J, Stohr T, Feldon J. Long-term effects of prenatal stress experiences and postnatal maternal separation on emotionality and attentional processes. Behav Brain Res. 2000;107:133–144. doi: 10.1016/s0166-4328(99)00122-9. [DOI] [PubMed] [Google Scholar]
- 8.Lippmann M, Bress A, Nemeroff CB, Plotsky PM, Monteggia LM. Long-term behavioural and molecular alterations associated with maternal separation in rats. Eur J Neurosci. 2007;25:3091–3098. doi: 10.1111/j.1460-9568.2007.05522.x. [DOI] [PubMed] [Google Scholar]
- 9.Lehmann J, Feldon J. Long-term biobehavioral effects of maternal separation in the rat: consistent or confusing? Rev Neurosci. 2000;11:383–408. doi: 10.1515/revneuro.2000.11.4.383. [DOI] [PubMed] [Google Scholar]
- 10.Biagini G, Pich EM, Carani C, Marrama P, Agnati LF. Postnatal maternal separation during the stress hyporesponsive period enhances the adrenocortical response to novelty in adult rats by affecting feedback regulation in the CA1 hippocampal field. Int J Dev Neurosci. 1998;16:187–197. doi: 10.1016/s0736-5748(98)00019-7. [DOI] [PubMed] [Google Scholar]
- 11.Hilakivi-Clarke LA, Turkka J, Lister RG, Linnoila M. Effects of early postnatal handling on brain beta-adrenoceptors and behavior in tests related to stress. Brain Res. 1991;542:286–292. doi: 10.1016/0006-8993(91)91580-t. [DOI] [PubMed] [Google Scholar]
- 12.Vig R, Gordon JR, Thebaud B, Befus AD, Vliagoftis H. The effect of early-life stress on airway inflammation in adult mice. Neuroimmunomodulation. 2010;17:229–239. doi: 10.1159/000290039. [DOI] [PubMed] [Google Scholar]
- 13.Loria AS, Pollock DM, Pollock JS. Early life stress sensitizes rats to angiotensin II-induced hypertension and vascular inflammation in adult life. Hypertension. 2010;55:494–499. doi: 10.1161/HYPERTENSIONAHA.109.145391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrario CM. Angiotensin I, angiotensin II and their biologically active peptides. J Hypertens. 2002;20:805–807. doi: 10.1097/00004872-200205000-00004. [DOI] [PubMed] [Google Scholar]
- 15.Toko H, Zou Y, Minamino T, Masaya M, Harada M, Nagai T, Sugaya T, Terasaki F, Kitaura Y, Komuro I. Angiotensin II type 1a receptor is involved in cell infiltration, cytokine production, and neovascularization in infarcted myocardium. Arterioscler Thromb Vasc Biol. 2004;24:664–670. doi: 10.1161/01.ATV.0000122361.63827.ab. [DOI] [PubMed] [Google Scholar]
- 16.Kaschina E, Unger T. Angiotensin AT1/AT2 receptors: regulation, signalling and function. Blood Press. 2003;12:70–88. doi: 10.1080/08037050310001057. [DOI] [PubMed] [Google Scholar]
- 17.Garrido AM, Griendling KK. NADPH oxidases and angiotensin II receptor signaling. Mol Cell Endocrinol. 2009;302:148–158. doi: 10.1016/j.mce.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siragy HM. The potential role of the angiotensin subtype 2 receptor in cardiovascular protection. Curr Hypertens Rep. 2009;11:260–262. doi: 10.1007/s11906-009-0044-3. [DOI] [PubMed] [Google Scholar]
- 19.Saito S, Hirata Y, Emori T, Imai T, Marumo F. Angiotensin II activates endothelial constitutive nitric oxide synthase via AT1 receptors. Hypertens Res. 1996;19:201–206. doi: 10.1291/hypres.19.201. [DOI] [PubMed] [Google Scholar]
- 20.Boulanger CM, Caputo L, Levy BI. Endothelial AT1-mediated release of nitric oxide decreases angiotensin II contractions in rat carotid artery. Hypertension. 1995;26:752–757. doi: 10.1161/01.hyp.26.5.752. [DOI] [PubMed] [Google Scholar]
- 21.Siragy HM, Carey RM. The subtype 2 (AT2) angiotensin receptor mediates renal production of nitric oxide in conscious rats. J Clin Invest. 1997;100:264–269. doi: 10.1172/JCI119531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 24.Kang KT, Sullivan JC, Sasser JM, Imig JD, Pollock JS. Novel nitric oxide synthase--dependent mechanism of vasorelaxation in small arteries from hypertensive rats. Hypertension. 2007;49:893–901. doi: 10.1161/01.HYP.0000259669.40991.1e. [DOI] [PubMed] [Google Scholar]
- 25.Sullivan JC, Pollock DM, Pollock JS. Altered nitric oxide synthase 3 distribution in mesenteric arteries of hypertensive rats. Hypertension. 2002;39:597–602. doi: 10.1161/hy0202.103286. [DOI] [PubMed] [Google Scholar]
- 26.Matthews K, Robbins TW. Early experience as a determinant of adult behavioural responses to reward: the effects of repeated maternal separation in the rat. Neurosci Biobehav Rev. 2003;27:45–55. doi: 10.1016/s0149-7634(03)00008-3. [DOI] [PubMed] [Google Scholar]
- 27.O’Regan D, Kenyon CJ, Seckl JR, Holmes MC. Glucocorticoid exposure in late gestation in the rat permanently programs gender-specific differences in adult cardiovascular and metabolic physiology. Am J Physiol Endocrinol Metab. 2004;287:E863–870. doi: 10.1152/ajpendo.00137.2004. [DOI] [PubMed] [Google Scholar]
- 28.Moritz KM, Johnson K, Douglas-Denton R, Wintour EM, Dodic M. Maternal glucocorticoid treatment programs alterations in the renin-angiotensin system of the ovine fetal kidney. Endocrinology. 2002;143:4455–4463. doi: 10.1210/en.2002-220534. [DOI] [PubMed] [Google Scholar]
- 29.Ojeda NB, Grigore D, Alexander BT. Intrauterine growth restriction: fetal programming of hypertension and kidney disease. Adv Chronic Kidney Dis. 2008;15:101–106. doi: 10.1053/j.ackd.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bogdarina I, Welham S, King PJ, Burns SP, Clark AJ. Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circ Res. 2007;100:520–526. doi: 10.1161/01.RES.0000258855.60637.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 32.Siragy HM, Inagami T, Ichiki T, Carey RM. Sustained hypersensitivity to angiotensin II and its mechanism in mice lacking the subtype-2 (AT2) angiotensin receptor. Proc Natl Acad Sci U S A. 1999;96:6506–6510. doi: 10.1073/pnas.96.11.6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsutsumi Y, Matsubara H, Masaki H, Kurihara H, Murasawa S, Takai S, Miyazaki M, Nozawa Y, Ozono R, Nakagawa K, Miwa T, Kawada N, Mori Y, Shibasaki Y, Tanaka Y, Fujiyama S, Koyama Y, Fujiyama A, Takahashi H, Iwasaka T. Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J Clin Invest. 1999;104:925–935. doi: 10.1172/JCI7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh RR, Cullen-McEwen LA, Kett MM, Boon WM, Dowling J, Bertram JF, Moritz KM. Prenatal corticosterone exposure results in altered AT1/AT2, nephron deficit and hypertension in the rat offspring. J Physiol. 2007;579:503–513. doi: 10.1113/jphysiol.2006.125773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silva-Antonialli MM, Tostes RC, Fernandes L, Fior-Chadi DR, Akamine EH, Carvalho MH, Fortes ZB, Nigro D. A lower ratio of AT1/AT2 receptors of angiotensin II is found in female than in male spontaneously hypertensive rats. Cardiovasc Res. 2004;62:587–593. doi: 10.1016/j.cardiores.2004.01.020. [DOI] [PubMed] [Google Scholar]
- 36.Gao L, Wang W, Li YL, Schultz HD, Liu D, Cornish KG, Zucker IH. Superoxide mediates sympathoexcitation in heart failure: roles of angiotensin II and NAD(P)H oxidase. Circ Res. 2004;95:937–944. doi: 10.1161/01.RES.0000146676.04359.64. [DOI] [PubMed] [Google Scholar]
- 37.Touyz RM, Yao G, Quinn MT, Pagano PJ, Schiffrin EL. p47phox associates with the cytoskeleton through cortactin in human vascular smooth muscle cells: role in NAD(P)H oxidase regulation by angiotensin II. Arterioscler Thromb Vasc Biol. 2005;25:512–518. doi: 10.1161/01.ATV.0000154141.66879.98. [DOI] [PubMed] [Google Scholar]
- 38.Schuijt MP, Tom B, de Vries R, Saxena PR, Sluiter W, van Kats JP, Danser AH. Superoxide does not mediate the acute vasoconstrictor effects of angiotensin II: a study in human and porcine arteries. J Hypertens. 2003;21:2335–2344. doi: 10.1097/00004872-200312000-00023. [DOI] [PubMed] [Google Scholar]
- 39.Gardiner SM, Kemp PA, Bennett T, Palmer RM, Moncada S. Nitric oxide synthase inhibitors cause sustained, but reversible, hypertension and hindquarters vasoconstriction in Brattleboro rats. Eur J Pharmacol. 1992;213:449–451. doi: 10.1016/0014-2999(92)90636-i. [DOI] [PubMed] [Google Scholar]
- 40.Ribeiro MO, Antunes E, de Nucci G, Lovisolo SM, Zatz R. Chronic inhibition of nitric oxide synthesis. A new model of arterial hypertension. Hypertension. 1992;20:298–303. doi: 10.1161/01.hyp.20.3.298. [DOI] [PubMed] [Google Scholar]
- 41.Cunha RS, Cabral AM, Vasquez EC. Evidence that the autonomic nervous system plays a major role in the L-NAME-induced hypertension in conscious rats. Am J Hypertens. 1993;6:806–809. doi: 10.1093/ajh/6.9.806. [DOI] [PubMed] [Google Scholar]
- 42.Miller GE, Chen E, Fok AK, Walker H, Lim A, Nicholls EF, Cole S, Kobor MS. Low early-life social class leaves a biological residue manifested by decreased glucocorticoid and increased proinflammatory signaling. Proc Natl Acad Sci U S A. 2009;106:14716–14721. doi: 10.1073/pnas.0902971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.