

Abstract
T cells specific for the cytochrome c Ag are widely used to investigate many aspects of TCR specificity and interactions with peptide-MHC, but structural information has long been elusive. In this study, we present structures for the well-studied 2B4 TCR, as well as a naturally occurring variant of the 5c.c7 TCR, 226, which is cross-reactive with more than half of possible substitutions at all three TCR-sensitive residues on the peptide Ag. These structures alone and in complex with peptide-MHC ligands allow us to reassess many prior mutagenesis results. In addition, the structure of 226 bound to one peptide variant, p5E, shows major changes in the CDR3 contacts compared with wild-type, yet the TCR V-region contacts with MHC are conserved. These and other data illustrate the ability of TCRs to accommodate large variations in CDR3 structure and peptide contacts within the constraints of highly conserved TCR–MHC interactions.
In early studies of T cell recognition, a strongly immunodominant peptide from pigeon cytochrome c (PCC) emerged as an informative model Ag for studying the nature of MHC restriction (1–3). This has since been widely employed in studies of TCR genes and proteins to gain important insights into the many aspects of TCR specificity and function, including relating thermodynamic and kinetic properties to T cell responses (4). However, the structural foundations upon which these interactions are based have been elusive, despite efforts spanning decades.
The strength of this experimental system stems from the limited TCR usage of the CD4+ T cell population that responds to the single peptide epitope PCC88–104, or the related moth cytochrome c peptide (MCC88–103), presented by I-Ek (PCC/I-Ek or MCC/I-Ek). This population is strongly dominated by cells expressing TCRs encoded by the Vα11 (TRAV4) and Vβ3 (TRBV26) gene segments (5–7) that contain a number of reoccurring TCRα and TCRβ CDR3 junctional sequence features reported to be critical for clonal recruitment to this oligoclonal response (7–9). The frequency of cells bearing TCRs with the key junctional features that define the dominant PCC reactive clonotypes increases sharply over the course of the primary immune response and continues to focus in response to secondary challenge in a manner that correlates well with the relative affinity for MCC/I-Ek (7–9). Interestingly, this relationship between TCR CDR3 usage and affinity has provided important insights into what TCR characteristics facilitate recruitment from the CD4+ T cell repertoire to join a polyclonal response to Ag delivered by various adjuvants (10, 11) and those TCR features that promote differentiation to specific Th or CD25+ regulatory T cell subsets (12). Finally, variants of the MCC peptide have recently been used to address the relationship between potency and T cell responsiveness or differentiation in vivo (13, 14). Thus, the body of information that has been accumulated with this system has provided important insights into how the diverse CD4+ T cell repertoire is accessed and used in response to Ag.
Prior to the availability of TCR and TCR–peptide-MHC (pMHC) crystallographic information, several aspects of the structural basis for the preferred sequence characteristics of MCC/I-Ek–specific TCRs were deduced from experiments based on peptide (15) and MHC mutagenesis (16) combined with TCR sequence analysis. One such study clearly identified two conserved CDR3 residues (α93 Glu and β100 Asn, henceforth referred to as α89E and β97N using the same numbering system in the deposited Protein Data Bank [PDB] files) that appeared to be involved in reciprocal interactions with a critical lysine at the p5 position of the MCC peptide (MCC-99K, p5K) and a critical threonine at the p8 position (MCC-102T, p8T), respectively (17). In some T cells that reacted against MCC peptide with a lysine to glutamate substitution at p5 (MCC-99E), a charge reversal was observed at the CDR3α89 position, strongly suggesting that this position would be involved in a charge–charge interaction with the peptide (an example of one such TCR is the 202 TCR; see Supplemental Table II). However, cross-reactive TCRs capable of recognizing both versions of MCC (p5K and p5E) were also identified. In subsequent experiments with one of these cross-reactive TCRs, 226, it was found that p5K versus p5E recognition was differentially affected by MHC mutations at several positions on both the α- and β-chains of I-Ek, suggesting that pMHC binding was significantly altered depending on which peptide was being recognized (16). But, it was not clear how the 226 TCR recognizes these two dramatically different peptides (charge reversed at the centrally located p5 position) despite exhibiting the canonical features of a MCC/I-Ek–reactive TCR.
TCR degeneracy and polyspecificity have been the subjects of many studies (18–20). The cross-reactive potential of individual TCRs has been tested by systematically varying one amino acid at a time on the agonist peptide (altered peptide ligands [APLs]) (15, 21–23). Combinatorial approaches have also been taken, including the use of peptide pools fixed at particular residues or higher-throughput display methods, to look for alternate agonist peptides for a given TCR (24–26). The APL studies have revealed that most naturally derived TCRs can tolerate conservative mutations at a few TCR contact residues with occasional strong specificity for a particular residue at a couple key TCR contacts (15, 21, 22). Not surprisingly, certain experimental conditions can give rise to TCRs with drastically reduced specificity for peptide. For instance, TCRs engineered to have higher affinity for given Ag also lose specificity for that Ag and gain the ability to recognize a wide array of APLs (27, 28). Highly degenerate TCRs have also been derived from mice expressing MHC with tethered peptides to restrict the pMHC repertoire available for thymocyte selection (29), and, in some cases, these highly degenerate TCRs are autoreactive. The sum of these data suggests a division of labor for peptide and MHC recognition by TCRs.
From a series of studies on Vβ8.2-containing TCRs, we previously hypothesized that germline-encoded V-region gene segments have evolved to make conserved contacts on MHC, which we have termed recognition codons, that underlie the structural basis for TCR recognition of pMHC (25, 30, 31). Conserved Vβ8.2 interactions with a range of I-A allotypes presenting different peptides are also seen in other TCR–pMHC complexes (32), and this germline motif is critical for positive selection (33). However, this notion is complicated by several factors. One is the availability of a large number of MHC alleles, each of which needs to be compatible for a given species’ set of TCR V-regions. It is also evident that peptide binding in the groove of MHC can significantly alter the nonpeptide surface of the MHC, which may alter germline-encoded contacts. Lastly, a major technical challenge is that there are a large number of TCR Vα and Vβ pairings available, and each may use a different set of evolved codons to recognize each MHC allele. As of yet, the majority of crystallographic information has come from the mouse system and has focused mostly on comparing TCR–pMHC complex structures of Vβ8 TCRs. Although much has been learned about how the germline-encoded Vβ8.2 region contributes to binding various MHC class I and II molecules presenting a range of peptides, it is important to investigate the generality of these findings to other TCR V-regions and MHC molecules.
In this study, we have used the cytochrome c system to address the structural basis of TCR cross-reactivity using 226 as a naturally occurring example of a highly degenerate TCR. Previous data suggest that this TCR can use multiple modes of MHC recognition as part of its ability to recognize various ligands (16); therefore, this system serves as a stringent test of the recognition codon hypothesis (30, 31). Furthermore, we analyzed the previously unknown structures of Vα11+Vβ3+ TCRs that recognize the mouse MHC allele, I-Ek, which has yet to be crystallized as part of a TCR–pMHC complex. We determined the structures of the 2B4 and 226 TCRs in both the unliganded state as well as bound to an MCC/I-Ek complex and, for 226, bound to the charge-reversed p5E-MCC/I-Ek pMHC complex to assess the validity of previous mutational data, suggesting that germline-encoded MHC contacts were significantly altered between these two complexes. We report that all three Vα11/Vβ3 TCRs binding to MCC and MCC-p5E/I-Ek had nearly identical germline-encoded contacts with the MHC, despite gross differences in somatically derived CDR3-mediated contacts with the peptide.
Materials and Methods
Preparation and validation of proteins
For all three complexes, the TCR was expressed in bacteria as chimeric proteins made up of mouse 2B4, 5c.c7, or 226 VJ or VDJ regions fused to human constant regions [generously provided by Dr. J. McCluskey and Dr. J. Rossjohn (34) and refolded as described] (32, 35). In addition, the I-Ek–binding peptides (MCC or MCC-p5E) were tethered to the N terminus of the TCRβ-chain with a 10-residue flexible linker sequence as a tether with sequences MANERADLIAYLKQATKGGGGSGGGGG or MANE-RADLIAYLEQATKGGGGSGGGGG. His-tagged I-Ek with a β-chain N-terminally tethered thrombin-cleavable CLIP peptide was expressed in insect cells using a baculovirus expression system and dual promoter construct driving expression of both MHC chains modified from one generously provided by Dr. P. Allen as described (36). After Ni-NTA and size-exclusion purification, the MCC-tethered TCR and CLIP/I-Ek were subjected to an exchange reaction to form a stable tethered TCR–pMHC complex similar to that previously described (37, 38). The MCC-tether-TCR was mixed with CLIP/I-Ek at a 3:1 molar ratio at a final protein concentration of 4 mg/ml in PBS before addition of 3 U/ml thrombin and 1 h incubation at 37°C to remove the tethered CLIP peptide. A total of 30 mM sodium cacodylate (pH 6.2) was then added to adjust pH and facilitate peptide exchange during a 16-h incubation at 37°C. The stable tethered 100-kDa complex was then purified by size exclusion and purity and composition verified by SDS-PAGE electrophoresis in boiled, nonboiled, reducing, and nonreducing conditions.
Exogenous TCR expression in T cell hybridomas for functional studies
Wild-type and mutant 2B4 TCRs were expressed in the 58α−β− TCR− hybridomas for functional studies as described (39). In brief, ecotropic murine stem cell virus retrovirus containing full-length TCR chains as well as CD4 coreceptors were produced in the Phoenix retroviral expression system using FuGene transfection (Roche Molecular Biochemicals). 58α−β− T cell hybridomas were then infected with a combination of viral supernatants from phoenix cells transfected with TCR α, β, and CD4 expression constructs. Each TCR chain is equipped with either puromycin or zeocin antibiotic resistance under control of an internal ribosome entry sequence immediately downstream of the TCR sequence; infected 58α−β−T cell hybridomas were then selected on the basis of drug resistance (2.5 μg/ml puromycin, 50 μg/ml zeocin). To verify TCR (PE-conjugated anti-mouse TCR) and CD4 expression (allophycocyanin-Cy7–conjugated anti-CD4, both Abs obtained from BD Biosciences), the cells were stained with fluorescent Abs and run on a flow cytometer (Beckman Coulter).
To test the function of this exogenously expressed TCR, infected hybridomas were cocultured with various concentrations of peptide-pulsed APCs (CH27 B cells) both at 5 × 105 cells/well in 96-well plates. After ~16 h, supernatants were harvested and subjected to IL-2 ELISA assay. The concentrations of each peptide required to elicit production of either 10 or 50% of the maximal IL-2 produced by wild-type 2B4 transduced cells were determined from plots of IL-2 relative units versus the log of peptide concentration, as described (15).
226 hybridoma functional studies
MCC and altered peptide induced production of IL-2 by 226 hybridomas to assess the extent of 226 cross-reactivity against all possible amino acid substitutions at the three TCR contacts was performed exactly as described (15). For these experiments, irradiated B10.BR splenocytes were used as I-Ek–expressing APCs that were then pulsed with various concentrations of each MCC peptide mutant.
Crystallization and x-ray data collection
Protein crystals were grown via hanging-drop vapor diffusion at 22°C. 2B4-MCC/I-Ek complex crystals were grown in 50% PEG200, 0.2 M sodium chloride, and 0.1 M sodium potassium phosphate (pH 6.2). 226-MCC and 226-MCC–p5K/I-Ek complex crystals were grown in 15–20% PEG5000 MME and Bis Tris (pH 6.5–7). The unliganded 5c.c7 and 2B4 crystals were grown in 20% PEG3350, 0.1 M calcium chloride, and Tris (pH 7.7). Data sets were collected at Stanford Synchrotron Radiation Light Source (Stanford, CA) and Advanced Light Source (Berkeley, CA). Data were indexed, integrated, and scaled using either Mosflm (40) and SCALA from the CCP4 suite (41) or the HKL-2000 program package (42).
Structure determination and refinement
All structures were solved via molecular replacement using the program Phaser (43). The molecular replacement solution for the unliganded 2B4 structure was determined using the variable domains of the 2C TCR (PDB ID 1TCR) and the constant domains of the ELS4 TCR (PDB ID 2NW2). The refined 2B4 coordinates in conjunction with I-Ek (PDB ID 1FNE) were used as search models for the 2B4-MCC/I-Ek complex. The molecular replacement solutions for the 226–I-Ek complex structures were determined using the refined coordinates from the 2B4-MCC/I-Ek complex. To avoid model bias, peptides residues and CDR loops were removed from the search models. Manual model building was preformed in COOT followed by iterative rounds of coordinate and B-factor refinement in either the program CNS (44) or Phenix (45). The program PROCHECK (46) was used to monitor the geometry of the models. The CCP4 suite programs Contact and AreaIMol were used to assess the interface contacts and buried surface areas, respectively. Structural figures were generated using the program Pymol (Delano Scientific, San Carlos, CA).
Surface plasmon resonance
SPR data were collected on a BiaCORE 3000 system or a BiaCORE T100 system (GE Healthcare) as described (47). TCR was expressed as a hybrid receptor consisting of the clonotypic TCR Vα and Vβ domains fused to human LC13 TCR constant domain as described above (32). Escherichia coli inclusion bodies were refolded by fast dilution as described (34) and purified on an Äkta system (GE Healthcare) by anion exchange chromatography (MonoQ), and gel filtration (S200). For binding studies, MCC/I-Ek was expressed, refolded, purified, C-terminally biotinylated, and immobilized to a streptavidin-coated BiaCORE chip (GE Healthcare) as described (47).
Results
Structural overview
We determined the crystal structures of the 2B4 TCR bound to MCC/I-Ek (deposited at PDB [http://www.pdb.org], PDB ID 3QIB), the 226 TCR bound to MCC/I-Ek (PDB ID 3QIU), and the 226 TCR bound to p5E-MCC/I-Ek (PDB ID 3QIW) at a resolution of 2.7, 2.7, and 3.3 Å, respectively (Fig. 1, Supplemental Table I). We also determined structures of the unbound 2B4 (PDB ID 3QJF) and 5c.c7 (PDB ID 3QJH) TCRs (Supplemental Table I). On a gross level, the three bound structures are very similar in that they have buried surface areas of 2605, 2500, and 2494 Å2, respectively, and all have a common TCR diagonal docking angle on the pMHC of ~41° (48). The 2B4 and 226 TCRs have very similar V region segments, using Vα11.2 and Vα11.1, respectively (Supplemental Table II). They also use the same Vβ3 gene segment; however, they have dramatically different CDR3α sequences and moderately different CDR3β sequences, partially as a consequence of their different Jα and Jβ gene segments. As a large number of TCRs recognizing MCC/I-Ek have been sequenced and characterized, when comparing these TCRs, it is clear that 226, like 5c.c7, represents an example of the canonical MCC/I-Ek–reactive TCR in that it displays all eight previously identified preferred features of an MCC/I-Ek–reactive TCR (Table I). In contrast, 2B4 displays only three out of the eight possible preferred features. Aside from the use of Vα11, the noncanonical nature of this TCR is particularly evident for the α-chain, which has no other previously identified features. Despite the large junctional differences between these TCRs, both 2B4 and 226 TCRs bind to MCC/I-Ek in a similar manner. Furthermore, the recognition of a charge-reversed MCC (MCC-Kp5E) led to very little change in the overall docking orientation of 226 to MCC-p5E/I-Ek compared with the structure of this TCR bound to the wild-type MCC/I-Ek complex.
FIGURE 1.

Structural overview of the three TCR–pMHC extracellular domain complex structures solved in this study. For each, the TCR is a chimeric fusion of the mouse variable and human constant domains (α in light blue, β in dark blue) bound to the mouse MHC protein I-Ek (α in dark yellow, β in light yellow). A, The 2B4 TCR bound to MCC/I-Ek. B, The 226 TCR bound to MCC/I-Ek. C, The 226 TCR bound to MCC-p5E/I-Ek.
Table I.
A list of preferred features of TCRs specific for MCC/I-Ek
| No. | Feature | 2B4 | 226 | 5c.c7 |
|---|---|---|---|---|
| 1 | α89E (prev. α93E) | N (L) | Y | Y |
| 2 | 8 aa CDR3α | N (10) | Y | Y |
| 3 | α91S (prev. α95S) | N (A) | Y | Y |
| 4 | Jα 16, 17, 22, 34 | N (56) | Y (16) | Y (34) |
| 5 | β97N (prev. β100N) | Y | Y | Y |
| 6 | 9 aa CDR3β | Y | Y | Y |
| 7 | β99A/G (prev. β102A/G) | N (S) | Y (A) | Y (A) |
| 8 | Jβ1.2, Jβ2.5 | Y (2.5) | Y (1.2) | Y (1.2) |
| No. of preferred features | 3 | 8 | 8 |
A large number of studies on the evolution and properties of in vivo T cell responses to MCC/I-Ek have identified and subsequently relied on the strong bias for usage of eight common preferred features of MCC/I-Ek–reactive TCRs. These eight features are listed above. For all TCR residue numbers, we are now using the ImMunoGeneTics numbering system employed for numbering residues in all deposited PDB files. Residue numbers from previous cytochrome c-reactive TCRs are provided parenthetically.
Three TCRs with very different mechanisms of peptide recognition
The CDR3 interactions with the peptides in the three bound structures are illustrated in Fig. 2. Peptide recognition in each of these complexes is achieved by remarkably different chemistries. The main similarities between all three structures are the hydrogen bonds (H-bonds) made by the CDR3β3-N97 side-chain amide with the backbone of the peptide and between the MCC-T8 side-chain hydroxyl with the backbone carbonyl of CDR3β position 98 (N in 226, W in 2B4). This is not surprising given the conservation of sequences in this region. Contacts made by CDR3α differ the most among all three structures. Only 2B4 makes an H-bond with the side chain of MCC-Y3 using the backbone carbonyl of αG93 (Fig. 2A). Also, only 2B4 makes a β-chain–mediated (CDR3βD101) salt-bridge with the p5K position of MCC and makes an H-bond with the backbone carbonyl of p5K (via βS99, Fig. 2A). In contrast, 226 uses αE89 to salt-bridge with MCC-p5K and makes only one other H-bond using CDR3α (Fig. 2B), whereas the recognition of MCC-p5E by 226 is completely lacking in polar contacts using CDR3α (Fig. 2C).
FIGURE 2.
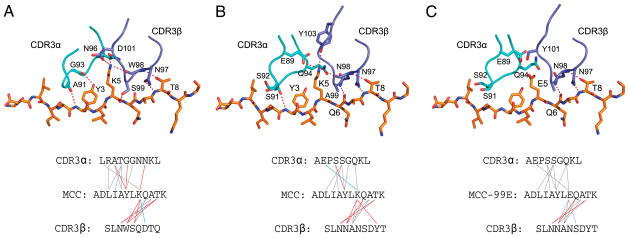
A comparison of the CDR3–peptide interactions of each TCR–pMHC complex. Only the CDR3 loops and MHC-bound peptide are considered for 2B4-MCC/I-Ek (A), 226-MCC/I-Ek (B), and 226-MCC-p5E/I-Ek (C). The top panel shows the arrangement of the CDR3 loops interacting with the MHC-bound peptide, shown in stick form. Side-chains involved in polar interactions are shown with the H-bonds/salt-bridges shown in red. The bottom panel shows the sequence of the CDR3s and the peptide. Lines connecting each residue represent van der Waals interactions (gray), H-bonds (red), and salt-bridges (blue).
Consistent docking mode despite large changes in CDR3 recognition
As described above, peptide recognition of MCC/I-Ek by the 2B4 and 226 TCR CDR3 loops is accomplished by different strategies. However, superimpositions of the three bound structures solved in this study show nearly identical docking with the MHC and precise superimposition of the germline-encoded HV4 loops (Fig. 3A). Inspection of individual contacting residues within the CDR1α, HV4α, CDR2α (Fig. 3B), and CDR2β (Fig. 3C) loops shows that, at most, very minor rearrangements are made between these three structures in side-chain conformation. Previous comparisons between several structures using the same V regions, such as the comparisons of many Vβ8.2-containing structures (30–32), have noted the striking conservation of contacts formed by tyrosine residues, namely Tyr48 in Vβ8.2 CDR2. For Vα11, it is possible that Tyr47 in CDR2 plays a similar role in that it is highly malleable and capable of forming a variety of different type of contacts, from polar to hydrophobic (Fig. 3B). None of the Vβ3-encoded contacts use aromatic residues, nor do any of these contacts appear to be playing a dominant role in MHC binding, as only a few van der Waals interactions and solvent exposed salt-bridge are seen. In contrast to the Vα-encoded contacts, we observe a small amount of wobble in the CDR2β contacts when comparing the 226-pMHC structures to the 2B4-pMHC structure (Fig. 3C).
FIGURE 3.
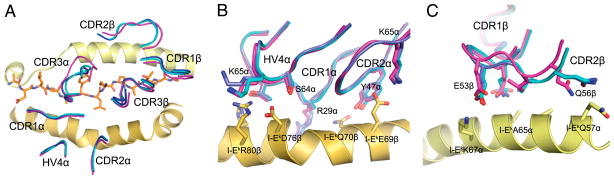
TCR docking comparison. In all panels, as in other figures, 2B4 is colored magenta, 226-binding MCC/I-Ek is cyan, and 226-binding MCC-p5E/I-Ek is colored blue. A, The ends of each CDR and HV4α loops are overlaid. B, Detailed illustration of germline-encoded TCRα contacts with I-Ekβ. Relevant side-chains for each structure are overlaid to compare the interface. Note extensive contacts between all three TRAV4 (Vα11) germline-encoded loops, CDR1, 2, and HV4, and no significant difference in conformation between the three structures solved in this study. C, TRBV26 (Vβ3) germline-encoded residues making contact with I-Ekα are compared. For these chains, only the CDR2β loop makes contact, and some small differences are seen when comparing the 2B4-bound structure with either 226-bound structure. No differences are seen between the two 226 structures.
Although it is striking that large differences in CDR3 do not require more significant adjustments to the germline-encoded contacts, it may not be necessarily surprising that these germ-line-encoded contacts, of which some may be conserved codons of recognition, strictly impose a docking mode for pMHC recognition for this pairing of Vα, Vβ, and MHC allele. It is also possible that different Vα and Vβ pairings may vary in their ability to accommodate multiple docking modes.
Germline-encoded contacts and dominance of Vα over Vβ
If it is true that this pairing of Vα11 and Vβ3 genes imposes a strict docking mode, does one V region predominate? Several metrics point at Vα11’s dominant role in contacting the I-Ek allele in this very specific way. For one, all three germline-encoded loops (CDR1α, CDR2α and HVα4 of Vα11) are making a large number of contacts of all types with the MHC (Fig. 3B, Supplemental Fig. 1, burying a total of 1045, 990, and 978 Å2 of surface in each of the structures). In contrast, the only Vβ3-encoded loop making a few contacts is CDR2β (Fig. 3C, Supplemental Fig. 1A, 1C, burying only 451, 447, and 433 Å2 of surface in each of the structures). This skewing toward Vα contacts in these structures is further illustrated by comparing the V region contacts made in another mouse MHC class II-restricted (MBP/I-Au) TCR, 1934.4, which has relatively equal contributions from Vα-versus Vβ-encoded contacts (Supplemental Fig. 1B, 1C). Furthermore, the strictness of the requirement for each V region in the mouse response to MCC in the context of I-Ek has been addressed by looking at the V region usages of 5c.c7α versus β hemitransgenic mice. In 5c.c7α transgenic mice, the frequency of non–Vβ3-expressing T cells responding to MCC is much greater than the frequency of non–Vα11-expressing T cells responding to MCC in 5c.c7β transgenic mice. Thus, one might predict that all Vα11 TCRs selected on the I-Ek MHC are making contact in this same docking mode. It is interesting to note that a TCR composed of 5c. c7α paired with a Vβ8.2 is capable of recognizing p8E-MCC/I-Ek (see Supplemental Table II). One might predict that Vβ8.2 is more flexible in this interaction, whereas Vα11 is more stringent.
Structural explanation for the preferred features of MCC/I-Ek recognition
The 226 TCR displays all of the canonical features of an MCC/I-Ek–reactive TCR (Table I, Supplemental Table II). Although numerous mutational and sequence comparisons have identified residues critical for recognition, the precise structural basis for each of these features and sequence requirements are not known. For example, MCC peptide charge-reversal experiments leading to a reciprocal charge reversal in CDR3α of responding TCRs strongly suggested that a charge–charge interaction is taking place in the bound structure, which would explain preferred feature 1 in Table I. This is confirmed in the crystal structure, where the central p5K position of MCC forms a salt-bridge with TCRα89E in the 226-MCC/I-Ek complex (Fig. 4A). There is further coordination of the positively charged amine group H-bonds from 226α94Q, 226β98N, 226β100N, and 226β103Y.
FIGURE 4.

Illustration of the conserved features of peptide recognition. The structural basis for each of the preferred sequence features of MCC/I-Ek reactive TCRs is illustrated in detail. A, The structural basis for recognition of MCC-p5K is illustrated. Several H-bonds coordinate the positive charge of this central lysine in addition to the predicted salt-bridge between α89E and p5K. B, The interactions made between 226 TCR α91S and MCC/I-Ek are shown. This residue hydrogen bonds both the peptide at p3Y as well as I-Ek at β77T. C, Reciprocal side-chain and main-chain H-bonds are shown for TCR β98N and the peptide p8T. D, The arrangement of p5K hydrogen-bonding 226 CDR3α residues is shown in the p5E-bound structure. E, The salt-bridge between MCC-p5K and the Jβ2.5-encoded aspartate of 2B4 is shown together with additional charge-coordinating H-bonds.
Preferred feature 2 in Table I is a conserved α91S that makes a backbone-carbonyl H-bond with the backbone amide of MCC-p3Y residue, positioned by side-chain hydroxyl H-bond with I-Ek β-T77 side-chain hydroxyl (Fig. 4B). In 2B4, this residue is replaced by an alanine residue, which lacks the H-bond with I-Ek β-T77. However, it does make a van der Waal’s contact with this same residue (not shown).
Sequence analysis of canonical MCC (wild-type p8T)-reactive TCRβ-chain sequences and comparison with MCC-p8E–reactive TCRs from 5c.c7α transgenic mice have repeatedly shown a strong conservation of CDR3β97N residue usage (preferred feature 5 in Table I) (17) and consistent changes in this residue for p8E-reactive TCRs. These data strongly suggested an interaction between these two residues on the TCR and pMHC. Indeed, all three structures from this study have a consistent mode of interaction between this CDR3β97N residue and MCC-p8T (Fig. 2); that is, hydrogen bonding between the TCRβ97N side-chain amide and the backbone carbonyl oxygen of MCC-p8T. At the same time, there is a precisely aligned H-bond between the backbone amide group of the following CDR3β98 position and the hydoxyl oxygen of MCC-p8T (Figs. 2, 4C). Slight alterations in this alignment in MCC-p8S peptide mutants are almost certainly the cause of the drastic reductions in binding affinity observed for all MCC/I-Ek–reactive TCRs tested thus far. Any other residue at this position is not tolerated.
In contrast, no salt-bridge is formed between the 226 TCR and the MCC-p5E residue (Fig. 4D). Instead, 226’s α89E residue forms an H-bond with the nearby 226α94Q [coded by Jα16 used by the AND, 226, and 202 TCRs, all of which have reduced specificity for MCC-p5K (17, 49)], but absent from 2B4 and 5c. c7, which absolutely require p5K for stimulatory activity (15) (Fig. 4D). It is interesting to note that with the exception of Jα22, all four preferred Jα gene segments, as well as the Jα56 gene segment used by 2B4, have an N/Q residue at position 94, which can H-bond to MCC-p5K (Fig. 4A) or, in the case of 226 and Jα16’s 94Q, make a H-bond with α98E, when bound to MCC-p5E (Fig. 4D). Thus, we have provided structural data that can be used to explain at least three of the eight preferred features involved MCC peptide recognition. These hypotheses as well as hypotheses for the structural basis for the remaining five related preferred features are summarized in Table II.
Table II.
Structural hypotheses explaining conserved features of MCC/I-Ek reactive TCRs
| No. | Feature | Structural Explanation |
|---|---|---|
| 1 | α89E | Forms salt bridge with central MCC-p5K residue (Fig. 4A). |
| 2 | 8 aa CDR3α | Allows appropriate spacing for germline encoding of Glu at position 89 by TRAV4 and germline encoding of N/Q at position 94 by the preferred Jα (feature 4). |
| 3 | α91S | Makes a backbone-carbonyl H-bond with the backbone amide of MCC-p3Y residue, positioned by side-chain hydroxyl H-bond with I-Ekβ-T77 hydroxyl side-chain (Fig. 4B). |
| 4 | Jα 16, 17, 22, 34 | Provide N/Q residue in position for H-bonding central MCC-p5K residue (Jα16 encoded Q appears to be required for MCC-p5K/E cross-reactivity; Fig. 4A, 4D). |
| 5 | β97N | An aligned pair of H-bonds of β96N side-chain amide with MCC-p8T backbone carbonyl and MCC-p8T side-chain hydroxyl with β95 backbone amide (Fig. 4C). |
| 6 | 9 aa CDR3β | |
| 7 | β99A/G | H-bond with I-Ekβ70Q and van der Waals interactions with I-Ekβ67F. Small to leave room for MCC-99K to protrude. β98A is at least partially coded by Jβ1.2. |
| 8 | Jβ1.2, Jβ2.5 | In Jβ1.2 there is a Tyr at the appropriate position (β103Y) for H-bonding MCC-p5K as in 226. In Jβ2.5, there is an Asp residue in place to make a salt-bridge with MCC-p5K as in 2B4 (Fig. 4A). |
Based on crystal structure information, we can now hypothesize the structural bases for each of the well-characterized preferred sequences features of MCC/I-Ek–reactive TCRs. These features are described in more detail in the Results section.
How then does 2B4 bind to the same positively charged residue, as there is no corresponding negatively charged residue in 2B4? In fact, this salt-bridge is replaced by a salt-bridge between MCC-p5K and 2B4βD101 coded by Jβ2.5 that is further coordinated by H-bonds with 2B4αN95, 2B4βW98, and 2B4βS99 (Fig. 4E).
CDR3 flexibility and MCC/I-Ek recognition
The unliganded structures of two MCC/I-Ek–reactive TCRs, 5c.c7 TCR and the 2B4 TCR, were also determined in this study. The CDR loop configurations of these two TCRs are overlaid with the MCC/I-Ek–bound version of 2B4 in Fig. 5A. As had been seen for other TCR structures, we see very similar CDR1 and -2 conformations, but quite different CDR3 conformations. For 5c.c7, the unliganded CDR3 loops would require large conformational changes to assume the structure expected for MCC/I-Ek binding, as is seen for its close relative 226. For 2B4, we see further evidence for CDR3 loop flexibility in that the majority of both CDR3α and β could not be resolved in the unbound structure. Thus, as has been seen for several other TCRs (31, 50–52), both 5c.c7 and 2B4 TCRs appear to have very flexible CDR3 loops.
FIGURE 5.

CDR3 flexibility and MCC/I-Ek recognition. A, In addition to the three bound structures, the unbound versions of both 2B4 and 5c.c7 TCRs were solved. These two unbound TCRs, 2B4-free in pink and 5c.c7-free in blue/purple, are aligned and overlaid with the bound 2B4 TCR in magenta. Note minimal conformational differences in the CDR1, 2, or HV4 loops. In contrast, large differences were seen for the CDR3 loops. In the case of the 2B4-free structure, neither loop could be resolved due to large b-factor probably caused by high mobility in the crystal. B and C, To reduce the mobility of 2B4 CDR3s, proline substitutions were made throughout these loops. These proline-mutant TCRs were then expressed in 58α−β− T cell hybridomas and tested for their ability to respond to MCC peptide stimulation in the presence of CH27 APCs (see Materials and Methods). The five residues able to accommodate proline substitution and maintain MCC reactivity are highlighted in B. Three out of these five TCR mutants had increased MCC sensitivity and are colored red; the other two had reduced sensitivity and are colored yellow. The sensitivity of each mutant-TCR–expressing hybridoma is shown in C. Stimulatory index is plotted for each hybridoma and represents the inverse peptide concentration required to stimulate 10 (EC10, gray bars) and 50% (EC50, black bars) of maximal IL-2 secretion (see Materials and Methods) (15). D, Three of the five MCC-reactive proline mutant TCRs were expressed as soluble proteins and tested for their ability to bind MCC/I-Ek in solution using SPR. Consistently with the hybridoma stimulation data, the two more sensitive TCRs had a higher affinity (lower KD) and/or slower off-rate (lower koff) than the wild-type 2B4 TCR, whereas the TCR displaying weaker responses to MCC peptide had a much lower affinity and faster off-rate than wild-type 2B4.
To address the role of this CDR3 loop flexibility in T cell recognition, we made a series of proline substitutions in the 2B4 TCR throughout both CDR3 loops. Proline has a cyclic side chain in which a backbone dihedral angle is fixed at approximately −75°, giving it unusual conformational rigidity compared with other amino acids. We postulated that if a given proline substitution did not destroy 2B4’s specificity, it might increase its affinity by freezing a CDR3 loop into its optimal conformation for binding MCC/I-Ek, similarly to the way somatic mutation rigidifies CDR loops of Abs (53). To test for the ability of the proline mutant TCRs to bind MCC/I-Ek, we expressed each mutant retrovirally in 58α−β− hybridomas and tested for the ability of these 2B4 TCR expressing hybridomas to produce IL-2 in response to MCC stimulation (see Materials and Methods). In this manner, 17 CDR3 proline mutants were tested, and 5 were found to maintain MCC/I-Ek reactivity (substituted positions are shown in Fig. 5B). These positions were not critical peptide contacts in the CDR3, but rather appear to play roles in supporting the backbone conformation of the CDR3s. Three out of the five had higher MCC sensitivity than the wild-type 2B4 TCR (Fig. 5C). We then made rTCR proteins for selected binding mutants. The affinities of these TCRs for MCC/I-Ek correlated well with Ag sensitivity seen for the hybridomas (Fig. 5D). These results indicate that 2B4 can accommodate rigidifying proline mutations and maintain MCC/I-Ek reactivity. In these cases, the affinity of the interaction can be improved, likely due to reduced entropic costs for binding. We did not observe any narrowing of fine specificity for recognition of MCC altered peptide ligands (not shown), which is consistent with the notion that gross CDR3 conformational changes are generally not always required for degenerate recognition of altered peptide ligands (see Discussion).
226 is a naturally occurring high-affinity, highly cross-reactive TCR
As described above, the 226 TCR was originally identified for its ability to react with both MCC-p5K/I-Ek and MCC-p5E/I-Ek, accommodating a charge reversal at a well-characterized critical TCR contact residue. To compare the affinities of each of these TCR–pMHC interactions, we subjected the soluble 226 and 2B4 TCR proteins to SPR analysis using SPR. Surprisingly, we found that 226 had an unusually high affinity for MCC/I-Ek at 25°C (KD = 321 nM) with very slow off-rate (koff = 0.0029, t1/2 = 239 s; Fig. 6A, 6B). In contrast, the values we obtained for 2B4 were similar to those previously described (54) (KD = 13.7 μM, koff = 0.032, t1/2 = 21 s). For 226 binding to MCC-p5E/I-Ek, we were also surprised to find that the binding was detectable (not shown) but extremely weak (KD > 500 μM, Fig. 6B). Thus, our use of tethered peptide strategy to crystallize 226 bound to MCC-p5E/I-Ek may represent one of the lowest affinity TCR–pMHC coc-rystals solved to date and implies that this strategy could be used to crystallize other low-affinity TCR–pMHC interactions such as positive-selecting or null peptides. To assess the extent of 226’s degeneracy for recognition of MCC TCR contact variants, IL-2 production by 226 hybridomas was used as readout. As positions 3, 5, and 8 had been previously described as critical for 5c.c7 and 2B4 recognition of MCC/I-Ek (15), we tested all 20 possible aa substitutions at each of these three positions. In contrast to 2B4 or 5c.c7, which have strict requirements at these three positions. 5c. c7 reacts well with F, W, or Y and weakly with P or C at p3, absolutely requires K at p5, and reacts well with only with T and weakly with V, S, W, H, or R at P8. 2B4 reacts well with F or Y and weakly with I, W, or C at p3, absolutely requires K at p5, and only reacts well with T and weakly with W, N, or S at p8. See figures 7 and 8 in Reay et al. (15). 226 reacts with more than half of the possible substitutions at all three of these positions individually. Given 226’s very weak recognition of the p5E variant of MCC, it is not surprising that this is an example of a weak agonist for 226 stimulation, despite the fact that 226 was originally cloned for its ability to recognize this peptide. It is also interesting to note that some substitutions such as MCC-p3F,-p5L, and -p8S were more potent than wild-type MCC (p3Y, p5K, p8T) for stimulating 226 even though 226 already binds MCC with submicromolar affinity, suggesting that in some instances, weaker binders may sometimes provide for a more optimal stimulus [as has been observed for AND (49); see Discussion]. Overall, these data indicate that 226’s very high affinity for MCC allows it to recognize a large number of MCC variants, such as MCC-p5E, albeit at a much lower affinity.
FIGURE 6.

226 is a peripherally derived TCR with high affinity and highly degenerate recognition of MCC-APLs. A, Kinetics of 226 versus 2B4 TCR binding are compared. Background subtracted binding SPR sensograms for injections of 226 TCR at 0.1, 0.2, 0.5, 1, 2, and 5 μM over MCC/I-Ek–immobilized surfaces are shown in comparison with 2B4 TCC applied at 0.39, 0.78, 1.56, 3.13, 6.25, and 12.5 μM in a different experiment. The 2B4 and 226 responses are plotted with the same scale time-axis. B, Kinetic parameters from these sensorgrams as well as those obtained from 226 applied to flow cells containing MCC-p5E and MCC-p5R/I-Ek are tabulated. 226 T cell responsiveness to MCC APLs at peptide positions 3 (C), 5 (D), and 8 (E) were tested using 226 hybridomas and CH27 cells in an IL-2 secretion assay. For each peptide, the inverse peptide concentration required to stimulate 10 (EC10, gray bars) and 50% (EC50, black bars) of maximal IL-2 secretion is plotted (see Materials and Methods).
Discussion
Since Schwartz and colleagues (2, 17) first developed it in the late 1970s, the murine CD4+ T cell response to the C-terminal portion of PCC has become one of the best-studied systems in immunology. Many important studies of T cell specificity, MHC restriction (1–3), gene structure (55), thymic selection (56–58), biochemistry (4, 39, 54, 59, 60), thermodynamics (47, 61, 62), and cellular biophysics (63–65) have used it (14, 49). A major problem however, has been the lack of any structural data on the TCRs or on any TCR–pMHC complexes. This has limited its value for detailed mechanistic studies and inhibited the resolution of some of the earlier findings. Now, three TCR–pMHC structures, as well as the unliganded structures of two of the TCRs in this family of well-studied TCRs, give us a framework to gain molecular insights into this system.
One of these longstanding issues has been the lack of any structural information concerning the findings of Jorgensen et al. (17), who reported that 5c.c7β TCR transgenic mice immunized with a cytochrome c peptide with a charge reversal (Lys to Glu) at the p5 position elicited a reciprocal charge reversal (Glu to Lys) on CDR3α at position 89. In this study, we can confirm that, as proposed in Jorgenesen et al. (17), the Glu residue of the closely related 226 TCR is involved in a salt-bridge with the central lysine (p5K) of the MCC peptide. We also found that this same central lysine is further coordinated by a series of H-bonds with several other residues on both CDR3α and β. Interestingly, we find the situation to be quite different from the 2B4 TCR–MCC/I-Ek complex, where a negatively charged residue (βD101) of the CDR3β forms a salt-bridge with the p5 lysine of the peptide. This is a further indication of the importance of charge–charge interactions in TCR recognition and also the shows the flexibility of these related TCRs in the way that their CDR3 residues can interact with the peptide.
As several studies from our laboratory and others aimed at understanding the biophysical basis for TCR recognition of pMHC have used the cytochrome system as a model system, there is large amount data that can now be reconciled based on the crystal structure of this interaction. For instance, mutagenesis studies aimed at identifying MHC residues involved in TCR contacts have used IL-2 production as readout for pMHC recognition by 2B4 and 226 T cell hybridomas (16, 17). Purified 2B4 TCR binding to MCC/I-Ek with various MHC and peptide residue substitution has also been investigated (4, 47, 62). In one such study, the kinetics of 2B4 binding to each of these altered ligands was used to determine the relative contribution of various residues to the binding transition state, using ϕ analysis (47). The residues making contact in the bound structures we have solved in this paper are all consistent with these data. That is, residues previously found to influence either T cell recognition or TCR binding are all making contacts in the appropriate structure (Supplemental Fig. 2 for graphical summaries of these data). It is interesting to note that only some of the residues found to be involved in the transition state are making contacts in the bound structure, consistent with the interpretations of Wu et al. (47) (Supplemental Fig. 2C). One residue (I-Ekα-K67), which has one of the highest ϕ values (and thus contributes mostly to the transition state), is involved in a solvent-exposed salt-bridge among 2B4-CDR2β-E53. Such a charge–charge interaction may help guide binding, but it is not surprising that it does not contribute significantly to the bound state, as it is solvent exposed. Particularly in the 2B4-MCC/I-Ek structure, these residues had high B factors, indicating that this interaction is not very stable.
Another goal of this study was to follow up the intriguing findings of Ehrich et al. (16), who used a panel of I-Ek mutants to characterize the MHC footprint of the various cytochrome c-specific TCRs, including 226. In this study, they found that 226 had a very different sensitivity to the I-Ek mutants when it recognized the wild-type MCC-p5K versus MCC-p5E. These different patterns of sensitivity suggested that 226 was binding in very different orientations depending on the ligand. But when we examine the structure of 226 complexed with either of these two ligands, we find no difference in the orientation of the TCR or in the way it binds I-Ek. What we do find is that 226 has a much higher affinity, >1000-fold, for MCC/I-Ek than for the p5E variant. Thus, it seems that this very large difference makes the 226 TCR much more tolerant of many of the MHC mutants when MCC is the peptide compared with the p5E variant (16). As to why 226 has such lower affinity for MCC-p5E, this can be explained by the lack of a number of TCR peptide contacts in the complex structure. In particular, the glutamate at p5 does not make any clear contacts with the TCR. Interestingly, the remaining TCR–pMHC interactions are maintained in the same configuration as the 226-MCC-p5K/I-Ek, indicating that the TCR does not use a completely different binding mode for recognition of these two ligands. Thus, even with poor quality peptide contacts, the binding footprint of the 226 TCR is essentially the same with both ligands and the same as the 2B4 TCR, suggesting that, at least with these closely related VαVβ heterodimers, this particular MHC binding site is highly favored.
Also reported by Ehrich et al. (16), the effect of I-Ek mutations on 226’s recognition of MCC-p5K/I-Ek versus MCC-p5E/I-Ek fell into three categories. In one category, I-Ek mutations caused decreased 226 IL-2 production when either peptide was presented (I-EkQβ64R, I-EkTβ77Q). This category of residues represents residues that are involved in 226’s binding of both pMHC complexes. It is interesting to note that neither of the I-Ek mutations completely abolished 226’s ability to respond to MCC-p5K/I-Ek, consistent with the notion that 226’s very high affinity for MCC-p5K/I-Ek allows it to accommodate less than ideal ligands. A second category of residues caused an increase in IL-2 production only for MCC-p5E presentation (I-EkAα65V, I-EkAα68V). This category could be explained if these two mutants caused an increase in affinity in either case. No change could be detected for MCC-p5K recognition because the affinity for this complex is already extremely high. The final type of mutant was observed for I-EkHβ81Y, which increased 226’s sensitivity for MCC-p5K and decreased it for MCC-p5E. This phenotype can be explained if 226’s sensitivity for MCC-p5K goes up when affinity is decreased. We think this is the case, as it can also be seen for several of the MCC peptide APLs (Fig. 6C); p3F, P5L, and p8S all had higher potency for 226 stimulation than p5K, but it is unlikely that these pMHC complexes would be higher affinity for the 226 TCR. Together, we take this as evidence for an incomplete detrimental effect of having TCR–pMHC interactions with too high an affinity. This effect has been seen in the AND system (49) and in vivo for the 5c.c7 system (14). However, it is also clear that there is no absolute ceiling on the functional affinity of TCR–pMHC interactions because even higher affinity TCRs have been shown to be functional when expressed in hybridoma T cell lines in vitro (66, 67).
As is clear from the structures presented in this study, the 226 TCR β-chain (same TCRβ as 5c.c7) also contributes to the coordination of the central p5 lysine of MCC. Thus, immunization of 5c.c7β hemitransgenic mice with MCC-p5E selected for TCR α-chains able to overcome a less than ideal TCRβ-chain sequence in order recognize this Ag, albeit very weakly (KD >500 μM). Consequently, the 226 TCR also has very high affinity for MCC-p5K/I-Ek, making it a robust responder to substitutions in contacting residues in the peptide and the MHC. As we have described in this paper, this TCR, which may be highest affinity and the most cross-reactive of the naturally occurring TCRs described to date, appears to accomplish this with very canonical peptide and MHC docking and without significant conformational changes.
Several lines of evidence suggest that cross-reactive T cells may underlie at least some forms of autoimmunity (68, 69). Indeed, there are clear examples of autoreactive TCRs that are also capable of recognizing viral (70) and bacterial peptides (71). Although there is no evidence that 226 is autoreactive in these mice, it is one of the few examples in which we have structural information of a naturally occurring dramatically cross-reative TCR, and so it should help us understand this phenomenon. Like the known autoimmune epitopes, the affinity of 226 for MCC-p5E/I-Ek is extremely low (72, 73) and is stimulatory despite suboptimal contacts lacking H-bonds between the TCR and the peptide (as reviewed in Ref. 74). Furthermore, a structure of the MBP-specific Ob.1A12 TCR (a DR2-restricted TCR clone) in complex with a bacterial mimitope shows nearly identical docking as it has with the MBP/HLA-DR2b complex (37, 71), as is seen in comparison of 226’s recognition of both p5K- and p5E-MCC. It is also interesting to compare the mechanism of peptide degeneracy for this TCR to structures of previously identified degenerate and auto-reactive TCRs. For instance, an extremely cross-reactive TCR isolated from mice expressing 3K-peptide–tethered/I-Ab (29), the Yae62 TCR, was later found to have reduced peptide contacts and a somewhat exceptional docking mode dominated by I-Abα α-helical contacts (32). In contrast, in an engineered high-affinity version of the 2C TCR, M6, which also has dramatically reduced peptide specificity and endogenous-peptide reactivity (27), docking with the MHC was nearly the same as wild-type 2C, and the affinity gains came from modest rearrangements of CDR3α, enhancing hydrophobic contacts with the peptide (75). Thus, in the case of the 226 TCR presented in this study, like the M6 TCR, 226’s MHC contacts are unchanged compared with its nondegenerate counterpart, 2B4. Similarly modest differences in CDR3α-mediated contacts are capable of dramatically increasing TCR affinity for MCC/I-Ek, allowing the recognition of a wide range of MCC-peptide variants.
In summary, after many years of effort, we have obtained structures of two cytochrome c-specific TCRs complexed to two different ligands. These structures have resolved and explained the results of a large number of cellular, molecular, and biochemical studies. They also represent the first structural examples of I-Ek–reactive TCRs, the first structures of the Vα11 and Vβ3 TCR gene segments, and, in the case of 226, the highest affinity and most degenerate of the naturally occurring TCRs identified to date. We have found the CDR3 regions of these TCRs can recognize peptide variants of MCC in quite different ways, despite strict constraints imposed by large numbers of Vα11-mediated conserved contacts with the MHC.
Supplementary Material
Table S1, related to Figure 1. Data collection and refinement statistics for the 5 structures solved in this study.
Table S2, related to Table 1. V D and J gene segment usage and junctional sequences of the TCRs used in this study as well as other notable I-Ek reactive TCRs. For each TCR, the Vα gene name, CDR3α sequence, Jα gene name, Vβ gene name, CDR3β sequence, Jβ gene name and peptide specificity is listed. IMGT nomenclature is used for all TCR gene names. In addition to 2B4 and 226 TCRs, crystallized in this study, the 5c.c7 and AND TCR sequences are shown for comparison purposes as they are widely studied MCC/I-Ek reactive TCRs. The 202 TCR shares the same β chain as 226 and 5c.c7 and reacts with only MCC-p5E not wildtype MCC. The E21 TCR shares the same α chain as 5c.c7 but reacts only with MCC-p8E not wildtype MCC. It is interesting to note that this TCR uses the Vβ8.2 gene segment, for which there is a great deal of structural information.
Figure S1, related to Figure 3. Contact surfaces for each loop and inter-residue contacts. (A) Here the pMHC residues bound by each CDR loop of the 2B4 and 226 TCR are highlighted for each CDR loop of each TCR chain. The upper plots show the residues bound by CDR1, CDR2, HV4 and CDR3 loops of both 2B4α and 226α. Underneath the only CDR2 and CDR3 made pMHC contacts from 2B4/226β. For each, residues colored in blue make contact in all three structures. Magenta indicates residues only contacted by 2B4. Cyan residues are contacted only by 226 in the p5K structure. Green residues make contacts with 226 in both the p5K and p5E structures. Red residues are contacted by both the 2B4 and 226 TCRs bound to MCC-p5K/I-Ek. (B, C) To illustrate the similarity in docking of Inter-residue contacts between the CDR1, CDR2 and HV4 loops of 2B4α and I-Ekβ (B) or CDR2 of 2B4β and I-Ekα (C) are illustrated. As in Figure 3, lines connecting each residue represent van der Waals interactions (black), hydrogen bonds (red) and salt-bridges (blue). For comparison purposes, the contacts between 1934.4 TCR CDR1 and 2α, and sequence-aligned I-Auβ are shown in panels B and C.
Figure S2, related to Figure 3. Summary of functional data and its relationship to the 2B4 and 226 complex crystal structures. (A) This panel summarizes mutational data, where Ala or Gly mutations were made in I-Ek as well as various MCC peptide mutations (Wu et al.). Each residue is colored by its effect on 2B4 TCR binding as measured by SPR where grey indicates a less than 3× decrease in steady-state affinity, pink indicates a 3–100× decrease and red indicates a greater than 100× decrease in steady-state affinity. (B) This panel summarizes functional data using a peptide dose-response curve for IL-2 cytokine production as a read-out for 2B4 hybridoma T cell activation using CHO cells expressing wild-type or mutant I-Ek as APCs for wild-type or mutant MCC peptides. In this panel grey indicates a less than 20× change in the EC50 for peptide elicited IL-2 production, pink indicates a 20–100× increase in EC50 for IL-2 production and red indicates a greater than 100× increase in peptide EC50 for IL-2 production (Ehrich et al., 1993; Reay et al., 1994). (C) Here each of the peptide and I-Ek residues tested are colored according to their measured phi value, a measurement of the contribution of the residue to the 2B4 TCR binding transition state. Residues colored grey were tested but had insignificantly small phi values, pink represents a significant phi value that is less than 0.3 and residues colored red have significant phi values greater than 0.3 (Wu et al.). (D, E) Alterations in stimulatory capacity of mutant I-Ek molecules expressed in CHO cells for 226, using MCC (D) or MCC-p5E (E) as a stimulatory peptide. In this panel, mutants of residues colored gray caused a less than 20× change in peptide potency for IL-2 production, mutants of residues colored pink caused a 20–100× increase in peptide EC50 and mutants of red residues caused a greater than 100× decrease in peptide potency. Green residues represent mutations that actually improved the peptide stimulatory capacity of MCC (D) or MCC-p5E (E) peptide with a 20–100× decrease in peptide EC50.
Table S1. Data collection and refinement statistics
Table S2. V, D, J and CDR3s of MCC- and other peptide-I-Ek reactive TCRs
Acknowledgments
We thank Natalia Goriatcheva for technical assistance. We also thank Fleur Tynan, Michael Birnbaum, and Jarrett Adams for helpful comments and suggestions, Jarrett Adams for providing some DNA cloning intermediates, and the beamline staff of the Stanford Synchrotron Radiation Lightsource and Advanced Photon Source for assistance with data collection.
Abbreviations used in this article
- APL
altered peptide ligand
- H-bond
hydrogen bond
- MCC
moth cytochrome c
- PCC
pigeon cytochrome c
- PDB
Protein Data Bank
- pMHC
peptide-MHC
- SPR
surface plasmon resonance
Footnotes
The structures and coordinates presented in this article have been submitted to the Research Collaboratory for Structural Bioinformatics Protein Data Bank (PDB) (http://www.pdb.org) under PDB ID 3QIB, 3QIW, 3QJF, and 3QJH.
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Solinger AM, Ultee ME, Margoliash E, Schwartz RH. T-lymphocyte response to cytochrome c. I. Demonstration of a T-cell heteroclitic proliferative response and identification of a topographic antigenic determinant on pigeon cytochrome c whose immune recognition requires two complementing major histocompatibility complex-linked immune response genes. J Exp Med. 1979;150:830–848. doi: 10.1084/jem.150.4.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwartz RH. T-lymphocyte recognition of antigen in association with gene products of the major histocompatibility complex. Annu Rev Immunol. 1985;3:237–261. doi: 10.1146/annurev.iy.03.040185.001321. [DOI] [PubMed] [Google Scholar]
- 3.Hedrick SM, Matis LA, Hecht TT, Samelson LE, Longo DL, Heber-Katz E, Schwartz RH. The fine specificity of antigen and Ia determinant recognition by T cell hybridoma clones specific for pigeon cytochrome c. Cell. 1982;30:141–152. doi: 10.1016/0092-8674(82)90020-4. [DOI] [PubMed] [Google Scholar]
- 4.Matsui K, Boniface JJ, Steffner P, Reay PA, Davis MM. Kinetics of T-cell receptor binding to peptide/I-Ek complexes: correlation of the dissociation rate with T-cell responsiveness. Proc Natl Acad Sci USA. 1994;91:12862–12866. doi: 10.1073/pnas.91.26.12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winoto A, Urban JL, Lan NC, Goverman J, Hood L, Hansburg D. Predominant use of a V alpha gene segment in mouse T-cell receptors for cytochrome c. Nature. 1986;324:679–682. doi: 10.1038/324679a0. [DOI] [PubMed] [Google Scholar]
- 6.Engel I, Hedrick SM. Site-directed mutations in the VDJ junctional region of a T cell receptor beta chain cause changes in antigenic peptide recognition. Cell. 1988;54:473–484. doi: 10.1016/0092-8674(88)90068-2. [DOI] [PubMed] [Google Scholar]
- 7.McHeyzer-Williams MG, Davis MM. Antigen-specific development of primary and memory T cells in vivo. Science. 1995;268:106–111. doi: 10.1126/science.7535476. [DOI] [PubMed] [Google Scholar]
- 8.Savage PA, Boniface JJ, Davis MM. A kinetic basis for T cell receptor repertoire selection during an immune response. Immunity. 1999;10:485–492. doi: 10.1016/s1074-7613(00)80048-5. [DOI] [PubMed] [Google Scholar]
- 9.Malherbe L, Hausl C, Teyton L, McHeyzer-Williams MG. Clonal selection of helper T cells is determined by an affinity threshold with no further skewing of TCR binding properties. Immunity. 2004;21:669–679. doi: 10.1016/j.immuni.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 10.Malherbe L, Mark L, Fazilleau N, McHeyzer-Williams LJ, McHeyzer-Williams MG. Vaccine adjuvants alter TCR-based selection thresholds. Immunity. 2008;28:698–709. doi: 10.1016/j.immuni.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol. 2009;10:375–384. doi: 10.1038/ni.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayashi Y, Tsukumo S, Shiota H, Kishihara K, Yasutomo K. Antigen-specific T cell repertoire modification of CD4+CD25+ regulatory T cells. J Immunol. 2004;172:5240–5248. doi: 10.4049/jimmunol.172.9.5240. [DOI] [PubMed] [Google Scholar]
- 13.Gottschalk RA, Corse E, Allison JP. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. J Exp Med. 2010;207:1701–1711. doi: 10.1084/jem.20091999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corse E, Gottschalk RA, Krogsgaard M, Allison JP. Attenuated T cell responses to a high-potency ligand in vivo. PLoS Biol. 2010;8:8. doi: 10.1371/journal.pbio.1000481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reay PA, Kantor RM, Davis MM. Use of global amino acid replacements to define the requirements for MHC binding and T cell recognition of moth cytochrome c (93–103) J Immunol. 1994;152:3946–3957. [PubMed] [Google Scholar]
- 16.Ehrich EW, Devaux B, Rock EP, Jorgensen JL, Davis MM, Chien YH. T cell receptor interaction with peptide/major histocompatibility complex (MHC) and superantigen/MHC ligands is dominated by antigen. J Exp Med. 1993;178:713–722. doi: 10.1084/jem.178.2.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jorgensen JL, Esser U, Fazekas de St Groth B, Reay PA, Davis MM. Mapping T-cell receptor-peptide contacts by variant peptide immunization of single-chain transgenics. Nature. 1992;355:224–230. doi: 10.1038/355224a0. [DOI] [PubMed] [Google Scholar]
- 18.Wucherpfennig KW, Allen PM, Celada F, Cohen IR, De Boer R, Garcia KC, Goldstein B, Greenspan R, Hafler D, Hodgkin P, et al. Polyspecificity of T cell and B cell receptor recognition. Semin Immunol. 2007;19:216–224. doi: 10.1016/j.smim.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Macdonald WA, Chen Z, Gras S, Archbold JK, Tynan FE, Clements CS, Bharadwaj M, Kjer-Nielsen L, Saunders PM, Wilce MC, et al. T cell allorecognition via molecular mimicry. Immunity. 2009;31:897–908. doi: 10.1016/j.immuni.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 20.Mazza C, Auphan-Anezin N, Gregoire C, Guimezanes A, Kellenberger C, Roussel A, Kearney A, van der Merwe PA, Schmitt-Verhulst AM, Malissen B. How much can a T-cell antigen receptor adapt to structurally distinct antigenic peptides? EMBO J. 2007;26:1972–1983. doi: 10.1038/sj.emboj.7601605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kersh GJ, Allen PM. Structural basis for T cell recognition of altered peptide ligands: a single T cell receptor can productively recognize a large continuum of related ligands. J Exp Med. 1996;184:1259–1268. doi: 10.1084/jem.184.4.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ausubel LJ, Bieganowska KD, Hafler DA. Cross-reactivity of T-cell clones specific for altered peptide ligands of myelin basic protein. Cell Immunol. 1999;193:99–107. doi: 10.1006/cimm.1998.1447. [DOI] [PubMed] [Google Scholar]
- 23.Jameson SC, Carbone FR, Bevan MJ. Clone-specific T cell receptor antagonists of major histocompatibility complex class I-restricted cytotoxic T cells. J Exp Med. 1993;177:1541–1550. doi: 10.1084/jem.177.6.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crawford F, Huseby E, White J, Marrack P, Kappler JW. Mimotopes for alloreactive and conventional T cells in a peptide-MHC display library. PLoS Biol. 2004;2:E90. doi: 10.1371/journal.pbio.0020090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maynard J, Petersson K, Wilson DH, Adams EJ, Blondelle SE, Boulanger MJ, Wilson DB, Garcia KC. Structure of an autoimmune T cell receptor complexed with class II peptide-MHC: insights into MHC bias and antigen specificity. Immunity. 2005;22:81–92. doi: 10.1016/j.immuni.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 26.Wilson DB, Pinilla C, Wilson DH, Schroder K, Boggiano C, Judkowski V, Kaye J, Hemmer B, Martin R, Houghten RA. Immunogenicity. I. Use of peptide libraries to identify epitopes that activate clonotypic CD4+ T cells and induce T cell responses to native peptide ligands. J Immunol. 1999;163:6424–6434. [PubMed] [Google Scholar]
- 27.Holler PD, Chlewicki LK, Kranz DM. TCRs with high affinity for foreign pMHC show self-reactivity. Nat Immunol. 2003;4:55–62. doi: 10.1038/ni863. [DOI] [PubMed] [Google Scholar]
- 28.Donermeyer DL, Weber KS, Kranz DM, Allen PM. The study of high-affinity TCRs reveals duality in T cell recognition of antigen: specificity and degeneracy. J Immunol. 2006;177:6911–6919. doi: 10.4049/jimmunol.177.10.6911. [DOI] [PubMed] [Google Scholar]
- 29.Huseby ES, White J, Crawford F, Vass T, Becker D, Pinilla C, Marrack P, Kappler JW. How the T cell repertoire becomes peptide and MHC specific. Cell. 2005;122:247–260. doi: 10.1016/j.cell.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 30.Garcia KC, Adams JJ, Feng D, Ely LK. The molecular basis of TCR germline bias for MHC is surprisingly simple. Nat Immunol. 2009;10:143–147. doi: 10.1038/ni.f.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng D, Bond CJ, Ely LK, Maynard J, Garcia KC. Structural evidence for a germline-encoded T cell receptor-major histocompatibility complex interaction ‘codon’. Nat Immunol. 2007;8:975–983. doi: 10.1038/ni1502. [DOI] [PubMed] [Google Scholar]
- 32.Dai S, Huseby ES, Rubtsova K, Scott-Browne J, Crawford F, Macdonald WA, Marrack P, Kappler JW. Crossreactive T Cells spotlight the germline rules for alphabeta T cell-receptor interactions with MHC molecules. Immunity. 2008;28:324–334. doi: 10.1016/j.immuni.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott-Browne JP, White J, Kappler JW, Gapin L, Marrack P. Germline-encoded amino acids in the alphabeta T-cell receptor control thymic selection. Nature. 2009;458:1043–1046. doi: 10.1038/nature07812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tynan FE, Reid HH, Kjer-Nielsen L, Miles JJ, Wilce MC, Kostenko L, Borg NA, Williamson NA, Beddoe T, Purcell AW, et al. A T cell receptor flattens a bulged antigenic peptide presented by a major histocompatibility complex class I molecule. Nat Immunol. 2007;8:268–276. doi: 10.1038/ni1432. [DOI] [PubMed] [Google Scholar]
- 35.Clements CS, Kjer-Nielsen L, MacDonald WA, Brooks AG, Purcell AW, McCluskey J, Rossjohn J. The production, purification and crystallization of a soluble heterodimeric form of a highly selected T-cell receptor in its unliganded and liganded state. Acta Crystallogr D Biol Crystallogr. 2002;58:2131–2134. doi: 10.1107/s0907444902015482. [DOI] [PubMed] [Google Scholar]
- 36.Kozono H, White J, Clements J, Marrack P, Kappler J. Production of soluble MHC class II proteins with covalently bound single peptides. Nature. 1994;369:151–154. doi: 10.1038/369151a0. [DOI] [PubMed] [Google Scholar]
- 37.Hahn M, Nicholson MJ, Pyrdol J, Wucherpfennig KW. Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune T cell receptor. Nat Immunol. 2005;6:490–496. doi: 10.1038/ni1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hennecke J, Carfi A, Wiley DC. Structure of a covalently stabilized complex of a human alphabeta T-cell receptor, influenza HA peptide and MHC class II molecule, HLA-DR1. EMBO J. 2000;19:5611–5624. doi: 10.1093/emboj/19.21.5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuhns MS, Girvin AT, Klein LO, Chen R, Jensen KD, Newell EW, Huppa JB, Lillemeier BF, Huse M, Chien YH, et al. Evidence for a functional sidedness to the alphabetaTCR. Proc Natl Acad Sci USA. 2010;107:5094–5099. doi: 10.1073/pnas.1000925107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leslie AGW. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography. 26. Daresbury Laboratory, Daresbury; Warrington, U.K: 1992. Recent changes to the MOSFLM package for processing film and image plate data. [Google Scholar]
- 41.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 42.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter ACW Jr, Sweet RM, editors. Methods in Enzymology, Vol. 276: Macro-molecular Crystallography Part. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 43.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brunger AT. Version 1.2 of the Crystallography and NMR system. Nat Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 45.Afonine PV, Grosse-Kunstleve RW, Adams PD. CCP4 Newsletter. Vol. 42. Daresbury Laboratory, Daresbury; Warrington, U.K: 2005. The Phenix refinement framework. [Google Scholar]
- 46.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 47.Wu LC, Tuot DS, Lyons DS, Garcia KC, Davis MM. Two-step binding mechanism for T-cell receptor recognition of peptide MHC. Nature. 2002;418:552–556. doi: 10.1038/nature00920. [DOI] [PubMed] [Google Scholar]
- 48.Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 49.Cemerski S, Das J, Locasale J, Arnold P, Giurisato E, Markiewicz MA, Fremont D, Allen PM, Chakraborty AK, Shaw AS. The stimulatory potency of T cell antigens is influenced by the formation of the immunological synapse. Immunity. 2007;26:345–355. doi: 10.1016/j.immuni.2007.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hare BJ, Wyss DF, Osburne MS, Kern PS, Reinherz EL, Wagner G. Structure, specificity and CDR mobility of a class II restricted single-chain T-cell receptor. Nat Struct Biol. 1999;6:574–581. doi: 10.1038/9359. [DOI] [PubMed] [Google Scholar]
- 51.Reiser JB, Darnault C, Grégoire C, Mosser T, Mazza G, Kearney A, van der Merwe PA, Fontecilla-Camps JC, Housset D, Malissen B. CDR3 loop flexibility contributes to the degeneracy of TCR recognition. Nat Immunol. 2003;4:241–247. doi: 10.1038/ni891. [DOI] [PubMed] [Google Scholar]
- 52.Armstrong KM, Piepenbrink KH, Baker BM. Conformational changes and flexibility in T-cell receptor recognition of peptide-MHC complexes. Biochem J. 2008;415:183–196. doi: 10.1042/BJ20080850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patten PA, Gray NS, Yang PL, Marks CB, Wedemayer GJ, Boniface JJ, Stevens RC, Schultz PG. The immunological evolution of catalysis. Science. 1996;271:1086–1091. doi: 10.1126/science.271.5252.1086. [DOI] [PubMed] [Google Scholar]
- 54.Matsui K, Boniface JJ, Reay PA, Schild H, Fazekas de StGroth B, Davis MM. Low affinity interaction of peptide-MHC complexes with T cell receptors. Science. 1991;254:1788–1791. doi: 10.1126/science.1763329. [DOI] [PubMed] [Google Scholar]
- 55.Hedrick SM, Cohen DI, Nielsen EA, Davis MM. Isolation of cDNA clones encoding T cell-specific membrane-associated proteins. Nature. 1984;308:149–153. doi: 10.1038/308149a0. [DOI] [PubMed] [Google Scholar]
- 56.Berg LJ, Fazekas de St Groth B, Pullen AM, Davis MM. Phenotypic differences between alpha beta versus beta T-cell receptor transgenic mice undergoing negative selection. Nature. 1989;340:559–562. doi: 10.1038/340559a0. [DOI] [PubMed] [Google Scholar]
- 57.Ebert PJ, Ehrlich LI, Davis MM. Low ligand requirement for deletion and lack of synapses in positive selection enforce the gauntlet of thymic T cell maturation. Immunity. 2008;29:734–745. doi: 10.1016/j.immuni.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ebert PJ, Jiang S, Xie J, Li QJ, Davis MM. An endogenous positively selecting peptide enhances mature T cell responses and becomes an autoantigen in the absence of microRNA miR-181a. Nat Immunol. 2009;10:1162–1169. doi: 10.1038/ni.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuhns MS, Davis MM. Disruption of extracellular interactions impairs T cell receptor-CD3 complex stability and signaling. Immunity. 2007;26:357–369. doi: 10.1016/j.immuni.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 60.Lyons DS, Lieberman SA, Hampl J, Boniface JJ, Chien Y, Berg LJ, Davis MM. A TCR binds to antagonist ligands with lower affinities and faster dissociation rates than to agonists. Immunity. 1996;5:53–61. doi: 10.1016/s1074-7613(00)80309-x. [DOI] [PubMed] [Google Scholar]
- 61.Boniface JJ, Reich Z, Lyons DS, Davis MM. Thermodynamics of T cell receptor binding to peptide-MHC: evidence for a general mechanism of molecular scanning. Proc Natl Acad Sci USA. 1999;96:11446–11451. doi: 10.1073/pnas.96.20.11446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krogsgaard M, Prado N, Adams EJ, He XL, Chow DC, Wilson DB, Garcia KC, Davis MM. Evidence that structural rearrangements and/or flexibility during TCR binding can contribute to T cell activation. Mol Cell. 2003;12:1367–1378. doi: 10.1016/s1097-2765(03)00474-x. [DOI] [PubMed] [Google Scholar]
- 63.Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM. Direct observation of ligand recognition by T cells. Nature. 2002;419:845–849. doi: 10.1038/nature01076. [DOI] [PubMed] [Google Scholar]
- 64.Lillemeier BF, Mörtelmaier MA, Forstner MB, Huppa JB, Groves JT, Davis MM. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat Immunol. 2010;11:90–96. doi: 10.1038/ni.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huppa JB, Axmann M, Mörtelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, Klein LO, Schütz GJ, Davis MM. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–967. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Holler PD, Lim AR, Cho BK, Rund LA, Kranz DM. CD8(−) T cell transfectants that express a high affinity T cell receptor exhibit enhanced peptide-dependent activation. J Exp Med. 2001;194:1043–1052. doi: 10.1084/jem.194.8.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weber KS, Donermeyer DL, Allen PM, Kranz DM. Class II-restricted T cell receptor engineered in vitro for higher affinity retains peptide specificity and function. Proc Natl Acad Sci USA. 2005;102:19033–19038. doi: 10.1073/pnas.0507554102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Benoist C, Mathis D. Autoimmunity provoked by infection: how good is the case for T cell epitope mimicry? Nat Immunol. 2001;2:797–801. doi: 10.1038/ni0901-797. [DOI] [PubMed] [Google Scholar]
- 69.Münz C, Lünemann JD, Getts MT, Miller SD. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol. 2009;9:246–258. doi: 10.1038/nri2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Harkiolaki M, Holmes SL, Svendsen P, Gregersen JW, Jensen LT, McMahon R, Friese MA, van Boxel G, Etzensperger R, Tzartos JS, et al. T cell-mediated autoimmune disease due to low-affinity crossreactivity to common microbial peptides. Immunity. 2009;30:348–357. doi: 10.1016/j.immuni.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 72.Appel H, Gauthier L, Pyrdol J, Wucherpfennig KW. Kinetics of T-cell receptor binding by bivalent HLA-DR. Peptide complexes that activate antigen-specific human T-cells. J Biol Chem. 2000;275:312–321. doi: 10.1074/jbc.275.1.312. [DOI] [PubMed] [Google Scholar]
- 73.Li Y, Huang Y, Lue J, Quandt JA, Martin R, Mariuzza RA. Structure of a human autoimmune TCR bound to a myelin basic protein self-peptide and a multiple sclerosis-associated MHC class II molecule. EMBO J. 2005;24:2968–2979. doi: 10.1038/sj.emboj.7600771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wucherpfennig KW, Call MJ, Deng L, Mariuzza R. Structural alterations in peptide-MHC recognition by self-reactive T cell receptors. Curr Opin Immunol. 2009;21:590–595. doi: 10.1016/j.coi.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Colf LA, Bankovich AJ, Hanick NA, Bowerman NA, Jones LL, Kranz DM, Garcia KC. How a single T cell receptor recognizes both self and foreign MHC. Cell. 2007;129:135–146. doi: 10.1016/j.cell.2007.01.048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, related to Figure 1. Data collection and refinement statistics for the 5 structures solved in this study.
Table S2, related to Table 1. V D and J gene segment usage and junctional sequences of the TCRs used in this study as well as other notable I-Ek reactive TCRs. For each TCR, the Vα gene name, CDR3α sequence, Jα gene name, Vβ gene name, CDR3β sequence, Jβ gene name and peptide specificity is listed. IMGT nomenclature is used for all TCR gene names. In addition to 2B4 and 226 TCRs, crystallized in this study, the 5c.c7 and AND TCR sequences are shown for comparison purposes as they are widely studied MCC/I-Ek reactive TCRs. The 202 TCR shares the same β chain as 226 and 5c.c7 and reacts with only MCC-p5E not wildtype MCC. The E21 TCR shares the same α chain as 5c.c7 but reacts only with MCC-p8E not wildtype MCC. It is interesting to note that this TCR uses the Vβ8.2 gene segment, for which there is a great deal of structural information.
Figure S1, related to Figure 3. Contact surfaces for each loop and inter-residue contacts. (A) Here the pMHC residues bound by each CDR loop of the 2B4 and 226 TCR are highlighted for each CDR loop of each TCR chain. The upper plots show the residues bound by CDR1, CDR2, HV4 and CDR3 loops of both 2B4α and 226α. Underneath the only CDR2 and CDR3 made pMHC contacts from 2B4/226β. For each, residues colored in blue make contact in all three structures. Magenta indicates residues only contacted by 2B4. Cyan residues are contacted only by 226 in the p5K structure. Green residues make contacts with 226 in both the p5K and p5E structures. Red residues are contacted by both the 2B4 and 226 TCRs bound to MCC-p5K/I-Ek. (B, C) To illustrate the similarity in docking of Inter-residue contacts between the CDR1, CDR2 and HV4 loops of 2B4α and I-Ekβ (B) or CDR2 of 2B4β and I-Ekα (C) are illustrated. As in Figure 3, lines connecting each residue represent van der Waals interactions (black), hydrogen bonds (red) and salt-bridges (blue). For comparison purposes, the contacts between 1934.4 TCR CDR1 and 2α, and sequence-aligned I-Auβ are shown in panels B and C.
Figure S2, related to Figure 3. Summary of functional data and its relationship to the 2B4 and 226 complex crystal structures. (A) This panel summarizes mutational data, where Ala or Gly mutations were made in I-Ek as well as various MCC peptide mutations (Wu et al.). Each residue is colored by its effect on 2B4 TCR binding as measured by SPR where grey indicates a less than 3× decrease in steady-state affinity, pink indicates a 3–100× decrease and red indicates a greater than 100× decrease in steady-state affinity. (B) This panel summarizes functional data using a peptide dose-response curve for IL-2 cytokine production as a read-out for 2B4 hybridoma T cell activation using CHO cells expressing wild-type or mutant I-Ek as APCs for wild-type or mutant MCC peptides. In this panel grey indicates a less than 20× change in the EC50 for peptide elicited IL-2 production, pink indicates a 20–100× increase in EC50 for IL-2 production and red indicates a greater than 100× increase in peptide EC50 for IL-2 production (Ehrich et al., 1993; Reay et al., 1994). (C) Here each of the peptide and I-Ek residues tested are colored according to their measured phi value, a measurement of the contribution of the residue to the 2B4 TCR binding transition state. Residues colored grey were tested but had insignificantly small phi values, pink represents a significant phi value that is less than 0.3 and residues colored red have significant phi values greater than 0.3 (Wu et al.). (D, E) Alterations in stimulatory capacity of mutant I-Ek molecules expressed in CHO cells for 226, using MCC (D) or MCC-p5E (E) as a stimulatory peptide. In this panel, mutants of residues colored gray caused a less than 20× change in peptide potency for IL-2 production, mutants of residues colored pink caused a 20–100× increase in peptide EC50 and mutants of red residues caused a greater than 100× decrease in peptide potency. Green residues represent mutations that actually improved the peptide stimulatory capacity of MCC (D) or MCC-p5E (E) peptide with a 20–100× decrease in peptide EC50.
Table S1. Data collection and refinement statistics
Table S2. V, D, J and CDR3s of MCC- and other peptide-I-Ek reactive TCRs