

Abstract
Within less than 10 years after the realization of the double helix of DNA, the ability of aminoglycosides to influence the misreading or readthrough of premature termination codons was discovered. It took another three decades to clone and sequence disease genes and appreciate the similarity of mutation spectra for most inborn errors. Nonsense mutations once again have become the target of readthrough compounds. In this brief review, we trace the development in our laboratory of the next generation of readthrough agents, small molecule readthrough (SMRT) drug-like chemicals, and assays for comparing their in vitro activity. Possible mechanisms of action and potential clinical applications are also considered.
Keywords: SMRT compounds, nonsense mutation, primary immunodeficiency, DNA
Introduction
In an attempt to address the treatment of ataxia-telangiectasia (A-T), a primary immunodeficiency disease with progressive cerebellar degeneration, cancer susceptibility and hypersensitivity to ionizing radiation, we searched for clues on the most fundamental disease-causing mechanism, the molecular pathogenesis of a DNA mutation. We examined how different types of mutations (i.e., frameshifting, splicing, nonsense, missense, and insertion/deletion) resulted in missing or non-functional proteins and on the spectrum of these mutations in the ATM gene (the gene mutated in A-T). We noted that splicing and nonsense mutations accounted for more than half of the mutations and that most A-T patients in the U.S. were compound heterozygotes, i.e., they inherit different mutations from each parent so that most American A-T patients carry at least one copy (allele) of the ATM gene with either a splicing or a nonsense mutation. Disappointedly, we further noted that at least five types of splicing mutations exist,(1) each with a different underlying molecular pathology and sequence specificity, making it unfeasible to prepare a single efficacious drug for patients with splicing mutations.(2,3) Nonsense mutations, on the other hand, are point mutations that result directly in a premature “stop” codon (e.g., CAG > TAG, when a cytosine has mutated to a thymine), and only three kinds of premature termination codons (PTCs) exist (UAG, UGA, and UAA) (Fig. 1). Thus, a single compound that would affect the readthrough of all three PTCs and would restore the translation of RNA , could be a candidate for correcting nonsense mutations—not only in the ATM gene but in many other genes as well. Approximately one-third of patients with A-T worldwide carry at least one nonsense mutation and would thus be SMRT drug candidates. Similarly, as a general rule, approximately one-third of patients with many other genetic disorders carry nonsense mutations.
Figure 1.

Readthrough of a ‘Stop’ codon (PTC) resulting directly froma point mutation (C to T) would not shift the codon frame (i.e., primary ‘Stop’ codon) whereas a ‘Stop’ codon resulting indirectly from the insertion of a nucleotide (N) would cause a frame shift and translation would result in jung protein both before and after the new ‘Stop’ codon (TAA) (i.e., secondary ‘Stop’ codon)
Aminoglycosides (e.g., streptomycin, neomycin, kanamycin, paromomycin, gentamicin) can cause phenotypic suppression of nonsense mutations in both prokaryotes and eukaryotes.(4–8) In 1964, Gorini and Kataja(4) reported that streptomycin interfered with accurate translation of the RNA code. This effect was confirmed in vitro with polynucleotide-directed polypeptide synthesis using ribosomes from E. coli (9) and later with rabbit nucleated erythrocytes.(8) Thus, what is referred to by microbiologists as “nonsense suppression” is no different than the later term of PTC “readthrough”. Perhaps Davies et al.(6) coined the best description in the title of their 1965 report: “misreading of RNA codewords induced by aminoglycoside”.
Our drug target organ for treating A-T patients is the degernerating cerebellum (Fig. 2). Because aminoglycosides are associated with many toxicities, are not easily modified, and do not cross the blood-brain barrier ( BBB) efficiently, we designed a high throughput screen (HTS) that would identify additional chemicals, non-aminoglycosides with readthrough activity, and we screened primarily libraries containing small molecules that would likely enter the brain, i.e., small molecules. To date, we have screened over 70,000 chemicals and have identified three chemical structure/activity related (SAR) groups: RTC13, GJ71, and GJ72 (Refs. 10,11) and we are now focused on the newly designed derivatives or analogues of the lead small molecule readthrough (SMRT) compounds, the next generation of readthrough drugs.
Figure 2.

Radiogram of an 8-year-old A-T patient with marked atrophy of cerebellum, the target of AMRT drug therapy.
PTT-ELISA high throughput screening assay
The HTS assay evolved from previous proof-of-concept experiments in our laboratory, which were using coupled transcription/translation (PTT) to efficiently scan about 9 kb of coding RNA from the large ATM gene (150 kb; 350kDa) to identify disease-causing mutations that resulted in truncated proteins. ATM mutations, like most genetic mutations, comprise four primary groups: nonsense, frameshifts, splice defects and missense.(12–14) Greater than 50% of ATM mutations truncate the protein and can be detected by PTT alone, although PTT then requires further characterization of each mutation by genomic DNA sequencing.(15) The PTT assay has also been defined in some literature as the Protein Truncation Test (PTT). It uses ribosomes in lysates from rabbit nucleated erythrocytes to translate RNA into protein.
To better quantify the output of PTT for high throughput testing, we used plasmids containing cloned fragments of the ATM gene from different A-T patients, each carrying a nonsense mutation that represented a different stop codon (PTC) and was located in a different part of the gene. Each isolated plasmid was also mutagenized back to wildtype to serve as an additional control for the readthrough studies. For more rapid throughput, the PTT assay was then converted to an ELISA readout by adding an “attaching” epitope to the 5’ end of the plasmid, from the myc gene, and a second “detection” epitope (V5) to the 3’ end. (16,17) In this way, the final protein resulting from full-length translation could bind to a polystyrene well surface pre-coated with anti-myc antibody, and translation of the 3’ end could be monitored by an antibody to the V5 viral epitope—which was tagged with horse radish peroxidase so that it could be detected with a proprietary luminescent substrate in the final step.(10) Thus, if an unknown chemical is able to “read through” a nonsense mutation in the plasmid template, the translated protein will contain the V5 epitope at the 3’ end and this activity is interpreted as preliminary evidence that the compound induced readthrough of the PTC.
In two screening cycles, using a robotic platform that harvests from 384-well trays, a total of roughly 70,000 chemicals from four libraries were characterized for readthough activity. Using somewhat arbitrary cutoff points, we identified about 50 “potential hits” for each batch. Manual screening with the same PTT-ELISA assay confirmed readthrough activity for 12 chemicals in the first batch and 14 chemicals in the second batch. EC50 dilution experiments identified two chemicals in the first batch as potentially “drug-like.” (10) These have been studied further (see below). The second batch contained several compounds that shared chemical characteristics and these are being studied as a single SAR group.
Cell-based assays for evaluating kinase activity
The single major function described to date for the ATM protein is that of a nuclear serine/threonine kinase activated in response to double strand breaks in DNA.(18) Two cell-based assays were selected for proof-of-principle that the SMRT-induced PTT-translated ATM protein was biologically active as an intranuclear kinase: (1) IRIF-pATM (irradiation(IR)-induced immunofluorescence of nuclear foci, using an ALEXA-fluor labelled antibody to phosphorylated Ser1981 ATM) and (2) FC-pATM (IR-induced flow cytometric detection of autophosphorylation at Serine1981 of the ATM protein), a modification of the FC-pSMC1 assay first developed in our laboratory for clinical detection of A-T homozygotes and heterozygotes.(19) It should be noted that each of these assays requires induction of double strand DNA breaks by ionizing radiation (IR) to activate ATM kinase activity. In general, lymphoblastoid cells were exposed to 1–100 μM of each chemical for 3–4 days prior to testing for ATM kinase activity, changing the drug-containing tissue culture medium at two days. Most compounds were dissolved in 1% DMSO (dimethyl sulfoxide); the vehicle alone was always included as an additional control. The results of the IRIF-pATM and FC-pATM (or FC-pSMC1) data have been comparable, within the limits of variability for each (Fig. 3).(11)
Figure 3.
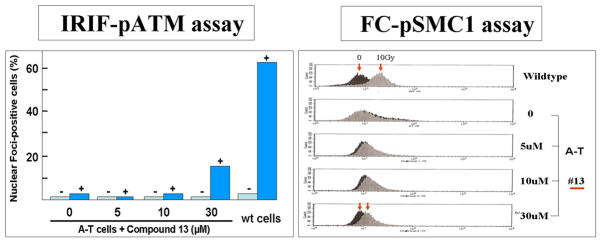
Two cell-based assays for evaluating SMRT drug effects on ATM kinase function of A-T cells: irradiation (1 hour post-2 Gy)-induced immunofluorescent nuclear foci (IRIFs), scored as % of cells containing at least four foci (left panel), and the shift in fluorescence measured by flow cytometry (1 hour post-10 Gy) (right panel).
IRIF-ATM measures whether the levels of ATM protein have been restored to an A-T cell line by readthrough and whether the restored full-length ATM protein is functional as a kinase. In these experiments, lymphoblastoid cell lines (LCLs) derived from A-T patients with a variety of nonsense mutations were prepared for ATM kinase assays after 3–4 day exposure to a chemical. LCLs are grown in suspension. At the time of study, they were irradiated with 2 Gray; after 1 hour, the cells were placed on glass slides, fixed, and stained with a fluor-tagged antibody to ATMpSer1981. Using a fluorescence microscope, the percentage of cells containing at least 4 foci per nucleus was scored manually. At least 100 cells were scored for each cell culture. Non-treated cells were used as controls in each experiment. Scores from 3 or 4 independent experiments were pooled to derive p values for differences observed.
Functional flow (FC-pATM or FC-pSMC1) is a second measure of ATM kinase activity of compounds screened.(19) It differs from the IRIF methods in that it is machine-driven and not prone to subjective interpretation and experimenter experience. The assay uses flow cytometry, with appropriate gating, to follow the appearance of ATM-dependent phosphorylated substrates, such as ATM itself or SMC1 (structural maintenance of proteins 1), during the DNA repair response to IR damage. Measurements are made one hour following 2 Gy. Thus, the phosphorylated proteins are compared for two cell populations, one irradiated and one not irradiated. The fluorescence index (delta FI) is derived from the peak FI of each curve at 1 hour after damage. Approximately 10,000 cells are counted for each sample.
Periodically, a third very similar assay, FC-pSMC1 (IR-induced phosphorylation of Serine966 of the SMC1 protein), was added to assess ATM transphosphorylation kinase activity because the activating mechanisms for ATM transphosphorylation are distinct from those for autophosphorylation of ATM. However, the antibody for SMC1-pS966 is expensive, precluding its use in evaluating multiple SMRT candidates, analogues, and limiting dilutions experiments. Unfortunately, none of these assays allowed for rapid throughput of samples or easy quantification, requiring repeated testing for statistically significant comparisons of leading chemicals—so that a single most promising drug could be selected for the subsequent, and costly, ADMET (absorption, distribution, metabolism, elimination, and toxicology) studies.
Attempts to measure intracellular ATM protein concentrations directly, using an ELISA assay,(20) have met with varying success, primarily because intracellular ATM concentrations are generally very low even in wild-type cells, thereby requiring large numbers of cells (~2 million cells per assay) for each variable tested. Lastly, lead compounds were tested on LCLs to measure abrogation of defective colony survival (CSA), a characteristic of AT cells.(21/22) This assay requires 10–12 days in culture after exposure to the SMRT compounds. Significant improvement of clonogenic activity has been induced by active SMRT drugs, sometimes abrogating CSA to normal levels.(8,10) However, this abrogation can also be taken to signify that CSA may lack the sensitivity necessary for making quantitative comparisons of lead SMRT candidates.
PTC target variants
Each active SMRT drug has been tested for ability to read through all three ‘stop’ (PTC) codons by selecting target cells from different A-T patients carrying different disease-causing nonsense mutations. For example, cell line TAT51 carries a nonsense mutation of TGA, whereas AT187LA carries one for a TAA “stop” codon. Although the first generation non-aminoglycoside readthrough drug approved for clinical trials, PTC124 (Ataluren), had its strongest activity for read through of UGA, and little activity for UAA or UAG,(23) all of our lead SMRT compounds to date have shown comparable activity against all three PTCs (Fig. 4). PTC124 also does not cross the BBB efficiently; this intensified our efforts to target our high throughput screening for small molecules.
Figure 4.

Efficacy of SMRT drugs RTC#13 and RTC#14, as measured by the IRIF-pATM assay. A-T cell used were homozygous for TGA A or TAG A stop codons. Note increase in ATM kinase activity from zero to 16% after 4-day exposure to RTC#13 and 18% after a similar exposure to RTC#14 with readthrough of TGA A. No appreciable difference was observed with readthrough of TAG A. The aminoglycoside G418 gave comparable results. (Modified from Ref. 10)
Because previous studies have suggested that the fourth nucleotide just after a PTC (e.g., UAG N) can have substantial influence on readthrough efficiency,(24, 25 ) i.e., “4+ wobble,” we continued to monitor this in our testing. We noted no distinctive differences. However, these effects are most likely very small and are unlikely to be detected by the sensitivity of our screening assays without optimizations. There may also be species specific differences. In any case, it is unlikely that these effects will be strong enough to be of clinical relevance.
Mechanisms of action of PTC read-through
Because our HTS-PTT assay detected the induction of full-length ATM protein in an in vitro system, to which only a reaction mixture of rabbit reticulocyte lysate, methionine, and a plasmid construct was added, it is difficult to escape the conclusion that ribosomes are the most likely primary target of SMRT compounds. Further, numerous reports describe elegant high resolution crystallography and scanning electron microscopy of various aminoglycosides binding to the decoding A-site in 16S rRNA of E. coli..(26–28) However, recent studies indicate that the target of a readthrough chemical is not always the small subunit of the ribosome. For example, macrolides such as tylosine inhibit protein synthesis and translation fidelity by binding to the large (50S) subunit of prokaryotes, while PTC124, a well-characterized readthrough compound that is in clinical trials for cystic fibrosis and hemophilia, binds to the large (28S) subunit.(29–31) Mechanistic studies for the SMRT compound RTC13 have not yet been done; however, hypothesizing that the thiazolidinone units of RTC13 might be binding to nucleobases in the ribosome via hydrogen bonding, we substituted a variety of pyrimidine bases. We found that almost all of the pyrimidinedione analogues showed good readthrough activity while neither of the corresponding cytosine analogues were active.(11) We interpret this as presumptive evidence for interaction of RTC13 with the ribosomal16S decoding region..
Potential therapeutic applications
To date, SMRT compounds have abrogated protein deficiencies in several genetic models. Approximately 5% of normal ATM protein levels were restored, as compared with 50–70% of normal in the colony survival assay, 20–60% abrogation of ATMpSer1981 IRIFs (one-third of normal values in Fig. 3), and 20–30% abrogation of ATMpSer1981 by flow cytometry (data not shown).(10) Although it is difficult to predict whether these increases in ATM protein or function would have clinical relevance, it is abundantly clear that A-T patients with missense mutations who have measurable residual kinase activity , despite very reduced (i.e., less than 15% of normal) ATM protein levels (Fig. 5, lane 9),(32) generally experience a milder disease with slower progression of neurological symptoms. Furthermore, A-T heterozygotes generally have 40–50% of normal ATM protein levels and remain asymptomatic throughout life.(33)
Figure 5.

Western blot demonstrates absence of ATM protein in nuclear extracts from three patients with A-T (lanes 2, 5, and 7). Lane 9 shows trace (4%) of protein in A-T patient with slower progression of neurological symptoms (~25 μg total protein loaded/lane).
Although attempting to predict appropriate clinical models for applying SMRT drugs is purely speculative, it might be expected that enzymatic deficiencies would be more appropriate disease candidates than diseases involving large structural molecules. However, quite to the contrary, preliminary collaborative data suggest that SMRT drugs may correct dystrophin deficiency in mice.(10) Dystrophin-deficient differentiated myotubes derived from mdx mice (UAA), a model of Duchenne muscular dystrophy, have been successfully abrogated by RTC13 and other SMRT compounds.(10) Muscle function appears to improve significantly following in vivo exposure to RTC13 (unpublished). These results are especially encouraging when one considers the structural, non-enzymatic, function of the dystrophin protein, its large size (427 kDa), and its general distribution throughout the body.
In other collaborative studies, using stably transfected fibroblasts and keratinocytes derived from patients with epidermolysis bullosum, a devastating skin disease in infants,(34,35) a readthrough effect for RTC13 has been demonstrated on over 20 disease-causing nonsense mutations (UAG and UGA)(unpublished). The mutations are located throughout the collagen VII gene. Further efforts to characterize derivatives of RTC13 are in progress.
Interestingly, very preliminary attempts to induce PHOX complex proteins in myeloid cells derived from patients with chronic granulomatous disease have not yielded readthrough, using RTC13 and two analogues (unpublished). Other unsuccessful but very preliminary attempts have been made to abrogate WASP protein function in cells from Wiscott-Aldrich patients (unpublished). Thus, in addition to the efficacy of the readthrough compounds on the gene(s) involved, clinical success can also reflect the pathophysiology of the disease, the non-reversibility of the pathology, and/or the pharmacokinetic properties of the protein being induced (e.g., stability, tissue distribution, complex formation). For these reasons, SMRT compounds are being tested ex vivo and in vivo in other genetic models as well. Once the most promising compounds have been selected, pharmacokinetic studies will be expanded. Blood-brain barrier penetration must also be evaluated for translational application to diseases involving the central nervous system.
Therapeutic applications of SMRT compounds need not be limited to inducing replacement protein for genetic diseases missing actual protein or functional protein. A much wider application can be imagined for prophylactic therapy. For example, if women with mutations in BRCA1 have a 100% life-time penetrance for developing breast cancer, those with nonsense mutations (16%) might benefit from long-term administration of SMRT drugs,(36) if they were proven safe for long-term use. Further, p53 (TP53) mutations are present in about half of BRCA1-positive woman with breast cancer and about half of these are carrying nonsense mutations and have low p53 tumor levels;(37) the p53 loss might be amenable to SMRT drug treatment. This same rationale could be applied to mothers of A-T patients carrying ATM nonsense mutations, who are at an increased risk of breast cancer,(36, 38–40) or to other at risk cancer patients with nonsense mutations in a tumor suppressor gene.
Acknowledgments
The studies described herein were partially funded by NIH grants NS052528 and AI067769, the A-T Medical Research Foundation, the Scott Richards Charitable Foundation, A-T Ease Foundation, APRAT, the A-T Medical Research Trust, and the AT Society. We thank Francesca Fike for assistance in the laboratory and in the preparation of this manuscript.
References
- 1.Eng L, Coutinho G, Nahas S, Yeo G, Tanouye R, Babaei M, Dork T, Burge C, Gatti RA. Non-classical splicing mutations in the coding and non-coding regions of the ATM gene: maximum entropy estimates of splice junction strengths. Hum Mutation. 2004;23:67–76. doi: 10.1002/humu.10295. [DOI] [PubMed] [Google Scholar]
- 2.Du L, Pollard J, Gatti RA. Correction of prototypic ATM splicing mutation and aberrant ATM function with antisense morpholino oligonucleotides. Proc Natl Acad Sci. 2007;104:6007–6012. doi: 10.1073/pnas.0608616104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakamura K, Du L, Tunuguntla R, Fike F, Cavalieri S, Morio T, Mizutani S, Brusco A, Gatti RA. Functional characterization and targeted correction of ATM mutations identified in Japanese patients with ataxia-telanigectasia. Hum Mut. 2011 doi: 10.1002/humu.21632. Online Oct 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gorini L, Kataja E. Phenotypic repair by streptomycin of defective genotypes in E. coli. Proc Natl Acad Sci US. 1964;51:487–93. doi: 10.1073/pnas.51.3.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pestka S, Marshall R, Nirenberg MW. RNA codewords and protein synthesis. V. Effects of streptomycin on the formation of ribosome-SRNA complexes. Proc Natl Acad Sci US. 1965;53:639–646. doi: 10.1073/pnas.53.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies J, Gorini L, Davis BD. Misreading of RNA codewords induced by aminoglycoside antibiotics. Mol Pharmacol. 1965;1:93–106. [PubMed] [Google Scholar]
- 7.Howard MT, Anderson CB, Fass U, Khatri S, Gesteland RF, Atkins JF, Flanigan KM. Readthrough of dystrophin stop codon mutations induced by aminoglycosides. Ann Neurol. 2004;55:422–426. doi: 10.1002/ana.20052. [DOI] [PubMed] [Google Scholar]
- 8.Lai C-H, Chun HH, Nahas SA, Mitui M, Gamo K, Du L-T, Gatti RA. Correction of ATM gene function by aminoglycoside-induced readthrough of premature termination codons. Proc Natl Acad Sci. 2004;101:15676–81. doi: 10.1073/pnas.0405155101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davies J, Gilbert W, Gorini L. Streptomycin, suppression, and the code. Proc Natl Acad Sci. 1964;51:883–90. doi: 10.1073/pnas.51.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Du L, Damoiseaux R, Nahas S, Gao K, Hu H, Pollard JM, Goldstine J, Jung ME, Henning SM, Bertoni C, Gatti RA. Nonaminoglycoside compounds induce readthrough of nonsense mutations. J Exp Med. 2009;206:2285–97. doi: 10.1084/jem.20081940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jung ME, Ku J-M, Du L, Hu H, Gatti RA. Synthesis and evaluation of compounds that induce readthrough of prenature termination codons. Bioorganic & Medicinal Chemistry Letters. 2011;21:58420–8. doi: 10.1016/j.bmcl.2011.07.107. [DOI] [PubMed] [Google Scholar]
- 12.Telatar M, Wang Z, Udar N, Liang T, Concannon P, Bernatovska-Matuszkiewicz E, Lavin M, Shiloh Y, Good RA, Gatti RA. Ataxia-telangiectasia: mutations in ATM cDNA detected by protein truncation screening. Amer J Hum Genet. 1996;59:40–44. [PMC free article] [PubMed] [Google Scholar]
- 13.Telatar M, Teraoka S, Wang Z, Chun HH, Liang T, Castellvi-Bel S, Udar N, Borresen-Dale A-L, Chessa L, Bernatowska-Matuszkiewicz E, Porras O, Watanabe M, Junker A, Concannon P, Gatti RA. Ataxia-telangiectasia: identification and detection of founder mutations in the ATM gene in ethnic populations. Am J Hum Genet. 1998a;62:86–97. doi: 10.1086/301673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Telatar M, Castellvi-Bel S, Tai L-Q, Wang Z, Regueiro JR, Porras O, Gatti RA. A model for ATM heterozygote identificiation in a large population: Four founder effect ATM mutations identify most of Costa Rican patients with ataxia-telangiectasia. Molec Genet Metab. 1998b;64:36–43. doi: 10.1006/mgme.1998.2693. [DOI] [PubMed] [Google Scholar]
- 15.FitzGerald MG, Bean JM, Hedge SR, Unsal H, MacDonald DJ, Harkin DP, et al. Heterozygous ATM mutations do not contribute to early onset of breast cancer. Nat Genet. 1997;15:307–10. doi: 10.1038/ng0397-307. [DOI] [PubMed] [Google Scholar]
- 16.Gite S, Lim M, Carlson R, Olejnik J, Zehnbauer B, Rothschild K. A high-throughput nonisotopic protein truncation test. Nat Biotechnol. 2003;21:194–197. doi: 10.1038/nbt779. [DOI] [PubMed] [Google Scholar]
- 17.Du L, Lai C-H, Concannon P, Gatti RA. Rapid screen for truncating ATM mutations by PTT-ELISA. Mutation Research. 2008;640:139–44. doi: 10.1016/j.mrfmmm.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 18.Ditch S, Paull T. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci. 2011 Nov 10; doi: 10.1016/j.tibs.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nahas SA, Butch AW, Du LT, Gatti RA. Rapid flow cytometry-based SMC1 phosphorylation assay for identification of ataxia-telangiectasia homozygotes and heterozygotes. Clin Chem. 2009;55:463–72. doi: 10.1373/clinchem.2008.107128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Butch AW, Chun HH, Nahas SA, Gatti RA. Development of an immunoassay to measure Ataxia-Telangiectasia Mutated (ATM) protein in cellular lysates. Clin Chem. 2004;50:2302–8. doi: 10.1373/clinchem.2004.039461. [DOI] [PubMed] [Google Scholar]
- 21.Huo YK, Wang Z, Hong J-H, Chessa L, McBride WH, Perlman SL, Gatti RA. Radiosensitivity of ataxia-telangiectasia, X-linked agammaglobulinemia and related syndromes. Canc Res. 1994;54:2544–2547. [PubMed] [Google Scholar]
- 22.Sun X, Becker-Catania S, Chun HH, Hwang MJ, Huo Y, Wang Z, Mitui M, Sanal O, Chessa L, Crandall B, Gatti RA. Early diagnosis of ataxia-telangiectasia using radiosensitivity testing. J Pediat. 2002;40:724–731. doi: 10.1067/mpd.2002.123879. [DOI] [PubMed] [Google Scholar]
- 23.Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, et al. 2008. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 24.Bidou L, Hatin I, Perez N, Allamand V, Pantheir JJ, Rousset IP. Premature stop codons involved in muscular dystrophies show a broad spectrum of readthorugh efficiencies in response to gentamicin treatment. Gene Therapy. 2004;11:619–627. doi: 10.1038/sj.gt.3302211. [DOI] [PubMed] [Google Scholar]
- 25.Manuvakhova M, Keeling K, Bedwell DM. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA. 2000;6:1044–1055. doi: 10.1017/s1355838200000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hobbie SN, Pfister P, Grull C, Westhof E, Bottger EC. Analysis of the contribution of individual subsittuents in 4,6-aminoglycoside-ribosome interaction. Antimicrobioal Agenets Chemother. 2005;49:5112–18. doi: 10.1128/AAC.49.12.5112-5118.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zingman LV, Park S, Olson TM, Alekseev Ae, Terzic A. Aminoglycoside-induced translational read-through in disease: overcoming nonsense mutations by pharmacogenetic therapy. Clin Pharmacol Ther. 2007;81:99–103. doi: 10.1038/sj.clpt.6100012. [DOI] [PubMed] [Google Scholar]
- 28.Kondo J, Westhof E. Structural comparisons between prokaryotic and eukaryotic ribosomal decoding A-sites free and complexed with aminoglycosides. In: Arya DP, editor. Aminoglycoside Antibiotics: From chemical biology to drug discovery. Hoboken, NJ: John Wiley & Sons, Inc; 2007. pp. 209–223. [Google Scholar]
- 29.Thompson J, Pratt CA, Dahlberg AE. Effects of a number of classes of 50S inhibitors on stop codon readthrough during protein syntehesis. Antimicrob Agenst Chemother. 2004;48:4889–4891. doi: 10.1128/AAC.48.12.4889-4891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Linde L, Kerem E. Introducing sense into nonsense in treatments of human genetic diseases. Trends Genet. 2008;24:552–563. doi: 10.1016/j.tig.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 31.Hamed SA. Drug evaluation: PTC-124 – a potential treat of cystic fibrosis and Duchenne muscular dystrophy. IDrugs. 2006;9:783–789. [PubMed] [Google Scholar]
- 32.Chun HH, Sun X, Nahas SA, Teraoka S, Lai C-h, Concannon P, Gatti RA. Improved diagnostic testing for ataxia-telangiectasia by immunblotting of nuclear lysates for ATM protein expression and PI-3 kinase activity. Mol Genetics & Metabolism. 2003;80:437–443. doi: 10.1016/j.ymgme.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 33.Gatti RA. Ataxia-Telangiectasia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8. Publ. McGraw-Hill; New York: 2001. pp. 705–732. [Google Scholar]
- 34.Christiano AM, Hoffman GG, Chung-Honet LC, Lee S, Cheng W, Iutto J, Greenspan DS. Structural organization of the human type VII collagen gene (COL7A1), composed of more exons than any previously characterized gene. Genomics. 1994;21:169–179. doi: 10.1006/geno.1994.1239. [DOI] [PubMed] [Google Scholar]
- 35.Woodley DT, You Y, Martin S, Li W, Chen M. Characterization of molecular mechanisms underlying mutations in dystrophic epidermolysis bullosa using site-directed mutagenesis. J Biol Chem. 2008;282:17838–45. doi: 10.1074/jbc.M709452200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tavtigian SV, Oerner PJ, Babikyan D, Hartmann A, Healey Sl, Le Calvez-Kelm Fl, Lesueur F, et al. Rare, evolutionarily unlikely missense substitutions in ATM confer increased risk of breast cancer. Amer J Human Genet. 2009;85:427–446. doi: 10.1016/j.ajhg.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holstege H, Joosse SA, van Oostrom CT, Nederlof PM, de Vries A, Jonkers J. High incidence of protein-truncating TP53 mutations in BRCA1-related breast cancer. Canc Res. 2009;69:3625–33. doi: 10.1158/0008-5472.CAN-08-3426. [DOI] [PubMed] [Google Scholar]
- 38.Swift A, Morrell D, Massey RB, Chase CL. Incidence of cancer in 161 families affected by ataxia-telangiectasia. New Engl J Med. 1991;325:1831–6. doi: 10.1056/NEJM199112263252602. [DOI] [PubMed] [Google Scholar]
- 39.Renwick A, Thompson D, Seal S, Kelly P, Chagtai R, Ahmed M, North B, Jayatilake R, Barfoot R, et al. ATM mutations that cause ataxia-telangiectasia are breast canceer susceptibility alleles. Nature Genet. 2006;38:873–75. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 40.Teraoka SN, Bernstein JL, Reiner AS, Haile RW, Brenstein L, Lynch CF, Malone KE, Stovall M, et al. Single nucleotide polymorphisms associated with risk for contralateral breast canceer in the Women’s Environment, Cancer, and Radiation Epidemiology (WECARE) Study. Breast Cancer Res. 2011;13(6):R114. doi: 10.1186/bcr3057. [DOI] [PMC free article] [PubMed] [Google Scholar]