

Abstract
Oxygen homeostasis represents an essential organizing principle of metazoan evolution and biology. Hypoxia-inducible factor 1 (HIF-1) is a master regulator of transcriptional responses to changes in O2 concentration. HIF-1 is a heterodimer of HIF-1α and HIF-1β subunits. O2-dependent degradation of the HIF-1α subunit is mediated by prolyl hydroxylase, von Hippel-Lindau protein (VHL)/Elongin-C E3 ubiquitin ligase, and the proteasome. O2-independent degradation of HIF-1α is regulated by the competition of RACK1 and HSP90 for binding to HIF-1α. RACK1 binding results in the recruitment of the Elongin-C E3 ubiquitin ligase, leading to VHL-independent ubiquitination and degradation of HIF-1α. In this report, we show that calcineurin inhibits the ubiquitination and proteasomal degradation of HIF-1α. Calcineurin is a serine/threonine phosphatase that is activated by calcium and calmodulin. The phosphatase activity of calcineurin is required for its regulation of HIF-1α. RACK1 binds to the catalytic domain of calcineurin and is required for HIF-1α degradation induced by the calcineurin inhibitor cyclosporine A. Elongin-C and HIF-1α each bind to RACK1 and dimerization of RACK1 is required to recruit Elongin-C to HIF-1α. Phosphorylation of RACK1 promotes its dimerization and dephosphorylation by calcineurin inhibits dimerization. Serine 146 within the dimerization domain is phosphorylated and mutation of serine 146 impairs RACK1 dimerization and HIF-1α degradation. These results indicate that intracellular calcium levels can regulate HIF-1α expression by modulating calcineurin activity and RACK1 dimerization.
All metazoan organisms possess physiological systems to regulate the delivery and/or utilization of O2. Hypoxia-inducible factor 1 (HIF-1)3 is a critical mediator of adaptive responses to reduced oxygen availability in many developmental, physiological, and pathological contexts through its transcriptional regulation of genes that encode proteins required for tissue oxygen delivery and energy metabolism (1–4). HIF-1 is required for embryonic vascularization. Mouse embryos with complete HIF-1α deficiency or expression of a dominant negative form of HIF-1 in endothelial cells fail to develop proper vascularization and die at embryonic days 10–11 (4–6). HIF-1-dependent induction of angiogenic factors promotes vascularization and cardiac function in the initial compensated phase of left ventricular hypertrophy caused by pressure overload (7). In contrast to this adaptive response, HIF-1 contributes to the pathogenesis of hypoxia-induced pulmonary hypertension and right ventricular hypertrophy (8). HIF-1 also plays an important pathogenic role in many critical aspects of cancer biology, including cell immortalization, energy metabolism, vascularization, invasion, metastasis, and resistance to radiation and chemotherapy (9).
HIF-1 is a heterodimeric transcription factor composed of a HIF-1β subunit, which is constitutively expressed, and a HIF-1α subunit, the expression and transcriptional activity of which are regulated by the cellular O2 concentration (10). Both subunits contain basic helix-loop-helix domains that mediate dimerization and DNA binding, as well as a second dimerization motif referred to as the PAS domain (10, 11). HIF-1α protein accumulates in response to hypoxia and is rapidly degraded upon reoxygenation (10, 12, 13). Under hypoxic conditions, HIF-1α dimerizes with HIF-1β and translocates into the nucleus. HIF-1 binds to its target DNA sequence within hypoxia response elements, recruits the coactivators p300 and CBP, and activates transcription (14, 15). An increasing number of HIF-1 target genes are known, including those encoding metabolic enzymes, cytokines, growth factors, receptors, and other signaling proteins (1–4).
The HIF-1α transactivation domain located at the carboxyl terminus is regulated by the binding of factor inhibiting HIF-1 (16). Under non-hypoxic conditions, factor inhibiting HIF-1 hydroxylates asparagine 803 of the 826-amino acid human HIF-1α using O2 and α-ketoglutarate. Asparagine hydroxylation blocks the binding of HIF-1α to coactivators p300 and CBP, thus disrupting its ability to activate target gene transcription (17). O2-dependent regulation of HIF-1α protein degradation is mediated by a 200-amino acid oxygen-dependent degradation domain that is located in the middle of the protein (18). HIF-1α prolyl hydroxylases (PHDs) also utilize O2 and α-ketoglutarate as substrates to hydroxylate proline residues 402 and/or 564 of human HIF-1α (19, 20). Osteosarcoma protein-9 binds to both PHDs and HIF-1α, leading to increased prolyl hydroxylation and proteasomal degradation (21). The von Hippel-Lindau tumor suppressor protein (VHL) is the substrate recognition subunit of an E3 ubiquitin ligase complex. Undernon-hypoxic conditions, VHL binds to both hydroxylated HIF-1α and to Elongin-C, which recruits Elongin-B and other subunits of the E3 ubiquitin ligase that targets HIF-1α for ubiquitination and subsequent degradation by the 26 S proteasome (22–25). Renal carcinoma cells lacking functional VHL have high HIF-1α expression even under non-hypoxic conditions (22).
HIF-1α protein stability is also regulated through an O2-independent pathway involving the interaction of HSP90 and RACK1 with the PAS domain (26–28). HSP90 and RACK1 compete for binding to the PAS-A subdomain of HIF-1α. When the interaction of HSP90 and HIF-1α is disrupted by the HSP90 inhibitor 17-allylaminogeldanamycin, RACK1 binds to HIF-1α and recruits an Elongin-C E3 ubiquitin ligase complex to HIF-1α, leading to HIF-1α ubiquitination and degradation. Thus, VHL and RACK1 recruit Elongin-C E3 ubiquitin ligase complexes to HIF-1α via O2-dependent and O2-independent pathways, respectively.
Calcineurin (also known as protein phosphatase 2B) is a calcium/calmodulin-dependent and serine/threonine-specific protein phosphatase that is conserved in all eukaryotes. It is composed of a catalytic subunit A (CnA) and a regulatory subunit B (CnB) (29, 30). The best characterized substrate of calcineurin is nuclear factor of activated T-cells (NFAT). Upon activation by calcium, calcineurin dephosphorylates NFAT at multiple residues, leading to its nuclear translocation and activation of gene expression (31, 32). The immunosuppressants cyclosporine A and FK506 bind to calcineurin and inhibit its phosphatase activity (33, 34). Calcineurin is involved in a number of physiological and pathological processes including T-cell activation and cardiac hypertrophy (35). In this study, we report that calcineurin promotes HIF-1α expression by dephosphorylating RACK1, blocking RACK1 dimerization, and inhibiting RACK1-mediated HIF-1α degradation.
EXPERIMENTAL PROCEDURES
Tissue Culture
Human HEK293T embryonic kidney and RCC4 renal carcinoma cells were cultured as described (1, 13). Cells were maintained at 37 °C in a 5% CO2, 95% air incubator. For hypoxic exposures, cells were placed in a modulator incubator chamber (Billups-Rothenberg) flushed with 1% O2, 5% CO2/balance N2 and incubated at 37 °C.
Chemicals
Ionomycin and cyclosporine A were from Sigma. [γ-32P]ATP was from GE Healthcare.
Transfection Assays
Construction of the FLAG epitope-tagged HIF-1α (27) and hemagglutinin (HA) epitope-tagged calcineurin (51) expression vectors was previously described. HEK293T cells were seeded onto 48-well plates and transfected with plasmid DNA using FuGENE-6 (Roche). Reporter plasmids pSV-Renilla (1 ng) and p2.1 (10 ng), empty vector (EV), and/or expression vectors were cotransfected (14). Cells were lysed and luciferase activities were determined in a Wallac Victor3 multiwell luminescence reader (PerkinElmer Life Sciences), using the Dual Luciferase Reporter Assay System (Pro-mega). Three independent transfections were performed for each experiment. For immunoblot (IB) and immunoprecipitation (IP) assays, HEK293T cells were seeded onto 6- or 10-cm culture dishes. The following day, the cells were cotransfected with expression vectors using FuGENE-6. 24 h after transfection, the cells were exposed to 20 or 1% O2 for 4 h and lysed in RIPA buffer with protease inhibitor mixture (Roche). For RNA interference assay, the expression vectors encoding short hairpin RNA for RACK1 and scrambled negative control were described previously (27).
IB and IP Assays
Cells were lysed in RIPA buffer (IB assays) or buffer Na150NP0.1 (50 mM Tris-Cl (pH 7.5), 150 mM NaCl, 0.1% Nonidet P-40) with 1 mM dithiothreitol and protease inhibitor mixture. 2 μg of antibody was incubated with 1 mg of whole cell lysates (WCL) overnight at 4 °C. 20 μl of protein G-Sepharose (GE Healthcare) was added to the samples and incubated for 2 h at 4 °C to capture IgG. Beads were washed 3 times with buffer Na150NP0.1. Proteins were eluted in Laemmli sample buffer and analyzed by SDS-PAGE. Antibodies used in IB assays were: FLAG and HA (Sigma), RACK1 (BD Bioscience), glutathione S-transferase (GST) (GE Healthcare), His (Santa Cruz), β-actin (Santa Cruz), Myc (Roche), and HIF-1α and CnA (Novus Biologicals).
Ubiquitination Assay
Cells were separately transfected with vectors encoding: (i) FLAG-HIF-1α-(P402A/P564A), (ii) His-tagged ubiquitin, (iii) HA-CnA-(1–401), or (iv) empty vector. The cells were lysed in phosphate-buffered saline, 1 mM CaCl2, 1 mM MgCl2, 0.1% Tween 20. Lysates were mixed, incubated, and precipitated with anti-FLAG antibody, followed by immunoblot analysis using anti-His antibody. The levels of ubiquitinated FLAG-HIF-1α were quantified by densitometric analysis.
GST Pull-down Assays
GST fusion protein was purified as described (21). [35S]Methionine-labeled proteins were generated in reticulocyte lysates using a T7-coupled in vitro transcription/translation (IVTT) system (Promega). 3 μg of GST-RACK1 fusion protein and 10 μl of IVTT product were incubated in 500 μl of buffer Na150NP0.1 at 4 °C for 2 h. 20 μl of glutathione-Sepharose 4B beads were added to the samples and incubated at 4 °C for 1 h to capture the GST fusion proteins. After washing 3 times with 1 ml of buffer Na150NP0.1, the proteins were eluted in Laemmli sample buffer and analyzed by SDS-PAGE followed by anti-GST IB assay and autoradiography.
In Vitro RACK1 Dimerization Assay
3 μg of GST-RACK1 fusion proteins were incubated with 1 mg of HEK293T WCL transfected with EV or CnA for 30 min at 4 or 37 °C for the phosphorylation reaction. ATP, [γ-32P]ATP, ionomycin, or CaCl2 was added to the reaction as indicated. The reaction volume was brought up to 500 μl with buffer Na150NP0.1 and the samples were incubated for 2 h at 4 °C for dimerization. 20 μl of glutathione-Sepharose 4B beads were added to the samples and incubated at 4 °C for 1 h to capture the GST fusion proteins. After washing 3 times with 1 ml of buffer Na150NP0.1, the proteins were eluted in Laemmli buffer and analyzed by SDS-PAGE. IB assay was used to detect dimerization of GST-RACK1 and endogenous RACK1. The gel was also stained with Coomassie Blue (Simple Blue Stain, Invitrogen) and ProQ-Diamond (Pierce), followed by autoradiography.
Two-dimensional PAGE
After incubation with WCL, GST-RACK1 was captured by glutathione-Sepharose 4B beads and eluted in 30 mM Tris-Cl (pH 8.5), 7 M urea, 2 M thiourea, 4% CHAPS. The proteins were loaded onto a 7-cm, pH 4–7 IPG strip (GE Healthcare), which was rehydrated for 10 h actively at 50 V. Isoelectric focusing was performed for 10 kV-h. The IPG strip was reduced, alkylated, equilibrated in Laemmli buffer, and second dimension SDS-PAGE (4–12% NuPAGE BisTris precast gel, Invitrogen) was performed. The gel was stained with Coomassie Blue.
Statistical/Computational Analyses
Data are presented as mean ± S.D. Differences between experimental conditions were analyzed for statistical significance (p <0.05) by Student’s t test. The NetPhos program was accessed at www.cbs.dtu.dk/services/NetPhos/.
RESULTS
Calcineurin A Increases HIF-1α Expression under Hypoxic and Non-hypoxic Conditions
The calcineurin inhibitor cyclosporine A was previously shown to inhibit HIF-1α protein expression, but the mechanism was not determined. D’Angelo et al. (36) reported that cyclosporine A activated HIF-1α proline 564 hydroxylation, whereas Kong et al. (37) concluded that cyclosporine A inhibited HIF-1α in a VHL- and hydroxylation-independent manner. The involvement of calcineurin was not extensively studied in these reports. We hypothesized that the calcineurin catalytic subunit CnA induces increased HIF-1α protein expression, and thus, inhibition of calcineurin by cyclosporine A would reduce HIF-1α levels. To test this hypothesis, we transfected HEK293T cells with an expression vector encoding HA-tagged full-length CnA. Overexpression of CnA modestly increases calcineurin phosphatase activity, which can be further increased by stimuli that increase intracellular calcium levels (50). Co-transfection of expression vectors encoding FLAG-tagged HIF-1α and HA-CnA led to increased FLAG-HIF-1α protein levels in cells cultured under non-hypoxic (20% O2) or hypoxic (1% O2) conditions (Fig. 1A).
FIGURE 1. Calcineurin A increases HIF-1αprotein levels under both hypoxic and non-hypoxic conditions.

A, CnA increases HIF-1α protein levels in co-transfected cells. HEK293T cells were co-transfected with EV or expression vector encoding FLAG-HIF-1α or HA-CnA. The cells were exposed to 20 or 1% O2 for 4 h. WCL were subjected to IB assay to detect FLAG-HIF-1α, HA-CnA, and β-actin. B, calcineurin A increases transcription mediated by cotransfected HIF-1α. HEK293T cells were cotransfected with pSV-Renilla, in which Renilla luciferase is expressed from an SV40 promoter; p2.1, which contains a hypoxia response element upstream of an SV40 promoter and firefly luciferase coding sequences; and EV or vector encoding FLAG-HIF-1α or HA-CnA. The cells were exposed to 20 or 1% O2 for 24 h. Cells were lysed and the ratio of firefly: Renilla luciferase activity was determined. The results were normalized to those from cells transfected with EV and incubated at 20% O2 (mean ±S.D. shown). *, p <0.05 for indicated comparison. C, calcineurin A increases endogenous HIF-1α protein levels. HEK293T cells were transfected with EV or vector encoding HA-CnA. The cells were exposed to 20 or 1% O2 for 4 h. WCL were subjected to IB assay to detect HIF-1α, HA-CnA, and β-actin. D, calcineurin A increases transcription mediated by endogenous HIF-1α. HEK293T cells were co-transfected with pSV-Renilla, p2.1, and EV or HA-CnA. The cells were exposed to 20 or 1% O2 for 24 h. Cells were lysed and the ratio of firefly: Renilla luciferase activity was determined. The results were normalized to those from cells transfected with EV and incubated at 20% O2 (mean ± S.D. shown). *, p <0.05 for indicated comparison. Inset, luciferase activity at 20% O2.
To determine whether HIF-1-dependent transcription was induced by CnA, cells were co-transfected with reporter plasmid p2.1, in which expression of firefly luciferase was driven by a HIF-1-dependent hypoxia response element upstream of an SV40 promoter; control reporter pSV-Renilla, in which expression of Renilla luciferase was driven by the SV40 promoter alone; and expression vector encoding HA-CnA and/or FLAG-HIF-1α. HIF-1-dependent reporter gene transcription mediated by FLAG-HIF-1α was increased by HA-CnA in cells incubated at 20 or 1% O2 (Fig. 1B).
We next investigated whether endogenous HIF-1α protein expression and endogenous HIF-1 transcriptional activity were also increased by HA-CnA under hypoxic conditions. The effect of HA-CnA on the levels of HIF-1α protein in cells cultured at 1% O2 was striking (Fig. 1C, compare lanes 3 and 4). HA-CnA had a similar effect on transcription of the HIF-1-dependent reporter gene in cells cultured at 1% O2 (Fig. 1D). Although endogenous HIF-1α protein levels in non-hypoxic cells were below the limits of detection by IB assay (Fig. 1C), in the more sensitive luciferase reporter gene assay, HA-CnA was shown to significantly increase HIF-1-dependent gene transcription 2-fold under non-hypoxic conditions (Fig. 1D, inset). Taken together, the results presented in Fig. 1 suggest that overexpression of CnA is sufficient to induce HIF-1α protein expression and HIF-1-dependent transcription in transiently transfected HEK293T cells.
Calcineurin activity requires expression of both calcineurin A and B subunits and the binding of Ca2+/calmodulin. We compared the effect of transiently co-transfecting various calcineurin expression vectors (51) into 293 cells along with the HIF-1-dependent reporter plasmid p2.1 or the reporter plasmid pNFAT-Luc, the transcription of which is dependent upon calcineurin-mediated dephosphorylation of NFAT. In each condition tested, the responses of the HIF-1-dependent reporter p2.1 were remarkably similar to those of the calcineurin-dependent reporter pNFAT-Luc (Fig. 2A). These results indicate that endogenous CnB levels in HEK293T cells were sufficient to form heterodimers with ectopically expressed CnA or its truncated, constitutively active form to reconstitute calcineurin activity; and that transient transfection was a sufficient stimulus for calcineurin activation.
FIGURE 2. Calcineurin has similar effects on HIF-1 and NFAT-dependent transcription.
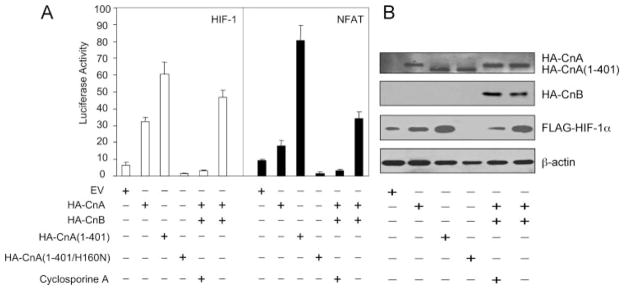
A, effect of calcineurin expression vectors on HIF-1 and NFAT-dependent transcription. HEK293T cells were co-transfected with control reporter plasmid pSV-Renilla and either HIF-1-dependent reporter plasmid p2.1 (left panel) or NFAT-dependent reporter plasmid pNFAT-Luc (right panel). Cells were co-transfected with FLAG-HIF-1α vector +additional expression vector(s) +treatment with 2.5 μM cyclosporine A as indicated. B, effect of HA-tagged calcineurin subunit expression on the levels of FLAG-HIF-1α and β-actin protein. The cells were transfected as in panel A except for the absence of reporter plasmids. IB assays were performed using aliquots of WCL prepared 24 h after transfection.
Transactivation of p2.1 by FLAG-HIF-1α was stimulated by overexpression of HA-CnA alone (column 1) or in combination with CnB (column 6) and by expression of a constitutively active form of CnA consisting of residues 1–401 (column 3). In contrast, reporter activity was eliminated by a dominant negative form of CnA containing the missense mutation H160N (column 4) and by cyclosporine A treatment (column 5). The effects of co-transfected CnA expression vectors on FLAG-HIF-1α protein levels (Fig. 2B) were in complete agreement with the results of the reporter assay (Fig. 2A), indicating that calcineurin activity stimulates HIF-1 transcriptional activity by increasing HIF-1α protein levels.
Calcineurin Phosphatase Activity Regulates HIF-1α
To determine the effect of inducing endogenous calcineurin, cells were treated with the calcium ionophore ionomycin, which induces calcineurin phosphatase activity by increasing the binding of calcium to calmodulin, which in turn binds to the carboxyl-terminal region of CnA, thereby disrupting the interaction of the autoinhibitory and catalytic domains; in addition, binding of calcium to CnB also occurs during calcineurin activation (31). Ionomycin increased FLAG-HIF-1α protein levels in a dose-dependent manner, whereas the endogenous CnA protein levels were unchanged (Fig. 3A). The data suggest that calcineurin phosphatase activity, rather than CnA protein levels, are the determining factor for HIF-1α regulation. In further support of this conclusion, cyclosporine A, which selectively inhibits calcineurin phosphatase activity, blocked the increase in HIF-1α levels that was induced by HA-CnA without affecting HA-CnA protein levels (Fig. 3B).
FIGURE 3. Calcineurin phosphatase activity regulates HIF-1α.

A, ionomycin increases HIF-1α protein levels. HEK293T cells were transfected with FLAG-HIF-1α expression vector. The cells were treated with vehicle or ionomycin at the indicated concentration for 6 h. WCL were subjected to IB assay to detect FLAG-HIF-1α, CnA, and β-actin. B, cyclosporine A decreases HIF-1α protein expression. HEK293T cells were co-transfected with FLAG-HIF-1α and EV or HA-CnA expression vector. The cells were treated with 2.5 μM cyclosporine A for 6 h. WCL were subjected to IB assay to detect FLAG-HIF-1α, HA-CnA, and β-actin.
PHD/VHL-independent and Ubiquitin/Proteasome-dependent Regulation of HIF-1α by CnA
The increased HIF-1α protein levels that were induced by calcineurin may result from increased synthesis or decreased degradation. The proteasome inhibitor MG132 equalized FLAG-HIF-1α levels in cells cotransfected with HA-CnA or empty vector (Fig. 4A, compare lanes 1 and 2 to lanes 3 and 4). These data suggest that CnA regulates HIF-1α levels by inhibiting proteasomal degradation. The proline to alanine mutation of residues 402 and 564 of human HIF-1α renders the protein resistant to PHD-dependent hydroxylation and subsequent VHL-dependent ubiquitination and degradation (21). HA-CnA also increased the levels of FLAG-HIF-1α-(P402A/P564A) (Fig. 4A, lanes 5 and 6), an effect that was mimicked by MG132 treatment in the absence of HA-CnA (lane 7). HA-CnA also potentiated the activation of reporter gene transcription by wild type or mutant FLAG-HIF-1α (Fig. 4B). The finding that CnA had similar effects on the wild type and mutant forms of FLAG-HIF-1α indicates that CnA regulates HIF-1α independently of the PHD pathway.
FIGURE 4. Calcineurin A regulates HIF-1α in a PHD/VHL-independent, ubiquitin/proteasome-dependent manner.

A, calcineurin A increases the levels of both wild type and hydroxylation resistant mutant forms of HIF-1α protein by inhibiting their degradation in a proteasome dependent manner. HEK293T cells were co-transfected with EV, wild-type FLAG-HIF-1α, FLAG-HIF-1α-(P420A/P564A), or HA-CnA. The cells were treated with vehicle or 10 μM MG132 for 4 h. WCL were subjected to IB assay to detect FLAG-HIF-1α, HA-CnA, and β-actin. B, calcineurin A increases transcription mediated by both wild type and hydroxylation resistant mutant forms of HIF-1α. HEK293T cells were co-transfected with pSV-Renilla, p2.1, wild-type FLAG-HIF-1α or FLAG-HIF-1α(P420A/P564A), and EV or HA-CnA. Cells were lysed and the ratio of firefly: Renilla luciferase activity was determined. The results were normalized to those from cells transfected with EV and incubated at 20% O2 (mean ± S.D. shown). *, p < 0.05 for indicated comparison. C, calcineurin increases HIF-1α protein expression in a VHL independent manner. VHL-deficient RCC4 cells were treated with vehicle or the indicated concentration of ionomycin or cyclosporine A for 16 h. WCL were subjected to IB assay to detect HIF-1α, CnA, and β-actin. D, calcineurin A decreases the ubiquitination of HIF-1α. Aliquots of WCL containing FLAG-HIF-1α-(P402A/P564A) were incubated with WCL containing His-tagged ubiquitin with or without HA-CnA. IP of WCL was performed with anti-FLAG antibody. Anti-His and anti-FLAG antibodies were used to detect ubiquitinated and total FLAG-HIF-1α after FLAG IP.
RCC4 renal carcinoma cells do not express functional VHL and have high endogenous HIF-1α levels even under non-hypoxic conditions (22). Ionomycin dose-dependently increased HIF-1α protein levels in RCC4 cells, whereas cyclosporine A decreased HIF-1α protein levels (Fig. 4C). The data indicate that CnA regulates HIF-1α independently of VHL.
To determine whether CnA affected the ubiquitination of HIF-1α, cells were transfected with expression vectors encoding FLAG-HIF-1α-(P402A/P564A), His-tagged ubiquitin, and either empty vector or vector encoding HA-CnA-(1–401). Analysis of anti-FLAG immunoprecipitates for ubiquitination using anti-His antibodies revealed that HA-CnA-(1–401) decreased the ubiquitination of HIF-1α-(P402A/P564A) 2-fold (Fig. 4D). Taken together, the data presented in Fig. 4 demonstrate that CnA inhibits HIF-1α ubiquitination and proteasomal degradation by an O2/PHD/VHL-independent pathway.
RACK1 Binds to CnA and Is Required for Cyclosporine A-induced HIF-1α Degradation
Previously we identified RACK1 as a critical factor in an O2/PHD/VHL-independent HIF-1α degradation pathway. RACK1 competes with HSP90 for binding to the PAS domain of HIF-1α. RACK1 recruits an Elongin-C-containing E3 ubiquitin ligase complex to HIF-1α, thereby increasing HIF-1α ubiquitination and proteasomal degradation (27). RACK1 was reported to bind to PP2A, a protein phosphatase that has high sequence similarity to CnA (38). We therefore investigated the interaction of RACK1 and CnA. Purified GST-RACK1 bound to in vitro translated full-length CnA and amino acids 1– 401, which encompass the phosphatase catalytic domain of CnA (Fig. 5A). HA-CnA co-immunoprecipitated with endogenous RACK1 in HEK293T cells (Fig. 5B).
FIGURE 5. RACK1 binds to calcineurin A and is required for cyclosporine A-induced HIF-1α degradation.

A, RACK1 binds to both full-length calcineurin A and the isolated catalytic domain of calcineurin A in vitro. Purified GST or GST-RACK1 was incubated with in vitro transcribed, translated, and 35S-labeled full-length or the amino-terminal 401 amino acids of calcineurin A (IVTT-CnA(FL) or IVTT-CnA(1– 401), respectively); captured on glutathione (GSH)-Sepharose beads; and analyzed by SDS-PAGE followed by autoradiography (top and middle panels) and IB assay with anti-GST (bottom panel). An aliquot of the IVTT product was also applied directly to the gel (Input). B, co-IP of endogenous RACK1 and transfected HA-CnA. WCL were prepared from HEK293T cells transfected with EV or HA-CnA. IP was performed using anti-HA antibody. WCL and IP products were subject to IB assays to detect HA-CnA, RACK1, and β-actin. C, RACK1 WD4 domain binds to full-length and the amino-terminal catalytic domain of calcineurin A in vitro. Purified GST or GST-RACK1-WD4 were incubated with IVTT-CnA(FL) or IVTT-CnA(1– 401), captured on GSH-Sepharose beads, and analyzed by SDS-PAGE followed by autoradiography (top and middle panels) and IB assay with anti-GST (bottom panel). An aliquot of the IVTT product was also applied directly to the gel (Input). D, RACK1 knockdown blocks HIF-1α degradation induced by cyclosporine A. HEK293T cells were co-transfected with FLAG-HIF-1α and short hairpin RNA directed against RACK1 mRNA (shRNA-RACK1) or a scrambled negative control (shRNA-SNC). Cells were treated with vehicle or 2.5 μM cyclosporine A for 6 h. WCL were subjected to IB assays to detect FLAG-HIF-1α, CnA, β-actin, and RACK1. FLAG-HIF-1α levels were quantified by densitometric analysis (band intensity).
RACK1 consists of seven tryptophan-aspartate-rich WD40 domains and the fourth of these domains (WD4) mediates homodimerization. The isolated WD4 domain bound to both full-length CnA and amino acids 1–401 (Fig. 5C). Based on this finding of physical interaction between RACK1 and CnA, we investigated whether RACK1 was required for the reduction in HIF-1α protein levels in cells treated with the calcineurin inhibitor cyclosporine A. RACK1 knockdown by RNA interference increased HIF-1α protein levels by 2-fold and blocked the degradation of HIF-1α in cyclosporine-treated cells (Fig. 5D). The data in Fig. 5 demonstrate that RACK1 binds to CnA both in vitro and in human cells and is required for cyclosporine A-induced HIF-1α degradation.
CnA Dephosphorylates RACK1 and Blocks RACK1 Dimerization
We have previously shown that RACK1 recruits Elongin-C to HIF-1α and that they both bind to the 48-amino acid WD6 domain of RACK1 (27). Two models were proposed for the mechanism by which RACK1 promotes the ubiquitination of HIF-1α: HIF-1α and Elongin-C may bind to two separate subdomains within the WD6 domain of monomeric RACK1; alternatively, RACK1 may form a homodimer in which one monomer binds to HIF-1α and the other binds to Elongin-C. RACK1 was shown to dimerize within its WD4 domain both in vitro and in vivo (39). We constructed a T7-tagged RACK1 expression vector, which only contained the carboxyl-terminal WD5–7 domains of RACK1. If the first model were correct, WD5–7 would have the same effect as full-length RACK1 to degrade HIF-1α. If the second model were correct, WD5–7 would not be able to degrade HIF-1α because it lacks the WD4 dimerization domain. The data in Fig. 6A support the dimerization model because RACK1-WD5–7 failed to induce HIF-1α degradation.
FIGURE 6. Dephosphorylation of RACK1 by calcineurin A blocks dimerization and HIF-1α degradation.

A, RACK1 carboxyl-terminal domain is not sufficient to degrade HIF-1α. HEK293T cells were cotransfected with EV, FLAG-HIF-1α, full-length T7-RACK1-WD1–7, or carboxyl-terminal T7-RACK1-WD5–7. WCL were subjected to IB assay to detect FLAG-HIF-1α, β-actin, and RACK1. B, full-length GST-RACK1-WD1–7, but not GST-RACK1-WD56, can pull down endogenous RACK1 through dimerization only when incubated at 37 °C. GST-RACK1-WD1–7 or GST-RACK1-WD56 were incubated with WCL from HEK293T cells at 4 or 37 °C for 30 min. GST fusion proteins were captured on GSH-Sepharose beads and analyzed by SDS-PAGE followed by IB assays to detect GST-RACK1 with anti-GST antibody (top panel) and endogenous RACK1 with anti-RACK1 antibody (bottom panel). C, pI of GST-RACK1 was reduced upon incubation with WCL at 37 °C in the presence of ATP. GST-RACK1 was incubated with WCL from HEK293T cells at 4 or 37 °C with 1 mM ATP. GST-RACK1 was captured on GSH-Sepharose beads and analyzed by two-dimensional PAGE followed by Coomassie Blue staining. D, regulation of RACK1 dimerization by ATP and calcium. GST-RACK1 was incubated with WCL from HEK293T cells at 37 °C with 1 mM ATP or 10 μM ionomycin. GST-RACK1 was captured on GSH-Sepharose beads. The pull-down products or WCL were analyzed by IB assays using anti-GST or anti-RACK1 antibody. E, calcineurin A and calcium decrease RACK1 phosphorylation and inhibit RACK1 dimerization. GST-RACK1 was incubated with WCL prepared from HEK293T cells transfected with EV or HA-CnA expression vector, at 37 °C with or without 1 mM CaCl2. GST-RACK1 was captured on GSH-Sepharose beads. The pull-down products were analyzed by IB assays to detect GST-RACK1 with anti-GST (top panel) or RACK1 with anti-RACK1 (second panel from bottom) antibody and by SDS-PAGE followed by ProQ-Diamond phosphoprotein stain (bottom panel). WCL were subject to IB assay to detect HA-CnA (second panel from top) and RACK1 (middle panel). F, HEK293T cells were co-transfected with FLAG-HIF-1α and Myc-Elongin-C. Cells were treated with vehicle or 1 μM ionomycin in the presence of 10 μM MG132 for 6 h before WCL were collected. IP was performed with anti-FLAG antibody. The WCL and IP products were subjected to IB assays to detect FLAG-HIF-1α, CnA, β-actin, and Myc-Elongin-C.
RACK1 dimerization is based on non-covalent interactions, which are disrupted by the denaturing conditions used for SDS-PAGE. To study RACK1 dimerization, we developed an in vitro assay to measure the ability of purified GST-RACK1 to pull down endogenous RACK1 from cell lysates. Because GST-RACK1 and RACK1 have different molecular masses, we could analyze them individually by immunoblot assay. GST did not bind to RACK1 (Fig. 5C and data not shown), indicating that GST-RACK1 pulled down endogenous RACK1 by dimerization. This conclusion was supported by the finding that GST-RACK1-WD56, which lacked the WD4 dimerization domain, did not pull down endogenous RACK1 (Fig. 6B, lanes 1 and 2). Full-length GST-RACK1 (WD1–7) purified from E. coli pulled down endogenous RACK1 when incubated with cell lysates at 37 °C but not at 4 °C (Fig. 6B, lanes 3 and 4).
These results suggested that an enzymatic reaction, which occurs at 37 °C but not at 4 °C, is required to modify GST-RACK1 prior to dimerization with endogenous RACK1. Two-dimensional gel electrophoresis revealed that incubation of GST-RACK1 with cell lysates and ATP at 37 °C reduced the pI of the protein without changing its apparent molecular mass (Fig. 6C), which is consistent with a post-translational modification, such as phosphorylation, that introduces a negative charge. The ability of GST-RACK1 to pull down endogenous RACK1 through dimerization was promoted in the presence of ATP and inhibited in the presence of ionomycin (Fig. 6D).
The data suggested that phosphorylation promotes RACK1 dimerization and that ionomycin activates a calcium-dependent phosphatase such as calcineurin, which dephosphorylates RACK1, and inhibits RACK1 dimerization. In support of these conclusions, RACK1 dimerization was reduced in the presence of overexpressed HA-CnA and/or added calcium (Fig. 6E). ProQ-Diamond phosphoprotein staining revealed that phosphorylation of GST-RACK1 was also reduced by HA-CnA or calcium. Thus, GST-RACK1 phosphorylation correlated with competence for dimerization. The data suggest that phosphorylation of RACK1, perhaps within the WD4 dimerization domain, is required for the RACK1 dimer formation and that calcineurin inhibits this process by dephosphorylating RACK1. Furthermore, activation of calcineurin by ionomycin decreased the interaction between HIF-1α and Elongin-C (Fig. 6F). Thus, calcineurin blocks RACK1 dimerization, which is required for HIF-1α/Elongin-C interaction.
Serine 146 Is Phosphorylated and Is Required for RACK1 Dimerization and HIF-1α Degradation
We used the NetPhos phosphorylation prediction software (40) to identify potential phosphorylated residues within the WD4 dimerization domain of RACK1. Scores >0.5 indicate residues that are possibly phosphorylated, with 1.0 representing the highest possible score. Serine 146 (Ser146) and Ser157 were assigned scores of 0.99 and 0.97, respectively (Fig. 7A), and were therefore strong candidate sites for phosphorylation. We investigated the effect of serine to glycine substitution at amino acid residue 146 of RACK1. In HEK293T cells, the T7 epitope-tagged RACK1(S146G) mutant protein demonstrated impaired ability to reduce the levels of co-expressed FLAG-HIF-1α compared with wild type T7-RACK1 or T7-RACK1 with other serine mutations (Fig. 7B). Remarkably, GST-RACK1(S146G) was also less effective in pulling down endogenous RACK1 compared with wild type GST-RACK1 in the dimerization assay (Fig. 7C). Furthermore, when GST-RACK1 was incubated with cell lysates in the presence of [γ-32P]ATP, the incorporation of radioactive ATP was decreased in the S146G mutant compared with the wild type protein (Fig. 7D). The data in Fig. 7 indicate that Ser146 is a phosphorylation site and that mutation of this residue results in reduced RACK1 phosphorylation, reduced RACK1 dimerization, and reduced RACK1-mediated degradation of HIF-1α.
FIGURE 7. Serine 146 is a target for phosphorylation and is required for efficient RACK1 dimerization and HIF-1α degradation.

A, identification of candidate phosphorylation sites in the WD4 domain of RACK1 based on scores assigned by NetPhos. B, serine 146 is critical for RACK1-mediated HIF-1α degradation. HEK293T cells were co-transfected with vectors encoding FLAG-HIF-1α and wild type or the indicated mutant form of T7-RACK1. WCL were subjected to IB assays to detect FLAG-HIF-1α, RACK1, and β-actin. C, effect of serine 146 mutation on RACK1 dimerization. The wild type (WT) or S146G mutant form of GST-RACK1 was incubated with WCL at 4 or 37 °C. GST fusion proteins were captured on GSH-Sepharose beads. The pull-down products were analyzed by IB assays using anti-GST (middle panel) or anti-RACK1 (bottom panel) antibody. Endogenous RACK1 in WCL was also analyzed by IB assays (top panel). D, effect of serine 146 mutation on RACK1 phosphorylation. The WT or S146G mutant form of GST-RACK1 was incubated with WCL at 4 or 37 °C in the presence of 0.3 mCi of [γ-32P]ATP. GST fusion proteins were captured on GSH-Sepharose beads. The pull-down products were subjected to SDS-PAGE followed by Coomassie Blue staining and autoradiography.
DISCUSSION
In this study, we further investigated the previously reported observation that cyclosporine A inhibits HIF-1α protein expression (36, 37). We have demonstrated that promoting calcineurin phosphatase activity through ionomycin treatment or overexpression of CnA is sufficient to increase HIF-1α protein levels and HIF-1 transcriptional activity. Calcineurin promotes HIF-1α expression by inhibiting its proteasomal degradation through a ubiquitin-dependent and O2/PHD/VHL-independent pathway.
We investigated the involvement of RACK1 in this process based on our previous discovery that RACK1 is an important mediator of O2/PHD/VHL-independent HIF-1α degradation (27) and a recent report that RACK1 interacts with PP2A, a protein phosphatase with high sequence similarity to calcineurin (38). We found that RACK1 binds to CnA both in vitro and in human cells. Furthermore, RACK1 is required for cyclosporine A-induced degradation of HIF-1α protein. These results provide a molecular basis for the recent demonstration that cyclosporine administration blunted the development of hypoxia-induced pulmonary hypertension (41), which has previously been shown to involve HIF-1 (8).
We focused on the effect of calcineurin on RACK1 dimerization after determining that RACK1 dimer formation is required for HIF-1α degradation. Enhancing calcineurin activity through calcium addition and/or CnA overexpression decreased RACK1 phosphorylation and dimerization, leading to reduced Elongin-C E3 ubiquitin ligase recruitment and subsequent HIF-1α degradation (Fig. 8). We identified Ser146 in the WD4 dimerization domain as a phosphorylation site and demonstrated that mutation of this residue results in impaired RACK1 dimerization and impaired ability to induce HIF-1α degradation.
FIGURE 8. Calcineurin dephosphorylates RACK1, inhibits RACK1 dimerization, and blocks RACK1-mediated ubiquitination and degradation of HIF-1α.

Phosphorylation of the WD4 domain promotes RACK1 dimerization. The WD6 domain of each monomer binds to either HIF-1α or Elongin-C. HIF-1α is ubiquitinated by the E3 ubiquitin ligase complex (composed of Elongin-B (B), Cullin-2 (CUL2), ring box protein 1 (RBX1), and an E2 ubiquitin-conjugating enzyme (E2), which is associated with Elongin-C. Ubiquitination (Ubi) of HIF-1α targets the protein for degradation by the 26 S proteasome. The calcium-dependent phosphatase calcineurin dephosphorylates RACK1, inhibits RACK1 dimerization, and thereby reduces the association of HIF-1α with the Elongin-C E3 ubiquitin ligase complex. As a result, HIF-1α is protected from ubiquitination and proteasomal degradation. Cyclosporin A inhibits calcineurin activity, thereby increasing RACK1 dimerization and HIF-1α degradation.
Our data indicate that phosphorylated Ser146 is likely to be the direct target for calcineurin-mediated dephosphorylation. An antibody that specifically recognizes RACK1 phosphorylated at Ser146 would provide a means to definitively prove this hypothesis. Further studies are required to determine which kinase is responsible for the phosphorylation of RACK1 that is required for its dimerization. Although the experimental approach to answer this question is beyond the scope of the current study, we note that Ser146 is present within a context that matches the consensus sequence (SXXE/D) for phosphorylation by casein kinase II.
RACK1 dimer-mediated HIF-1α degradation requires that each monomer bind to either HIF-1α or Elongin-C. This strategy may seem inefficient because by chance only half of the RACK1 dimers will co-recruit HIF-1α and Elongin-C, whereas the other half will bind two molecules of HIF-1α or Elongin-C. However, this process provides a mechanism for regulation by controlling RACK1 dimer formation. In this report, we have shown that calcineurin inhibits RACK1 dimerization, thereby linking the regulation of HIF-1α expression to intracellular calcium levels and calcium-activated signal transduction cascades. These findings complement previous studies, which demonstrated that HIF-1α translation is calcium-regulated by protein kinase C-α signaling (42) and HIF-1α transactivation function is regulated by calcium/calmodulin kinase-mediated phosphorylation of p300 (43). In some cell types, hypoxia induces increased intracellular calcium levels (42), which may amplify the induction of HIF-1α in these cells.
RACK1 was originally identified as an anchoring protein for activated protein kinase C (44). However, RACK1 is now recognized as a multifunctional scaffold protein that plays an important role in diverse biological processes including intra-cellular signal transduction (45) and assembly of the 80 S ribosome from 40 S and 60 S subunits (46). Some processes that are mediated by RACK1, such as HIF-1α regulation, require RACK1 dimerization, whereas other processes, such as ribosome assembly, involve RACK1 monomers (47). For most of the processes in which RACK1 has been implicated, it is not known whether dimers or monomers are utilized. Calcium-activated calcineurin signaling is likely to regulate many physiological pathways that involve RACK1 dimerization. Furthermore, RACK1 dimers increase calcium release by binding to inositol 1,4,5-triphosphate receptors (48). Thus, a feedback loop involving RACK1 dimerization and calcium influx may serve to tightly control the RACK1 dimer:monomer equilibrium. HIF-1 may contribute to the complexity of this regulatory circuit by mediating transcriptional activation of the TRPC1 and TRPC6 genes, which encode store-operated calcium channels (49). Calcineurin plays a key role in physiological responses such as T lymphocyte activation and cardiac hypertrophy. Further studies are required to determine whether these responses are dependent upon modulation of the RACK1-HIF-1α regulatory pathway by calcineurin.
Acknowledgments
We thank Karen Padgett (Novus Biologicals Inc.) for providing CnA antibody.
Footnotes
This work was supported by National Institutes of Health NHLBI Grant N01-HV28180.
The abbreviations used are: HIF-1, hypoxia-inducible factor 1; RACK, receptor for activated C kinase; PHD, prolyl hydroxylase domain protein; VHL, von Hippel-Lindau protein; HSP, heat shock protein; CBP, CREB-binding protein; CREB, cyclic AMP response element-binding protein; PAS, PER-ARNT-SIM homology domain; PAS-A, subdomain A of PAS; CnA, calcineurin A; NFAT, nuclear factor of activated T cells; GST, glutathione S-transferase; IVTT, in vitro transcription/translation; WD, tryptophanaspartate-rich; IB, immunoblot; EV, empty vector; WCL, whole cell lysates; SV40, simian virus 40; IP, immunoprecipitation; sh, short hairpin; HA, hemagglutinin; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid; BisTris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxy-methyl)propane-1,3-diol.
References
- 1.Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Mol Cell Biol. 2003;23:9361–9374. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 3.Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J, Gleadle JM. J Biol Chem. 2006;281:15215–15226. doi: 10.1074/jbc.M511408200. [DOI] [PubMed] [Google Scholar]
- 4.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryan HE, Lo J, Johnson RS. EMBO J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Licht AH, Muller-Holtkamp F, Flamme I, Breier G. Blood. 2006;107:584–590. doi: 10.1182/blood-2005-07-3033. [DOI] [PubMed] [Google Scholar]
- 7.Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T, Hamada H, Tomita S, Molkentin JD, Zou Y, Komuro I. Nature. 2007;446:444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- 8.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT, Semenza GL. J Clin Investig. 1999;103:691–696. doi: 10.1172/JCI5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Semenza GL. Expert Opin Ther Targets. 2006;10:267–280. doi: 10.1517/14728222.10.2.267. [DOI] [PubMed] [Google Scholar]
- 10.Wang GL, Jiang BH, Rue EA, Semenza GL. Proc Natl Acad Sci U S A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang BH, Rue E, Wang GL, Roe R, Semenza GL. J Biol Chem. 1996;271:17771–17778. doi: 10.1074/jbc.271.30.17771. [DOI] [PubMed] [Google Scholar]
- 12.Jiang BH, Semenza GL, Bauer C, Marti HH. Am J Physiol. 1996;271:C1172–C1180. doi: 10.1152/ajpcell.1996.271.4.C1172. [DOI] [PubMed] [Google Scholar]
- 13.Salceda S, Caro J. J Biol Chem. 1997;272:22642–22647. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- 14.Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. J Biol Chem. 1996;271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- 15.Ebert BL, Bunn HF. Mol Cell Biol. 1998;18:4089–4096. doi: 10.1128/mcb.18.7.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahon PC, Hirota K, Semenza GL. Genes Dev. 2001;15:2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. Genes Dev. 2002;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang LE, Gu J, Schau M, Bunn HF. Proc Natl Acad Sci U S A. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruick RK, McKnight SL. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 20.Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 21.Baek JH, Mahon PC, Oh J, Kelly B, Krishnamachary B, Pearson M, Chan DA, Giaccia AJ, Semenza GL. Mol Cell. 2005;17:503–512. doi: 10.1016/j.molcel.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 22.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 23.Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW. Proc Natl Acad Sci U S A. 2000;97:10430–10435. doi: 10.1073/pnas.190332597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ivan M, Kaelin WG., Jr Curr Opin Genet Dev. 2001;11:27–34. doi: 10.1016/s0959-437x(00)00152-0. [DOI] [PubMed] [Google Scholar]
- 25.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 26.Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM. J Biol Chem. 2002;277:29936–29944. doi: 10.1074/jbc.M204733200. [DOI] [PubMed] [Google Scholar]
- 27.Liu YV, Baek JH, Zhang H, Diez R, Cole RN, Semenza GL. Mol Cell. 2007;25:207–217. doi: 10.1016/j.molcel.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu YV, Semenza GL. Cell Cycle. 2007;6:656–659. doi: 10.4161/cc.6.6.3981. [DOI] [PubMed] [Google Scholar]
- 29.Klee CB, Crouch TH, Krinks MH. Proc Natl Acad Sci U S A. 1979;76:6270–6273. doi: 10.1073/pnas.76.12.6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klee CB, Ren H, Wang X. J Biol Chem. 1998;273:13367–13370. doi: 10.1074/jbc.273.22.13367. [DOI] [PubMed] [Google Scholar]
- 31.Rao A, Luo C, Hogan PG. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- 32.Hogan PG, Chen L, Nardone J, Rao A. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 33.Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 34.Huai Q, Kim HY, Liu Y, Zhao Y, Mondragon A, Liu JO, Ke H. Proc Natl Acad Sci U S A. 2002;99:12037–12042. doi: 10.1073/pnas.192206699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heineke J, Molkentin JD. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 36.D’Angelo G, Duplan E, Vigne P, Frelin C. J Biol Chem. 2003;278:15406–15411. doi: 10.1074/jbc.M211293200. [DOI] [PubMed] [Google Scholar]
- 37.Kong X, Lin Z, Caro J. FEBS Lett. 2006;580:6182–6186. doi: 10.1016/j.febslet.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 38.Kiely PA, O’Gorman D, Luong K, Ron D, O’Connor R. Mol Cell Biol. 2006;26:4041–4051. doi: 10.1128/MCB.01868-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thornton C, Tang KC, Phamluong K, Luong K, Vagts A, Nikanjam D, Yaka R, Ron D. J Biol Chem. 2004;279:31357–31364. doi: 10.1074/jbc.M402316200. [DOI] [PubMed] [Google Scholar]
- 40.Blom N, Gammeltoft S, Brunak S. J Mol Biol. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 41.Koulmann N, Novel-Chate V, Peinnequin A, Chapot R, Serrurier B, Simler N, Richard H, Ventura-Clapier R, Bigard X. Am J Respir Crit Care Med. 2006;174:699–705. doi: 10.1164/rccm.200512-1976OC. [DOI] [PubMed] [Google Scholar]
- 42.Hui AS, Bauer AL, Striet JB, Schnell PO, Czyzyk-Krzeska MF. FASEB J. 2006;20:466–475. doi: 10.1096/fj.05-5086com. [DOI] [PubMed] [Google Scholar]
- 43.Yuan G, Nanduri J, Bhasker CR, Semenza GL, Prabhakar NR. J Biol Chem. 2005;280:4321–4328. doi: 10.1074/jbc.M407706200. [DOI] [PubMed] [Google Scholar]
- 44.Ron D, Chen CH, Caldwell J, Jamieson L, Orr E, Mochly-Rosen D. Proc Natl Acad Sci U S A. 1994;91:839–843. doi: 10.1073/pnas.91.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCahill A, Warwicker J, Bolger GB, Houslay MD, Yarwood SJ. Mol Pharmacol. 2002;62:1261–1273. doi: 10.1124/mol.62.6.1261. [DOI] [PubMed] [Google Scholar]
- 46.Ceci M, Gaviraghi C, Gorrini C, Sala LA, Offenhauser N, Marchisio PC, Biffo S. Nature. 2003;426:579–584. doi: 10.1038/nature02160. [DOI] [PubMed] [Google Scholar]
- 47.Sengupta J, Nilsson J, Gursky R, Spahn CM, Nissen P, Frank J. Nat Struct Mol Biol. 2004;11:957–962. doi: 10.1038/nsmb822. [DOI] [PubMed] [Google Scholar]
- 48.Patterson RL, van Rossum DB, Barrow RK, Snyder SH. Proc Natl Acad Sci U S A. 2004;101:2328–2332. doi: 10.1073/pnas.0308567100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Circ Res. 2006;98:1528–1537. doi: 10.1161/01.RES.0000227551.68124.98. [DOI] [PubMed] [Google Scholar]
- 50.Lin X, Sikkink RA, Rusnak F, Barber DL. J Biol Chem. 1999;274:36125–36131. doi: 10.1074/jbc.274.51.36125. [DOI] [PubMed] [Google Scholar]
- 51.Sun L, Youn HD, Loh C, Stolow M, He W, Liu JO. Immunity. 1998;8:703–711. doi: 10.1016/s1074-7613(00)80575-0. [DOI] [PubMed] [Google Scholar]