

Abstract
Background
The tissue accumulation of protein-bound advanced glycation endproducts (AGE) may be involved in the etiology of diabetic chronic complications, including osteopenia. The aim of this study was to investigate the effect of an AGE-modified type I collagen substratum on the adhesion, spreading, proliferation and differentiation of rat osteosarcoma UMR106 and mouse non-transformed MC3T3E1 osteoblastic cells. We also studied the role of reactive oxygen species (ROS) and nitric oxide synthase (NOS) expression on these AGE-collagen mediated effects.
Results
AGE-collagen decreased the adhesion of UMR106 cells, but had no effect on the attachment of MC3T3E1 cells. In the UMR106 cell line, AGE-collagen also inhibited cellular proliferation, spreading and alkaline phosphatase (ALP) activity. In preosteoblastic MC3T3E1 cells (24-hour culture), proliferation and spreading were significantly increased by AGE-collagen. After one week of culture (differentiated MC3T3E1 osteoblasts) AGE-collagen inhibited ALP activity, but had no effect on cell number. In mineralizing MC3T3E1 cells (3-week culture) AGE-collagen induced a decrease in the number of surviving cells and of extracellular nodules of mineralization, without modifying their ALP activity. Intracellular ROS production, measured after a 48-hour culture, was decreased by AGE-collagen in MC3T3E1 cells, but was increased by AGE-collagen in UMR106 cells. After a 24-hour culture, AGE-collagen increased the expression of endothelial and inducible NOS, in both osteoblastic cell lines.
Conclusions
These results suggest that the accumulation of AGE on bone extracellular matrix could regulate the proliferation and differentiation of osteoblastic cells. These effects appear to depend on the stage of osteoblastic development, and possibly involve the modulation of NOS expression and intracellular ROS pathways.
Background
Recent research indicates that an increase in the steady-state levels of highly reactive dicarbonylic compounds ("carbonyl stress") may lead to the formation of advanced glycation endproducts or AGE. These AGE products could be involved in the etiology of the long-term complications of several human afflictions, such as Diabetes mellitus, ageing, uremia and Alzheimer's disease [1,2]. In diabetic patients with long-standing hyperglycemia, an increase in the generation of AGE products can be partly explained by the process of non-enzymatic glycosylation of proteins. In this reaction, glucose reacts reversibly with the free amino groups of proteins to form unstable Schiff bases, which may undergo an intramolecular rearrangement giving rise to the more stable Amadori product. A further modification of this product originates 3-deoxyglucasone, a reactive dicarbonylic non-protein-bound molecule, which can then form adducts with nearby proteins and irreversibly progress to the formation of a family of compounds collectively known as AGE, many of which are fluorescent and originate protein-proteins crosslinks [2]. Most studies have demonstrated that AGE can induce structural and functional alterations of various proteins.
Cell surface receptors for AGE moieties have been detected on different cell types. These proteins appear to contribute to diverse cellular functions, such as the specific recognition and degradation of AGE-modified proteins [3]. Depending on the cell type involved, cellular recognition of AGE can be associated with chemotaxis, angiogenesis, oxidative stress, and either cell proliferation or apoptosis. Recent studies have suggested that the nitric oxide synthase (NOS) / nitric oxide (NO) pathway may be involved in the AGE-mediated alterations observed in several cell types, such as retinal vascular endothelial cells and macrophages [4,5]. In addition, NO-mediated vasodilation has been reported to be impaired in diabetes [6].
Collagenous proteins play important roles in the function of several tissues, including bone. They have a long half-life and various free amino groups which are exposed to ambient glucose levels [7]. An increase in AGE-modified collagen has been detected in the cortical bone of diabetic and ageing rats [8-10]. The alterations caused by AGE-modifications of collagen may play a role in the pathogenesis of the osteopenia present in poorly controlled diabetic patients [11]. This osteopenia has been attributed to a long-term decrease in the recruitment and bone-forming activity of osteoblasts [12].
In our laboratory we have previously demonstrated that soluble AGE-modified proteins can regulate the growth of mouse MC3T3E1 and rat UMR106 osteoblast-like cells:while initially eliciting an increase in cellular proliferation and differentation, a relatively long-term incubation of these cells with AGE-modified bovine serum albumin (BSA) induces a significant decrease in both parameters [13]. We have also described the presence of membrane-associated receptors in both cell lines, which specifically participate in the recognition and degradation of AGE-modified proteins. The expression of these receptors is differentially regulated according to the osteoblastic stage of development [14]. Furthermore, we have observed in this system [15] that AGE-BSA can modify the secretion of insulin-like growth factor (IGF)-I, one of the major growth factors involved in bone development [16]. In addition, the levels of IGF-binding proteins present in the osteoblastic conditioned media are also regulated by AGE-BSA and the stage of osteoblast development [15].
The aim of the present study was to investigate the effect of an AGE-modified type I collagen matrix obtained in vitro, on the adhesion, spreading, proliferation, differentiation and mineralization of rat osteosarcoma UMR106 and mouse non-transformed MC3T3E1 calvaria-derived cells in culture, throughout their different stages of osteoblastic development. In addition, we also studied the role of reactive oxygen species (ROS) and NOS expression in the possible alterations of osteoblastic growth induced by the glycated collagen matrix.
Results
Effect of an AGE-modified collagen matrix on cell adhesion
Non-enzymatic glycosylation of the type I collagen matrix induced cell-line specific alterations in cellular adhesion (Fig. 1). As can be seen, the adhesion of osteosarcoma UMR106 cells was significantly inhibited by the AGE-modified collagen matrix (40 % inhibition). However, the formation of AGE-products on the collagenous matrix caused no effect on the adhesion of non-transformed MC3T3E1 cells.
Figure 1.

Effect of an AGE-modified type I collagen matrix on cell adhesion. UMR106 and MC3T3E1 osteoblast-like cells were incubated on an unmodified collagen matrix or an AGE-collagen matrix for 1 hour at 37°C. Cells adhering to the matrices were fixed, stained with Giemsa and counted microscopically at a magnification of 400X. Results are given as the number of cells counted per field, and are expressed as the mean ± SEM. Differences between collagen vs. AGE-collagen are as follows: * p < 0.001
Effect of AGE-collagen on cell proliferation
The effect of an AGE-modified collagen matrix on UMR106 proliferation was evaluated by measurement of cellular growth and spreading after 24 hours of incubation. When the cells were plated on an unmodified type I collagen gel, they proliferated as expected, and about 70 % of them showed spreading (Fig. 2). However, when cells were grown on AGE-modified collagen their proliferation was significantly decreased (60 % of control), as was the relative number of cells which showed spreading (40 % of attached cells). The non-transformed MC3T3E1 cells were cultured on either control or glycated collagen for different periods of time, in order for them to attain different stages of osteoblastic development. In the proliferating stage (24 hours) cells were found to grow as expected on the untreated collagen matrix, and about 30 % of them showed spreading (Fig. 3). However, when they were cultured on AGE-modified collagen for 24 hours, a significant increase was observed both in cell proliferation (124 % of control) and in cellular spreading (42 % of attached cells). After one week of culture (differentiated stage), there was no statistically significant effect of AGE-collagen on cell growth (Fig. 4). Finally, after three weeks of culture (mineralizing stage), AGE-modified collagen induced a statistically significant decrease in the number of surviving cells (65 % of control).
Figure 2.

Effect of an AGE-modified type I collagen matrix on UMR106 cell proliferation and spreading. UMR106 osteosarcoma cells were cultured on an unmodified collagen matrix or an AGE-collagen matrix for 24 hours at 37°C. Cell proliferation was evaluated by microscopically counting cells stained with Giemsa, at a magnification of 400X. Cellular spreading was also estimated microscopically, as the number of cells possessing at least two comers. Results are given as the number of cells counted per field, and are expressed as the mean ± SEM. Differences between collagen vs. AGE-collagen are as follows: * p < 0.001
Figure 3.

Effect of an AGE-modified type I collagen matrix on MC3T3E1 cell proliferation and spreading. MC3T3E1 osteoblastic cells were cultured on an unmodified collagen matrix or an AGE-collagen matrix for 24 hours at 37°C. Cell proliferation was evaluated by microscopically counting cells stained with Giemsa, at a magnification of 400X. Cellular spreading was also estimated microscopically, as the number of cells possessing at least two corners. Results are given as the number of cells counted per field, and are expressed as the mean ± SEM. Differences between collagen vs. AGE-collagen are as follows: * p<0.01; ** p<0.001.
Figure 4.

Effect of an AGE-modified type I collagen matrix on MC3T3E1 cell growth. MC3T3E1 calvaria-derived osteoblastic cells were cultured on control collagen or AGE-modified collagen at 37°C for one week (differentiated osteoblasts) or three weeks (mineralizing cultures). The quantity of cells growing on the corresponding collagenous matrices at each timepoint, was evaluated by trypsinization and cell counting with a haemocytometer. Results are given as the number of cells counted in each case, and are expressed as the mean ± SEM. Differences between collagen vs. AGE-collagen are as follows: ** p<0.001.
Effect of AGE-collagen on osteoblastic differentiation and mineralization
In our culture system, cellular differentiation was evaluated using specific ALP activity as a marker of mature osteoblastic phenotype. UMR106 osteosarcoma cells expressed high levels of ALP (168 ± 8 nmol pNP / min × mg protein) when grown on an unmodified collagen matrix. However, when they were cultured on an AGE-modified collagenous substratum, a significant decrease of about 15 % was observed in their specific ALP activity (Fig. 5). The non-transformed MC3T3E1 osteoblasts grown on unmodified collagen, which initially (at 24 hours) expressed undetectable levels of ALP activity, increased this expression to 2.77 ± 0.33 nmol pNP / min × mg protein after one week of culture in a medium supplemented with ascorbic acid and β-glycerol phosphate, and reached a plateau of 3.05 ± 0.23 nmol pNP/ min × mg protein after a three-week culture. When these cells were cultured for one week on an AGE-modified collagenous matrix, a 30 % decrease in ALP activity was observed (Fig. 5). However, after three weeks of culture (mineralizing stage), no significant differences in specific ALP activity were observed between cells grown on either kind of matrix. The AGE-modified matrix was also found to inhibit mineralization, the end point of MC3T3E1 osteoblastic differentiation. Cells growing for three weeks on control type I collagen developed large areas of mineralization, with microscopically prominent mineral nodules (43 ± 5 calcified nodules per well). On the other hand, AGE-modified collagen significantly inhibited the formation of mineral nodules, which showed a much more patchy distribution over the MC3T3E1 culture surface area (27 ± 4 calcified nodules per well; p < 0.05 vs. control collagen).
Figure 5.

Effect of an AGE-modified type I collagen matrix on the ALP activity of UMR106 and MC3T3E1 cells. Osteoblastic cells were cultured on control or AGE-modified type I collagen at 37°C either for 24 hours (UMR106 cells), or for one and three weeks (MC3T3E1 cells). At the end of all incubations, the cell monolayer was lysated in 0.1 % Triton X-100. Alkaline phosphatase activity was determined in the lysates using p-nitro-phenyl-phosphate as a substrate, and was normalized for cellular protein content. Results are given as a percentage of the corresponding basal condition (unmodified collagen), and are expressed as the mean ± SEM. Differences between collagen vs. AGE-collagen are as follows: * p<0.01. Basal ALP activity was 168 ± 8 nmol pNP / min x mg protein for UMR106 cells; 2.77 ± 0.33 nmol pNP / min x mg protein for a one-week culture of MC3T3E1 cells; and 3.05 ± 0.23 nmol pNP / min x mg protein for a three-week culture of MC3T3E1 cells.
Role of ROS formation on AGE-induced osteoblastic development
In order to investigate the role of ROS as possible mediators of the AGE-collagen-induced osteoblast alterations, proliferating osteoblast-like cells were loaded with DHR and the intracellular content of the fluorescent oxidation product rhodamine was assessed. Fig. 6 shows that AGE-modified collagen caused an increase in ROS formation after 48 hours of UMR106 cell culture. On the contrary, a statistically significant decrease in ROS formation was detected in MC3T3E1 cells growing on AGE-modified collagen versus control collagen.
Figure 6.

Effect of an AGE-modified type I collagen substratum on the generation of intracellular reactive oxygen species by MC3T3E1 and UMR106 cells. Osteoblasts were cultured at 37°C either on control collagen or on AGE-modified collagen. After 48 hours, media were replaced by phenol red-free DMEM with 10 μM dihydro-rhodamine, and cells were incubated for an additional 4 hours. At the end of this incubation the cell monolayers were lysated with 0.1 % Triton X-100, and the levels of the oxidation product rhodamine were measured in the lysates by the determination of fluorescence intensity (excitation wavelength 495 nm, emission wavelength 532 nm). Results are given as a percentage of the corresponding basal condition (unmodified collagen), and are expressed as the mean ± SEM. Differences between collagen vs. AGE-collagen are as follows: * p<0.01; ** p<0.001
Effect of AGE-collagen on the expression of NOS in osteoblast-like cells
We next examined the effect of an AGE-modified matrix on the expression of endothelial constitutive (eNOS) and inducible (iNOS) nitric oxide synthases, as evaluated by Western blot analysis. The analysis of eNOS revealed a band corresponding to a relative molecular mass of 135 kDa (Fig. 7). The expression of eNOS was found to be significantly increased both in UMR106 and MC3T3E1 cells after 24 hours of culture on AGE-collagen versus control collagen. In addition, by using a specific anti-iNOS antibody, a protein of 130 kDa was detected (Fig. 8). The AGE-modified collagen matrix versus control collagen also significantly increased the intensity of this band in both cell lines, after 24 hours of culture.
Figure 7.
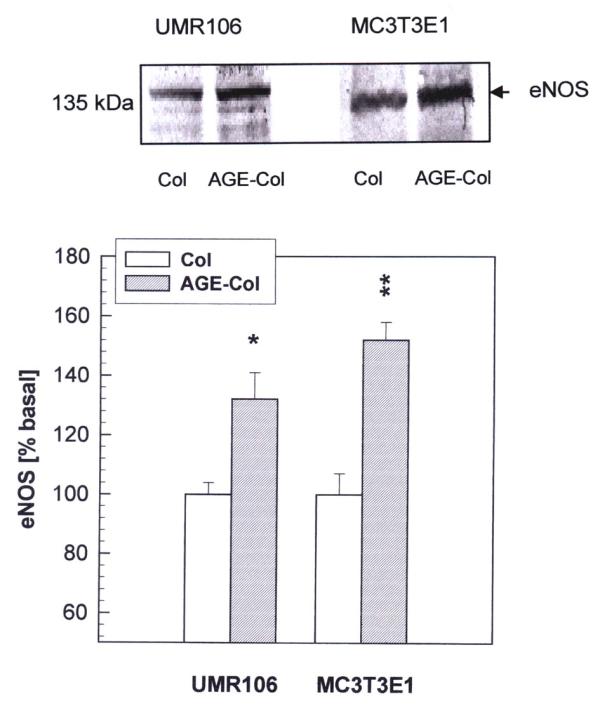
Effect of AGE-modified type I collagen on the expression of eNOS in osteoblast-like cells. UMR106 and MC3T3E1 cells were cultured on control or AGE-modified type I collagen matrices at 37ºC for 24 hours. After this incubation, cell monolayers were lysated in Laemmli's buffer and electrophoresed under reducing conditions on an 8 % SDS-PAGE. Western blotting was performed, and specific bands were detected using a rabbit polyclonal antibody against eNOS. A representative blot from three independent experiments is presented (top figure). Images were scanned and analyzed by densitometry. The relative intensity of each band is shown (bottom figure) as a percentage of the corresponding basal condition (unmodified collagen), and represents the mean ± SEM. Differences between collagen vs. AGE-collagen are as follows: * p<0.02; ** p<0.002
Figure 8.
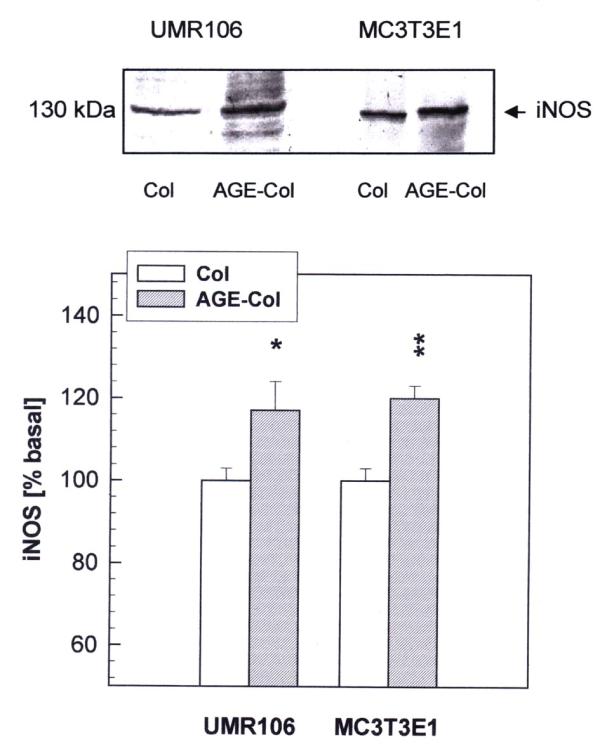
Effect of AGE-modified type I collagen on the expression of iNOS in osteoblast-like cells. UMR106 and MC3T3E1 cells were cultured on control or AGE-modified type I collagen matrices at 37°C for 24 hours. After this incubation, cell monolayers were lysated in Laemmli's buffer and electrophoresed under reducing conditions on an 8 % SDS-PAGE. Western blotting was performed, and specific bands were detected using a rabbit polyclonal antibody against iNOS. A representative blot from three independent experiments is presented (top figure). Images were scanned and analyzed by densitometry. The relative intensity of each band is shown (bottom figure) as a percentage of the corresponding basal condition (unmodified collagen), and represents the mean ± SEM. Differences between collagen vs. AGE-collagen are as follows: * p<0.05; ** p<0.002
Discussion
It has been established that sustained hyperglycemia can cause tissue damage through several mechanisms, including the accelerated formation of AGE on proteins, phospholipids and DNA [17]. An important consequence of this non-enzymatic modification involves the recognition of the AGE moieties by specific plasma membrane receptors, which can in turn induce alterations in cellular functions by ROS-mediated mechanisms. The accumulation of AGE occurs on both intracellular and extracellular structures (such as plasma and extracellular matrix proteins [ECM]), altering their bioactivity and associative properties. In particular, the accumulation of AGE on bone type I collagen, which constitutes over 85% of bone ECM proteins, has been previously described in both diabetes and ageing [8-10]. The integrity of the ECM is essential to maintain the normal function of osteoblasts, the main cell type involved in the formation of bone [18]. Thus, AGE-modified ECM may have important consequences on the normal bone-forming activity of osteoblasts, possibly through changes in cell-matrix interactions. Our present study confirms a previous report [10] showing that the formation of AGE on type I collagen can alter osteoblastic growth and inhibit osteoblast differentiation. In addition, we have further extended that study by investigating the effect of AGE-modified collagen on cell adhesion, its relationship to the stage of osteoblastic development, and the possible involvement of oxidative stress and NOS expression as mediators of the action of AGE on osteoblastic cells.
Our results show that UMR106 osteosarcoma cells tend to adhere less to AGE-collagen than to unmodified collagen. However, no differences were found in the case of the non-transformed MC3T3E1 line. These observations seem to indicate that the effect of AGE modification of ECM on cell attachment and growth could depend on cell type. This concept is supported by reports from other investigators who have found either a decrease in cell adhesion induced by AGE collagen in osteosarcoma MG63 cells, fibrosarcoma cells [19] and rat embryo 3Y1 fibroblasts [20], or no effect of AGE on adhesion as in the case of the osteosarcoma sub-clone UMR106-06 [21]. In this context, the observed differences could be due to different subsets of adhesion molecules involved in the attachment to ECM by different cell types, for example in non-transformed as opposed to tumorigenic cells. It has recently been demonstrated that type I collagen interacts with the α2β1 cellular integrin receptor [22,23]. The sequence Asp-Gly-Glu-Ala (DGEA) present in type I collagen serves as the main recognition site for α2β1 integrin, which has been dubbed the collagen-specific receptor. In recent studies, Paul and Bailey [19] found that α2β1 is probably involved in the attachment of MG63 osteosarcoma cells to a collagenous matrix, since they were able to inhibit cellular adhesion by an incubation with blocking anti-α2β1 antibodies. They also suggested, though did not directly prove, that glycation of type I collagen with methylglyoxal (a potent dicarbonylic AGE precursor) modifies arginine residues present in its Arg-Gly-Asp (RGD) sequence which is common to several other extracellular matrix proteins, and normally is recognized by the α5β1 integrin receptor [22]. Thus, the RGD sequence is present in the procollagen molecule and could in theory mediate the interaction between bone cells and type I collagen. In actual fact, since anti-α2 (but not anti-α5) blocking antibodies inhibit cellular adhesion to a type I collagen substratum, RGD-α5β1 interaction does not appear to be normally involved in the cellular attachment to type I collagen [22,23]. However, it is conceivable that different adhesion sequences of collagen could become either exposed or masked for interaction with several kinds of cellular receptors, as a consequence of molecular alterations of ECM such as AGE modification. Further experiments are in progress in our laboratory to investigate this hypothesis.
In this study we also examined the effect of AGE-modified collagen on osteoblastic proliferation and spreading, differentiation (ALP activity) and mineralization (nodule formation). The effect of AGE on cellular phenotypic expression was found to depend on cell type and/or the stage of osteoblast development. AGE-modified collagen not only decreased ALP activity in phenotypically mature osteoblastic cells, but also diminished nodule formation in post-mitotic osteoblasts in their mineralizing stage of development. AGE-collagen also induced changes in cell proliferation and spreading in a cell-type-specific manner. Thus, after an incubation of 24 hours the glycated matrix significantly inhibited growth and spreading in osteosarcoma UMR106 cells. These observations are in general agreement with the report of Paul and Bailey [19], who found that AGE-collagen decreased the number of matrix-attached MG63 osteosarcoma cells, after an incubation of 20 hours. On the contrary, in the case of non-transformed MC3T3E1 osteoblasts in their proliferating stage of development (24-hour incubation), AGE-modified collagen induced a significant increase in cell growth and spreading. Similarly, in a recent report [10] Katayama found an AGE-collagen-induced increase of DNA synthesis in a primary culture of rat osteoblasts after an incubation of 48 hours. The apparently discordant results obtained in the two cell lines which we studied, may merely be the consequence of dissimilar patterns of intracellular signaling between tumorigenic and non-transformed cells, possibly as a reflection of different receptors involved in their recognition of control or glycated matrices. In post-mitotic mineralizing MC3T3E1 osteoblasts (3 weeks of culture), AGE-collagen was associated with a significant decrease in the number of surviving cells. However, since cells at this stage of development do not divide, the observed effect may be explained by an AGE-induced acceleration of necrosis or apoptosis in the mineralizing cultures. In fact, apoptosis is the endpoint of most recruited osteoblasts in vivo, and it has been shown that the products of different genes are involved in the growth, differentiation and apoptosis of osteoblast-like cells in culture [24].
We have previously described that in UMR106 and MC3T3E1 cells, soluble AGE-BSA initially induces an increase in cellular proliferation and differentiation, while a relatively long-term incubation of these cells with AGE-BSA elicits a significant decrease in both parameters [13]. In addition, we have also shown that soluble AGE-modified BSA alters the secretion of IGF-I and IGFBPs-one of the main growth factor systems involved in osteoblastic progression and apoptosis- in a manner that is dependent on the stage of osteoblastic development [15]. All of these AGE-BSA induced effects are most probably a consequence of the recognition of soluble AGE structures by their receptors, which we have previously described for both these cell lines [14]. On the other hand, the relatively different osteoblastic response to AGE-collagen which we report in the present study, may not only be a consequence of the signal transduction mechanisms induced by the occupancy of AGE-receptors, but also be mediated by integrin or growth factor receptors, which are intimately associated with the process of osteoblast growth and differentiation. In summary, both previous reports and our present study suggest a deleterious effect of AGE-modified proteins on osteoblastic development.
To unravel the mechanisms by which AGE-modified collagen may affect osteoblast development, we studied ROS formation and NOS expression in our culture model. The formation of ROS was inversely correlated with the AGE-collagen-induced changes in cellular proliferation. Thus, an increase in ROS formation was observed in parallel with an inhibition of osteosarcoma UMR106 cell growth. On the contrary, an AGE-mediated increase in MC3T3E1 proliferation was associated with a decrease in ROS formation. ROS have been found to be central to the early stages of the transduction-signaling mechanism initiated as a consequence of the recognition of AGE moieties by their specific receptor RAGE [25,26]. This signaling involves oxidation of p21ras by ROS, and subsequent activation of MAP kinases and NF-κB (a nuclear transcription factor). In this sense, the AGE-induced modulation of intracellular ROS levels may also be an important regulator in the expression of certain subsets of genes in osteoblastic cells.
We also assessed the effect of AGE-modified collagen on the expression of NOS. There are a number of studies that have linked an increase in the release of NO with hyperglycemia and diabetes [27-29]. In UMR106 as well as in MC3T3E1 osteoblasts we have previously shown that the release of NO is very low [30], and that eNOS and iNOS are enhanced after culturing the cells with vanadate, an agent that induces cytotoxicity in both cell lines. The present study provides evidence which suggests that eNOS and iNOS may be involved in the AGE-collagen-induced events observed in osteoblastic cells. Culturing UMR106 and MC3T3E1 osteoblasts for 24 hours on an AGE-modified type I collagen matrix, significantly enhanced the expression of both NOS isoforms. Previous studies have shown that pretreatment of macrophages with AGE-BSA provokes an increase in the levels of LPS- and IFN-γ-induced iNOS mRNA, an effect which is accompanied by an enhanced production of NO [6]. However, this effect is only observed in the presence of cytokines: the incubation of macrophages with AGE-BSA or unmodified BSA in the absence of cytokines does not produce any increase in the nitrite levels. To our knowledge, our present findings are the first to document an effect of AGE-modified collagen on the expression of non-cytokine stimulated iNOS in osteoblastic cells. We also detected an even greater increment in the expression of the constitutive isoform eNOS after culturing osteoblast-like cells on an AGE-modified collagenous matrix. These observations are in contrast with a previous report describing an inhibition of eNOS expression in retinal vascular endothelial cells after their incubation for 5 days with soluble glycated albumin [5]. The observed differences may be due to the cell system under study, to the time of AGE exposure, or more likely to the cell surface receptors involved in the AGE-modified protein effects and the signaling pathways evoked after their occupation. Nevertheless, the NO/NOS system very probably represents a relevant regulatory mechanism compromised in the cellular consequences of the accumulation of AGE on extracellular matrix molecules.
Conclusions
The present study shows that the non-enzymatic glycosylation of a type I collagen matrix induces alterations on bone cells in culture, which are dependent on (a) cell type and (b) the stage of osteoblastic development. In the osteosarcoma UMR106 cell line, AGE-collagen decreases cellular attachment, spreading, proliferation, and ALP activity. In pre-osteoblastic MC3T3E1 cells, AGE transiently increases cell proliferation and spreading. When these cells attain a mature osteoblastic phenotype, AGE-modified collagen inhibits the expression of ALP, the formation of calcified nodules of mineralization, and the long-term survival of cells. Overall, these results seem to indicate that AGE-collagen induces deleterious effects on osteoblastic growth and function. If these mechanisms were relevant in vivo, the accumulation of AGE on bone extracellular matrix could contribute to the decrease in osteoblastic function which has been described in diabetic patients with osteopenia. Furthermore, we have presented evidence for the involvement of ROS and NOS in the AGE-mediated effects on bone-producing cells.
Materials and Methods
Materials
Rat tail acid-soluble type I collagen was purchased from Sigma (St Louis, MO, USA). Dulbecco's modified Eagle's medium (DMEM), trypsine-EDTA and fetal bovine serum (FBS) were obtained from Gibco (Life Technology, Argentina). Tissue culture disposable material was from Nunc. Dihydrorhodamine 123 (DHR) was from Molecular Probes (Eugene, OR, USA). Polyclonal endothelial and inducible nitric oxide synthase (eNOS and iNOS) antibodies were from Research Biochemicals International (RBI, MA, USA). All other chemicals and reagents were obtained from commercial sources and were of analytical grade.
Preparation of AGE-modified type I collagen for cell matrices
Collagen was solubilized in sterile 0.02 N acetic acid (2.5 mg/ml) (pH 3.0), poured into plastic dishes (50 μg/cm2) and incubated at 37°C. Collagen formed a thin film during incubation [10]. The film was washed with phosphate-buffered saline (PBS) and further incubated with or without 100 mM ribose in PBS at 37°C for 3 weeks in sterile conditions [20]. Finally, the plates were extensively washed with DMEM to eliminate excess ribose, and the cells were plated for different experiments. Ribose was used instead of glucose as the glycating sugar to speed up nonenzymatic glycosylation. Kawano et al [20] have shown that the sugar content in ribose-glycated collagen increased 10-fold after 2 weeks of incubation, and that the lysine and arginine content of collagen decreased 42 and 26 % respectively. In the present study, AGE-formation on collagen was monitored by measuring AGE-specific fluorescence emission (356 nm excitation wavelength and 440 nm emission wavelength), as we have previously described [13]. After incubating 3 weeks with ribose, collagen contained more AGE-associated fluorescence than control collagen incubated with PBS alone (30 versus 7.5 relative fluorescence units / mg protein, for AGE-modified and control collagen respectively). In addition, the content of potentially reactive free amino groups, estimated according to Lavy et al with tri-nitro-benzene-sulphonic acid [31], decreased to 72 % of control in the case of glycated collagen.
Cell culture
UMR106 rat osteosarcoma-derived cells were grown in DMEM-10 % FBS and antibiotics. This cell line has been shown to conserve certain characteristics of differentiated osteoblastic phenotype [32]. After 5–7 days, cells were sub-cultured using trypsin / EDTA and replated on control and glycated collagen matrices to begin the experiments. MC3T3E1 osteoblastic mouse calvaria-derived cells were grown in 75 cm3 plastic flasks at 37°C in a humidified 5 % CO2 atmosphere in DMEM supplemented with 10 % FBS, 100 U/mL penicillin and 100 mg/L streptomycin, and passaged every 4–6 days. Previous studies have demonstrated that expression of osteoblastic markers begins after culturing these cells with medium supplemented by β-glycerol-phosphate and ascorbic acid [22,33]. When cells are cultured on collagen-coated plates, ALP is expressed after 5 days. In addition, mineralization is achieved with these cells after extending this culture to 20 days. However, the cells only undergo active replication during the first 5 days of incubation. To study the effect of AGE-collagen on cell growth in the different stages of MC3T3E1 osteoblastic development, cells were grown under the conditions that we have previously reported [14] for the times indicated in each experiment.
Cell adhesion assay
Osteoblast-like cells were plated on control or AGE-modified collagen-coated 24 well plates at a seeding density of 2.5 × 105 cells/ml. Cells were allowed to adhere for 1 hour. Each well was then washed with PBS, fixed with methanol for 5 minutes and stained with Giemsa for 10 minutes. Cells adhered to the collagen or AGE-collagen matrices were evaluated microscopically by counting several representative fields per well. A linear regression of the adhesion assay occurred between 1 × 105 and 1 × 106 cells/ml (y = 0.826 + 0.759 x, r= 0.993).
Cell proliferation assay
Cells were grown at 37°C on control or AGE-modified collagen-coated 24 well plates (1 × 104 cells/ml). UMR106 cell line was cultured for 24 hours, while MC3T3E1 cells were incubated for 24 hours (proliferating cells), one week (differentiated cells) or 3 weeks (mineralizing cells), changing the supplemented medium every two days. Cell proliferation was assessed by counting the cells as described above (for short periods of culture) or by trypsinization and cell counting with a haemocytometer (for 1 and 3 weeks of culture). Spreading was assessed by counting the proportion of stained cells which presented more than one corner, in several representative microscopic fields per well.
Cell differentiation. Alkaline phosphatase activity (ALP) determination
Cells were plated in 24-well plates coated with control or AGE-modified collagen, and were incubated at 37°C as described above for 24 hours (both cell lines), one week (MC3T3E1 cells) or three weeks (MC3T3E1 cells). At the end of these incubation periods, each cell monolayer was washed with PBS and solubilized in 0.1 % Triton X-100. The ALP specific activity (a marker of osteoblastic phenotype) was assessed as has been previously described [13,34].
Mineralization assay
To assess the mineralizing potential of MC3T3E1 cultures, cells were grown for three weeks on control or AGE-modified collagen matrices as described above. Cell monolayers were stained using the von Kossa method as we have previously described [15], and the number of calcified nodules per dish was counted using a microscope.
Determination of reactive oxygen species (ROS) formation
Intracellular ROS generation in osteoblast-like cells was measured by oxidation of DHR to rhodamine as we have previously described [35]. Osteoblasts were cultured on either control or AGE-modified collagen at 37°C for 48 hours. Media were replaced by phenol red-free DMEM with 10 μM DHR and the cells were further incubated for 4 hours. After washing with PBS, the monolayer was lysated in 0.1 % Triton X-100. The oxidized product present in the cell extract (rhodamine), was analysed by measuring fluorescence (excitation wavelength 495 nm, emission wavelength 532 nm).
Western blot analysis of nitric oxide synthases (NOS)
In order to evaluate the expression of NOS and the possible effect of AGE-collagen on these parameters, cells growing on control or AGE-modified collagen matrices were lysated in Laemmli buffer [36] and the protein content was evaluated by the method of Lowry [37]. The lysate was heated at 100°C for 3 min and 15 μg of protein subjected to 8% SDS-PAGE. The separated proteins were then transferred to nitrocellulose membranes. After washing and blocking, the membranes were incubated with anti-iNOS or eNOS polyclonal antibodies as we have previously described [30]. The intensity of the iNOS or eNOS specific bands was quantified by densitometry after scanning of the nitrocellulose paper. Images were analysed using the Scion-beta 2 program.
Statistical analysis
Three independent experiments were run for each experimental condition. Results are expressed as mean ± SEM. Statistical analysis of the data was performed by Student's t test.
Acknowledgments
Acknowledgements
This study was partly supported by grants from Universidad Nacional de La Plata, CICPBA, CONICET (PIP1044/98), Agencia Nacional de Promoción Científica (PICT 00375) and Ministerio de Salud y Acción Social de la Nación (Subsecretaria de Investigación y Tecnología, Beca Ramón Carrillo-Arturo Oñativia). AC is a member of the Carrera del Investigador, CICPBA, and SE is a member of the Carrera del Investigador, CONICET. DB is a fellow of the Colegio de Farmacéuticos de la Provincia de Buenos Aires, Argentina.
Contributor Information
Antonio D McCarthy, Email: mccarthy@biol.unlp.edu.ar.
Susana B Etcheverry, Email: etcheverry@biol.unlp.edu.ar.
Liliana Bruzzone, Email: lbruzzone@biol.unlp.edu.ar.
Gabriela Lettieri, Email: mglettieri@yahoo.com.
Daniel A Barrio, Email: dbarrio@biol.unlp.edu.ar.
Ana M Cortizo, Email: cortizo@biol.unlp.edu.ar.
References
- Vlassara H, Bucala R, Striker L. Pathogenic effects of advanced glycosylation: biochemical, biologic and clinical implications for diabetes and ageing. Lab Invest. 1994;70:138–151. [PubMed] [Google Scholar]
- Baynes JW, Thorpe SR. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes. 1999;48:1–9. doi: 10.2337/diabetes.48.1.1. [DOI] [PubMed] [Google Scholar]
- Thornalley PJ. Cell activation by glycated proteins. Age receptors, receptor recognition factors and functional classification of AGEs. Cell Mol Biol (Noisy-le-Grand) 1998;44:1013–1033. [PubMed] [Google Scholar]
- Chakravarthy U, Hayes RG, Stitt AW, McAuley E, Archer DB. Constitutive nitric oxide synthase expression in retinal vascular endothelial cells is supported by high glucose and advanced glycation end products. Diabetes. 1998;47:945–952. doi: 10.2337/diabetes.47.6.945. [DOI] [PubMed] [Google Scholar]
- Rojas A, Caveda L, Romay C, López E, Valdés S, Padrón J, Glarái L, Martínez 0, Delgado R. Effect of advanced glycation end products on the induction of nitric oxide synthase in murine macrophages. Biochem Biophys Res Comm. 1996;225:358–362. doi: 10.1006/bbrc.1996.1180. [DOI] [PubMed] [Google Scholar]
- Pieper GM. Acute amelloration of diabetes endothelial dysfunction with a derivative of the nitric oxide synthase cofactor, tetrahydrobiopterin. J Cardiovasc Pharm. 1997;29:8–11. doi: 10.1097/00005344-199701000-00002. [DOI] [PubMed] [Google Scholar]
- Reiser KM. Nonenzymatic glycation of collagen in aging and diabetes. Proc Soc Biol Med. 1998;218:23–37. doi: 10.3181/00379727-218-44264. [DOI] [PubMed] [Google Scholar]
- Tomasek JJ, Meyers SW, Basinger JB, Green DJ, Shew RL. Diabetic and age-related enhancement of collagen-linked fluorescence in cortical bones of rats. Life Sci. 1994;55:855–861. doi: 10.1016/0024-3205(94)90041-8. [DOI] [PubMed] [Google Scholar]
- Locatto ME, Abrazon H, Caferra D, Fernanadez MC, Alloatti R, Puche RC. Growth and development of bone mass in untreated alloxan diabetic rats. Effects of collagen glycosylation and parathyroid activity on bone turnover. Bone Miner. 1993;23:129–144. doi: 10.1016/s0169-6009(08)80049-9. [DOI] [PubMed] [Google Scholar]
- Katayama Y, Akatsu T, Yamamoto M, Kugai N, Nagata N. Role of nonenzymatic glycosylation of type I collagen in diabetic osteopenia. J Bone Miner Res. 1996;11:931–937. doi: 10.1002/jbmr.5650110709. [DOI] [PubMed] [Google Scholar]
- Levin ME, Boisseau BC, Avioli LVL. Effects of Diabetes mellitus on bone mass in juvenile and adult-onset Diabetes. New Engl J Med. 1976;294:241–245. doi: 10.1056/NEJM197601292940502. [DOI] [PubMed] [Google Scholar]
- Krakauer JC, McKenna MJ, Buderer NF, Rao DS, Whitehouse FW, Parfitt AM. Bone loss and bone turnover in diabetes mellitus. Diabetes. 1995;44:775–782. doi: 10.2337/diab.44.7.775. [DOI] [PubMed] [Google Scholar]
- McCarthy AD, Etcheverry SB, Bruzzone L, Cortizo AM. Effects of advanced glycation end-products on the proliferation and differentiation of osteoblast-like cells. Mol Cell Biochem. 1997;170:43–51. doi: 10.1023/A:1006816223292. [DOI] [PubMed] [Google Scholar]
- McCarthy AD, Etcheverry SB, Cortizo AM. Advanced glycation endproduct-specific receptors in rat and mouse osteoblast-like cells: regulation with stages of differentiation. Acta Diabetol. 1999;36:45–52. doi: 10.1007/s005920050144. [DOI] [PubMed] [Google Scholar]
- McCarthy AD, Etcheverry SB, Cortizo AM. Involvement of the insulin-like growth factor(IGF)system in the advanced glycation end product(AGE)-induced modulation of osteoblast development. Diabetes Res Clin Pract. 2000;50(Suppl1):S358. doi: 10.1016/S0168-8227(00)81219-X. [DOI] [Google Scholar]
- McCarthy TL, Centrella M, Canalis E. Insulin-like growth factor (IGF) and bone. Connect Tissue Res. 1989;20:277–282. doi: 10.3109/03008208909023897. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, Edelstein D, Brownlee M. The missing link: A single unifying mechanism for diabetic complications. Kidney Int. 2000;58(Supp 77):S26–S30. doi: 10.1046/j.1523-1755.2000.07705.x. [DOI] [PubMed] [Google Scholar]
- Ng KW, Romas E, Donnan L, Findlay DM. Bone biology. Bailliere's Clinical Endocrinology and Metabolism. 1997;11:1–21. doi: 10.1016/s0950-351x(97)80473-9. [DOI] [PubMed] [Google Scholar]
- Paul RG, Bailey AJ. The effect of advanced glycation end-product formation upon cell-matrix interactions. Int J Biochem Cell Biol. 1999;31:653–660. doi: 10.1016/S1357-2725(99)00023-0. [DOI] [PubMed] [Google Scholar]
- Kawano E, Takahashi S-I, Sakano Y, Fujimoto D. Non enzymatic glycosylation alters properties of collagen as a substratum for cells. Matrix. 1990;10:300–305. doi: 10.1016/s0934-8832(11)80185-x. [DOI] [PubMed] [Google Scholar]
- Celic S, Katayama Y, Chilco PJ, Martin TJ, Findlay DM. Type I collagen influence on gene expression in UMR106-06 osteoblast-like cells is inhibited by genistein. J Endocrinol. 1998;158:377–388. doi: 10.1677/joe.0.1580377. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Nakayama K, Matsumoto T. Differentiation and cell surface expression of transforming growth factor-β receptors are regulated by interaction with matrix collagen in murine osteoblastic cells. J Biol Chem. 1996;271:3938–3944. doi: 10.1074/jbc.271.7.3938. [DOI] [PubMed] [Google Scholar]
- Staatz ND, Fok KF, Zutter MM, Adams SF, Rodriguez BA, Santoro SA. Identification of a tetrapeptide recognition sequence for the alpha-2 beta-1 integrin in collagen. J Biol Chem. 1991;266:7363–7367. [PubMed] [Google Scholar]
- Lynch MP, Capparelli C, Stein JL, Stein GS, Lian JB. Apoptosis during bone-like tissue development in vitro. J Cell Biochem. 1998;68:31–49. doi: 10.1002/(SICI)1097-4644(19980101)68:1<31::AID-JCB4>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Lander HM, Tauras JM, Ogiste JS, Hori 0, Moss RA, Schmidt AM. Activation of the receptor for advanced glycation end products triggers a p21ras-dependent – mitogen-activated protein kinasce pathway regulated by oxidant stress. J Biol Chem. 1997;272:17810–17814. doi: 10.1074/jbc.272.28.17810. [DOI] [PubMed] [Google Scholar]
- Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zoc YS, Pinsky D, Stern D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their specific receptors/binding proteins. J Biol Chem. 1994;269:9889–9897. [PubMed] [Google Scholar]
- Eizirik DL, Leijerstam F. The inducible form of nitric oxide synthase (iNOS) in insulin-producing cells. Diabetes Metab. 1994;20:116–122. [PubMed] [Google Scholar]
- Sharma K, Danoff TM, Depiero A, Ziyadeh FN. Enhanced expression of inducible nitric oxide synthase in murine macrophages and glomerular mesangial cells by elevated glucose levels; possible mediation by protein kinasce C. Biochem Biophys Res Comm. 1995;207:80–88. doi: 10.1006/bbrc.1995.1156. [DOI] [PubMed] [Google Scholar]
- Schonfelder G, John M, Hopp H, Fuhr N, van Der Giet M, Paul M. Expression of inducible nitric oxide synthase in placenta of women with gestational diabetes. FASEB J. 1996;10:777–784. doi: 10.1096/fasebj.10.7.8635695. [DOI] [PubMed] [Google Scholar]
- Cortizo AM, Caporossi M, Lettieri G, Etcheverry SB. Vanadate-induced nitric oxide production: role in osteoblast growth and differentiation. Eur J Pharmacol. 2000;400:279–285. doi: 10.1016/S0014-2999(00)00356-3. [DOI] [PubMed] [Google Scholar]
- Lavy A, Brook GJ, Dankner G, Amotz AB, Aviram M. Enhanced in vitro oxidation of plasma lipoproteins derived from hypercholesterolemic patients. Metabolism. 1991;40:794–799. doi: 10.1016/0026-0495(91)90005-H. [DOI] [PubMed] [Google Scholar]
- Partridge NC, Alcorn D, Michelangeli VP, Ryan G, Martin TJ. Morphological and biochemical characterization of four clonal osteogenic sarcoma cell lines of rat origin. Cancer Res. 1983;43:4308–4312. [PubMed] [Google Scholar]
- Quarles LD, Yahay DA, Lever LW, Caton R, Wenstrup RJ. Distinct proliferative and differentiated stages of murine MC3T3E1 cells in culture: an in vitro model of osteoblast development. J Bone Miner Res. 1992;7:683–692. doi: 10.1002/jbmr.5650070613. [DOI] [PubMed] [Google Scholar]
- Bradford M. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Cortizo AM, Bruzone L, Molinuevo MS, Etcheverry SB. A possible role of oxidative stress in the vanadium-induced cytotoxicity in the MC3T3E1 osteoblast and UMR106 osteosarcoma cell lines. Toxicology. 2000;147:A89–99. doi: 10.1016/S0300-483X(00)00181-5. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural protein during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AJ, Randall RJ. Protein measurement with Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]