

Abstract
There are seemingly conflicting data in the literature regarding the role of serotonin (5-HT) 5-HT2C receptors in the mouse head-twitch response (HTR) elicited by the hallucinogenic 5-HT2A/2B/2C receptor agonist 2,5-dimethoxy-4-iodoamphetamine (DOI). Namely, both 5-HT2C receptor agonists and antagonists, regarding 5-HT2C receptor-mediated Gq-phospholipase C (PLC) signaling, reportedly attenuate the HTR response. The present experiments tested the hypothesis that both classes of 5-HT2C receptor compounds could attenuate the DOI-elicited-HTR in a single strain of mice, C57Bl/6J. The expected results were considered in accordance with ligand functional selectivity. Commercially-available 5-HT2C agonists (CP 809101, Ro 60-0175, WAY 161503, mCPP, and 1-methylpsilocin), novel 4-phenyl-2-N,N-dimethyl-aminotetralin (PAT)-type 5-HT2C agonists (with 5-HT2A/2B antagonist activity), and antagonists selective for 5-HT2A (M100907), 5-HT2C (SB-242084), and 5-HT2B/2C (SB-206553) receptors attenuated the DOI-elicited-HTR. In contrast, there were differential effects on locomotion across classes of compounds. The 5-HT2C agonists and M100907 decreased locomotion, SB-242084 increased locomotion, SB-206553 resulted in dose-dependent biphasic effects on locomotion, and the PATs did not alter locomotion. In vitro molecular pharmacology studies showed that 5-HT2C agonists potent for attenuating the DOI-elicited-HTR also reduced the efficacy of DOI to activate mouse 5-HT2C receptor-mediated PLC signaling in HEK cells. Although there were differences in affinities of a few compounds at mouse compared to human 5-HT2A or 5-HT2C receptors, all compounds tested retained their selectivity for either receptor, regardless of receptor species. Results indicate that 5-HT2C receptor agonists and antagonists attenuate the DOI-elicited-HTR in C57Bl/6J mice, and suggest that structurally diverse 5-HT2C ligands result in different 5-HT2C receptor signaling outcomes compared to DOI.
Keywords: Head-twitch response, DOI, Hallucinogen, C57Bl/6J, Mouse serotonin 2C receptor, Mouse serotonin 2A receptor, Functional selectivity
1. Introduction
Serotonin (5-hydroxytryptamine, 5-HT) type 2 receptors (5-HT2A, 2B, and 2C) regulate many complex brain functions, including sleep, feeding, emotion, perception, cognition, reward, and memory (Jensen et al., 2010; Landolt and Wehrle, 2009; Millan, 2005; Nichols, 2004). The functions of 5-HT2 receptors implicate them as potentially useful targets for treating neuropsychiatric disorders, however, drug discovery programs have yielded only one approved 5-HT2-selective medicine, the 5-HT2C-preferring agonist lorcaserin (BELVIQ®), which was approved for obesity recently. A number of factors explain the lack of drugs targeting 5-HT2 receptors. The amino acid sequences of the transmembrane regions of 5-HT2 receptors are highly homologous, creating a challenge for designing and developing ligands that activate very selectively one but not the other 5-HT2 receptor subtypes. This is a serious issue, since most 5-HT2 receptor agonists that may have clinically useful effects also produce hallucinations, mediated primarily by activation of the 5-HT2A receptor subtype (Nichols, 2004). Furthermore, prolonged activation of 5-HT2B receptors on heart valve leaflets has been associated with the development of cardiac pulmonary valvulopathy, as seen with fenfluramine (Hutcheson et al., 2011). Finally, selective 5-HT2 antagonists including 5-HT2A antagonists proposed as novel medications to treat schizophrenia, have exhibited only modest antipsychotic effects in clinical trials (Ebdrup et al., 2011), yet are effective for treating L-DOPA-induced psychosis (Abbas and Roth, 2008). Recent clinical studies have shown that 5-HT2A antagonists also increase slow wave sleep, and thus may treat certain sleep disorders (Monti, 2010; Teegarden et al., 2008).
Specific (distinguished from “selective”) activation of 5-HT2C receptors may produce neuropharmacotherapeutic effects with few side-effects. For example, drugs specific for activating 5-HT2C receptors that have zero efficacy for activating 5-HT2A and/or 5-HT2B receptors may be useful for treating schizophrenia (Rosenzweig-Lipson et al., 2007) and substance abuse (Cunningham et al., 2011) by modulating central dopamine release. Extant 5-HT2C-selective agonists including BELVIQ®, however, produce 5-HT2C-dependent hypolocomotion effects in preclinical animal models and similarly can produce somnolence or fatigue in humans (Arena Pharmaceuticals, 2012; Halberstadt et al., 2009; Siuciak et al., 2007). Importantly, at higher concentrations, nearly all reported 5-HT2C selective agonists also activate 5-HT2A and/or 5-HT2B receptors, including BELVIQ® (Thomsen et al., 2008). Thus, the challenge remains to develop compounds that are specific 5-HT2C receptor agonists lacking sedative effects.
Most 5-HT2 receptor agonists (regardless of selectivity) produce a head-twitch response (HTR) in rodents (Canal and Morgan, 2012). Genetic ablation of 5-HT2A receptors or treatment with selective 5-HT2A antagonists abolishes the HTR in mice elicited by 5-HT2 agonist hallucinogens (Gonzalez-Maeso et al., 2007, 2003), such as 2,5-dimethoxy-4-iodoamphetamine (DOI), providing strong evidence that activation of 5-HT2A receptors is necessary for the HTR in rodents elicited by hallucinogenic 5-HT2 agonists. Several other neurotransmitter receptors also appear to modulate DOI-elicited-HTRs in mice, including 5-HT2C receptors (Canal et al., 2010; Fantegrossi et al., 2010). Interestingly, DOI produces HTRs at doses that also potently and efficaciously activate 5-HT2C receptors, yet selective 5-HT2C agonists, with lower potency and efficacy for activating 5-HT2A receptors, dose-dependently attenuate the DOI-elicited-HTR in rodents (Fantegrossi et al., 2010; Siuciak et al., 2007). Adding further intrigue, recent reports show that the selective 5-HT2C antagonist SB-242084 also attenuates the DOI-elicited-HTR in C57Bl/6J and DBA/2J mice (Canal et al., 2010). This effect was not observed, however, in NIH/Swiss mice, wherein SB-242084 actually enhanced the DOI-elicited-HTR (Fantegrossi et al., 2010).
One aim of the current studies was to resolve the seemingly conflicting observations in the literature by comparing directly the effects of multiple 5-HT2C agonists and antagonists on the DOI-elicited-HTR in a single animal model using identical experimental parameters and conditions. It was hypothesized that both classes of compounds could attenuate the DOI-elicited-HTR, and that the results could be in accordance with in vivo functional selectivity (Milligan, 1993; Moya et al., 2007; Urban et al., 2007). This hypothesis involves the notion that selective 5-HT2C agonists could activate 5-HT2C receptors in vivo in a different way compared to DOI, similar to reports of other 5-HT2 agonists acting at 5-HT2A receptors (Schmid and Bohn, 2010; Schmid et al., 2008). Therefore, like antagonists, selective 5-HT2C agonists could interfere with 5-HT2 receptor signaling that leads to the DOI-elicited-HTR by acting as functional antagonists of DOI in vivo.
Functional selectivity in vivo may indeed have therapeutic impact (Mailman, 2007). If 5-HT2C receptor ligands behave as functionally-selective agonists in vivo, then it may be feasible to develop therapeutic 5-HT2C receptor agonists that lack side-effects, such as sedation, by targeting specific 5-HT2C signaling patterns. To this end, in addition to effects on the DOI-elicited-HTR, several classes of selective 5-HT2C agonists were tested for their effects on locomotion following administration of DOI.
2. Materials and methods
2.1. Compounds and treatment doses
The chemical structures of all compounds used in the present studies are shown in Table 1. (±)-DOI (DOI), mCPP, SB-242084, SB-206553, and M100907 were purchased from Sigma-Aldrich (MO, USA). WAY 161503, Ro 60-0175, CP 809101, and 1-methylpsilocin were purchased from Tocris (Bristol, UK). Serotonin was purchased from Alfa Aesar (MA, USA). [3H]-mesulergine, [3H]-ketanserin, and [3H]-myo-inositol were purchased from Perkin-Elmer (MA, USA). The (+)-(2R, 4S)- and (−)-(2S, 4R)-trans enantiomers of 4-phenyl-2-N,N-dimethylaminotetralin (PAT) and (−)-(2S, 4R)-trans-4-(4′-methyl)-N,N-dimethylaminotetralin (methyl-PAT) were synthesized in our laboratories as racemates that were resolved by chiral stationary-phase HPLC and converted to hydrochloride salts as previously described (Booth et al., 2009; Bucholtz et al., 1999; Vincek and Booth, 2009). All compounds, with the exceptions of SB-242084 and 1-methylpsilocin, were dissolved in sterile MilliQ water which served as the vehicle control. SB-242084 was dissolved in sterile MilliQ water containing Tween-80 (6% final v/v), and 1-methylpsilocin was dissolved in sterile MilliQ water containing acetic acid (5% v/v). Separate MilliQ water vehicles containing these additives were used for control groups for these compounds. All compounds were administered subcutaneously (sc) at a volume of 10 mL/kg. A single dose of DOI (1 mg/kg) was used for all experiments, as it is known to reliably elicit a robust and consistent number of HTRs in C57Bl/6J mice (Canal et al., 2010; Fox et al., 2010). Two doses were used to test the efficacy of 5-HT2C selective agonists for attenuating the DOI-elicited-HTR 3 mg/kg and 5.6 mg/kg. Doses for antagonists were as follows: M100907, 0.0025, 0.025, and 0.25 mg/kg, SB-206553 and SB-242084, 0.03, 0.3, and 3 mg/kg. All compounds were weighed on a Mettler-Toledo (OH, USA) XP26 microanalytical balance, and solutions of all compounds were made fresh on the day of testing.
Table 1.
Approximate Ki values of 5-HT2 ligands at mouse vs. human 5-HT2A and 5-HT2C receptors. Ki values for novel 4-phenyl-2-aminotetralin ligands ((−)-PAT, (+)-PAT, methyl-PAT) and selective 5-HT2 antagonists (SB-242084, SB-206553, M100907) were obtained experimentally via competition with [3H]-ketanserin (5-HT2A) or [3H]-mesulergine (5-HT2C). Data are expressed as means (±SEM’s). Ki values of commercially available selective 5-HT2C agonists are from the literature; data from receptors labeled with ligands other than [3H]-ketanserin (5-HT2A) or [3H]-mesulergine (5-HT2C) are reported when these data were not available: CP 809101 (aSiuciak et al., 2007, [3H]-5-HT), Ro 60-0175 (bCussac et al., 2002; cBentley et al., 2004 and dKnight et al., 2004, [125I]-DOI), WAY 161503 (eWelmaker et al., 2000; fRosenzweig-Lipson et al., 2006, [125I]-DOI labeled), mCPP (g, hData from the NIMH Psychoactive Drug Screening Program), and 1-methylpsilocin (iSard et al., 2005). CLogP values were obtained from ChemBioDraw software version 12. *5-HT does not readily cross the bloodebrain barrier, and was not tested in vivo. m5-HT2A = C57Bl/6 mouse 5-HT2A receptor; m5-HT2C = C57Bl/6 mouse 5-HT2C-VNV receptor; h5-HT2A = human 5-HT2A receptor; h5-HT2C = human 5-HT2C-INI receptor. NR = not reported.
| Ligand | Structure | CLogP |
Ki, nM (SEM)
|
|||
|---|---|---|---|---|---|---|
| m5-HT2A | h5-HT2A | m5-HT2C | h5-HT2C | |||
| 5-HT |
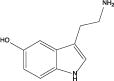
|
0.76* | 138 (27.7) | 128 (19.7) | 50.4 (10.2) | 15.4 (2.3) |
| (±)-DOI |
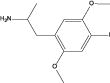
|
2.67 | 4.0 (1.2) | 3.2 (0.8) | 46.7 (4.4) | 19.1(1.7) |
| (−)-PAT |
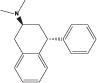
|
4.12 | 116 (18.7) | 102 (12.2) | 53 (2.7) | 29 (3.3) |
| (+)-PAT |
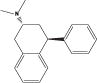
|
4.12 | 332 (46.8) | 463 (47.3) | 1734 (156.6) | 492 (73.0) |
| Methyl-PAT |
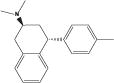
|
4.62 | 326 (29.9) | 179 (17.9) | 868 (86.8) | 128 (6.12) |
| SB-242084 |
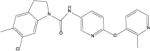
|
5.05 | 96 (20) | 146 (45.0) | 0.1 (0.03) | 1.5 (0.2) |
| SB-206553 |
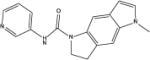
|
3.18 | 342 (44.4) | 676 (69.3) | 3.2 (0.5) | 3.1 (0.6) |
| M100907 |
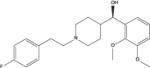
|
3.29 | 0.45 (0.05) | 0.2 (0) | 74.2 (12.2) | 36.6 (4.4) |
| CP 809101 |
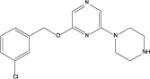
|
2.90 | NR | 6a | NR | 1.6a |
| Ro 60-0175 |
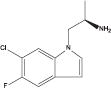
|
3.07 | NR | 37c,d | NR | 27b |
| WAY161503 |
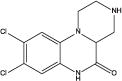
|
2.12 | NR | 18f | NR | 32e |
| mCPP |
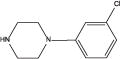
|
2.47 | NR | 143g | NR | 147h |
| 1-Methylpsilocin |
|
2.14 | NR | 900i | NR | 7i |
2.2. Animal subjects
Male C57Bl/6J mice, purchased at 60 days old from Jackson Laboratories (Bar Harbor, Maine) were used in all experiments. Mice were pair-housed in standard cages with ad libitum access to food and water, and were acclimated in the vivarium for at least 4 days prior to testing. Animal procedures were approved by the Institutional Animal Care and Use Committee and are in accordance with the principles in the National Institutes of Health Guide for Care and Use of Laboratory Animals (Publication No. 85-23, revised 1985).
2.3. Behavioral studies
Drug or vehicle was injected sc 10 min prior to injection of DOI. Mice were placed in an activity chamber (43 × 43 cm, Med Associates, Inc.) 10-min later, and head-twitches were counted for the next 10 min by an observer blind to experimental treatment. Locomotion (distance traveled, cm) was tracked with an overhead camera and Ethovision XT 7.0 software. A separate group of mice was administered DOI 10 min prior to injection of 0.25 mg/kg M100907 or 3 mg/kg SB-206553 to test whether the HTR becomes independent of 5-HT2 receptors after their activation, i.e. to test whether M100907 and/or SB-206553 remain effective at attenuating the DOI-elicited-HTR after 5-HT2 receptor activation.
2.4. Mouse 5-HT2A and 5-HT2C receptor constructs
Plasmid DNA containing either mouse htr2A or htr2C-vnv cDNA clones was purchased from Origene (MD, USA). cDNA clones were inserted between the EcoR 1 and Not 1 sites in pCMV6-Kan/Neo vectors. Plasmid DNA was transformed into Subcloning Efficiency DH5a competent cells following the manufacturer’s protocol (Invitrogen, CA, USA). Selections of individual colonies of kanamycin-resistant cells were grown in sterile LB broth overnight which served to generate glycerol stocks that were kept stored at −80 °C. Following purification of DNA using Wizard Plus SV minipreps (Promega, USA), samples of htr2A or htr2C DNA were sequenced by the University of Florida DNA sequencing core. Verification of the open-reading frame sequences for the mouse htr2A and mouse htr2C DNA was performed by comparing the results obtained to results published on Pubmed (http://www.ncbi.nlm.nih.gov/pubmed/). The sequences obtained were identical to the published sequences, and match the receptor htr2A and htr2C DNA sequences from the C57Bl/6 mouse. During the verification process, it was observed that the mouse htr2C cDNA from Origene coded for the VNV edited isoform of this receptor. cDNA coding for the human 5-HT2A or 5-HT2C-INI receptors were from previously transformed competent cells, as previously reported (Canal et al., 2011).
2.5. Radioreceptor assays
Transfection procedures, membrane receptor preparation, and binding assays were performed as previously described (Canal et al., 2011). Briefly, radioligand competitive displacement and saturation binding assays were performed in 96-well plates, using cell membranes from HEK-293 cells transiently expressing high femtomoles per mg protein mouse or human 5-HT2A or 5-HT2C receptors. All competition and saturation binding experiments involved the use of triplicates or quadruplicates of samples for each dose of unlabeled (competition) and radiolabeled (saturation) ligands, and each binding experiment was repeated a minimum of three times. Competitive displacement of 1–2 nM [3H]-mesulergine (5-HT2C) or [3H]-ketanserin (5-HT2A) was used to obtain approximate Ki values for unlabeled ligands. Mianserin (30 μM) was used to define non-specific binding. After a 90 min equilibration period at room temperature, incubation mixtures were rapidly passed through GF/B filters using a Mach 2 cell harvester (Tomtec, CT, USA) and subsequently washed with 50 mM Trise –HCl. Filter disks were placed in vials containing scintillation cocktail (ScintiVerse, Fisher Scientific, USA), allowed to equilibrate overnight, and then were counted for 3H-induced scintillation using a Beckman-Coulter LS6500 counter (Indianapolis, IN).
2.6. Phosphoinositide hydrolysis assay
Receptor-mediated phosphoinositide hydrolysis by phospholipase C (PLC) was measured as previously described (Canal et al., 2011), with a few changes. Briefly, HEK-293 cells, were grown in DMEM media containing 5% dialyzed fetal bovine serum (growth media) to 80% confluency in 10 cm plates in an incubator at 5% CO2, 37 °C. Cells were then transfected with 10 μg mouse 5-HT2A or mouse 5-HT2C-VNV pcDNA and 33 μL Lipofectamine 2000 reagent (Invitrogen, CA, USA) in 5 mL DMEM containing 5% dialyzed fetal bovine serum and 5 mL Opti-MEM (transfection media). After an overnight incubation, transfection media was removed and replaced with 16 mL inositol-free growth media containing 1.0 μCi/mL [3H]-myo-inositol. Cells were apportioned into 48 well CellBind® plates (Corning, MA, USA), and [3H]-myo-inositol incorporation continued for 24 h in an incubator. Cells were then acclimated to serum-free media, treated for 30 min with experimental drugs diluted in media containing a final concentration of 50 mM LiCl per well. Media was discarded, and 50 mM formic acid was added to lyse cells. Plates were stored at −80 °C for at least 24 h. After thawing, solution from each well was added to individual anion-exchange columns. Bound [3H]-inositol phosphates were eluted with 800 mM ammonium formate, and eluate was added to scintillation fluid. 3H-induced scintillations were counted with a Beckman-Coulter LS6500 counter. Experiments were performed with duplicates for 5-HT and triplicates for all other drugs, and each independent experiment was performed a minimum of three times.
2.7. Statistical analyses
Competition and saturation binding data were analyzed using nonlinear regression, curve-fitting algorithms in GraphPad Prism, 5.03 for Windows (CA, USA). Data points were limited (seven to nine points per experiment), thus Hill slopes were not calculated (Motulsky and Christopoulos, 2003); data were fit using the “one site fit-Ki” model that constrains the Hill slope to 1.0. Two-site curve-fitting did not result in an improved fit (data not shown). Approximate Ki values were determined by conversion of the IC50 data using the equation Ki= IC50/1 +L/KD where L is the concentration of radioligand (Cheng and Prusoff, 1973). Unpaired t-tests were used to compare Ki values of individual compounds for mouse vs. human 5-HT2A or 5-HT2C receptors. No statistical analyses were performed for results of functional assays characterizing compounds at mouse versus human 5-HT2 receptors, as a priori, they were conducted primarily for qualitative purposes. For experiment 6, individual t-tests were used to compare maximal functional responses between groups.
Separate ANOVAs were performed to analyze effects of experimental compounds on the DOI-elicited-HTR and on locomotion. Dunnett’s post-test compared the number of HTRs elicited by DOI after pretreatment with each experimental compound to the number of HTRs elicited by vehicle plus DOI. Dunnett’s post-test was used for analyzing the effects of each compound on locomotion relative to DOI or vehicle alone. Two-way ANOVA and Student-Newmane–Keuls comparisons were used to identify dose-dependent effects and differences in efficacy across test compounds.
3. Results
3.1. Experiment 1: effect of 5-HT2C agonists on the DOI-elicited-HTR
The effects of selective 5-HT2C agonists on the DOI-elicited-HTR are shown in Fig. 1a and b. In a 10 min period, vehicle plus 1 mg/kg DOI treated mice (N= 38) exhibited 40 ± 1.0 (mean ± S.E.M.) HTRs. There was a significant main effect of pretreatment with the selective 5-HT2C agonists (F10,76= 31.39, p < 0.001) and the novel PAT compounds (5-HT2C agonists/5-HT2A inverse agonist/antagonists) (F6,57= 86.03; p < 0.001) on the number of HTRs elicited by DOI. Both doses of each 5-HT2C-preferring agonist significantly attenuated the DOI-elicited-HTR (p-values < 0.05). Two-way ANOVA (F7,73= 2.85; p= 0.011 for the interaction) revealed that there was a significant dose-dependent effect of (+)-PAT and mCPP. WAY 161503, mCPP, Ro 60-0175, and CP 809101, (+)-PAT, and methyl-PAT were equally effective, attenuating the DOI-elicited-HTR by approximately 40%. (−)-PAT and 1-methylpsilocin were significantly more efficacious compared to all other 5-HT2C agonists at both doses tested (p-values < 0.05), decreasing the response by 84 and 82%, respectively, at the 5.6 mg/kg dose.
Fig. 1.
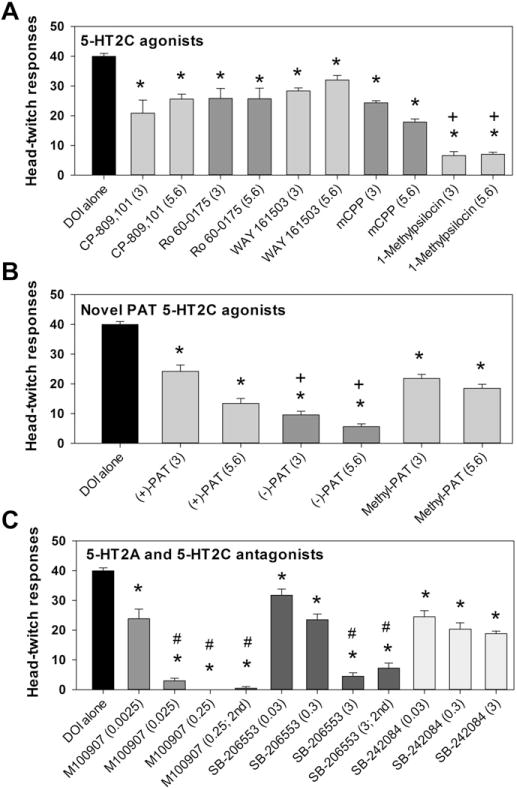
The effects of commercially available 5-HT2C agonists (A) 4-phenyl-2-aminotetralin 5-HT2C agonists (B) and selective 5-HT2 antagonists (C) on the HTR elicited by DOI (1 mg/kg). The dose (mg/kg) is indicated in parentheses. “2nd” refers to test sessions in which the antagonist was administered after DOI. *Indicates a statistically significant difference from DOI alone. +Indicates a statistically significant difference from the other 5-HT2C agonists. #Indicates a statistically significant difference from lower doses.
3.2. Experiment 2: effect of 5-HT2A and 5-HT2C antagonists on the DOI-elicited-HTR
The effects of pretreatment and post-treatment with selective 5-HT2 antagonists on the DOI-elicited-HTR data are shown in Fig. 1c. Two-way ANOVA examining the effects of the selective 5-HT2C/2B inverse agonist/antagonist, SB-206553, and the selective 5-HT2C antagonist, SB-242084 revealed a significant interaction (F2,34= 18.1 ; p < 0.001). SB-206553 dose-dependently attenuated the DOI-elicited-HTR: 0.03, 0.3, and 3.0 mg/kg significantly decreased the number of HTRs, by 20, 37, and 79%, respectively. SB-242084 significantly decreased the DOI-elicited-HTR at all doses tested. The 0.03 mg/kg dose, however, produced a larger, and the 3.0 mg/kg dose produced a smaller effect relative to SB-206553 (p-values <0.05). Unlike SB-206553, there was no dose-related difference in the efficacy of SB-242084 to attenuate the HTR; each dose decreased the number of HTRs by approximately 50%. The selective 5-HT2A antagonist, M100907, dose-dependently attenuated the DOI-elicited-HTR (F3,18 = 41.7; p < 0.001): 0.0025 mg/kg suppressed the response by 37%, 0.025 mg/kg by 92%, and 0.25 mg/kg by 100%, completely eliminating the HTR. Mice treated only with vehicle exhibit approximately 2–5 HTR’s in 10 min (unpublished observations), and thus the 0.25 mg/kg dose of M100907 blocked even the basal HTR in these mice. Finally, SB-206553 (3 mg/kg) and M100907 (0.25 mg/kg) administered 10 min after DOI retained their ability to suppress the HTR (p values < 0.05).
3.3. Experiment 3: effect of 5-HT2C agonists on locomotion
As shown in Fig. 2a, administration of DOI alone had no statistically significant effect on locomotion relative to vehicle administration (unpaired t-test, p = 0.083). Subsequent analyses compared the effects of 5-HT2C agonists (as a 10-min pretreatment to DOI) to either vehicle/vehicle or DOI/vehicle. Both doses of each 5-HT2C agonist in combination with DOI resulted in lower levels of activity relative to DOI/vehicle administration (F10,85= 21.1; p < 0.001). The combinations of DOI and the 5-HT2C agonists CP 809101, Ro 60-0175, and WAY 161503 resulted in lower levels of activity relative to vehicle administration (F10,50= 8.6; p < 0.001). mCPP and 1-methylpsilocin showed dose-dependent effects in that only the 5.6 mg/kg dose altered locomotion levels. In contrast to the 5-HT2C agonists described above, the novel 4-phenyl-2-aminotetralin compounds (+)-PAT, (−)-PAT, and methyl-PAT at both the 3.0 and 5.6 mg/kg doses failed to alter locomotion levels relative to either vehicle (F6,31= 0.63, p= 0.71) or DOI only (F6,66= 1.22; p= 0.30) administration (Fig. 2b). A two-way ANOVA across all agonists demonstrated that Ro 60-0175 was most effective in suppressing locomotion (F7,71= 19.8; p < 0.001).
Fig. 2.
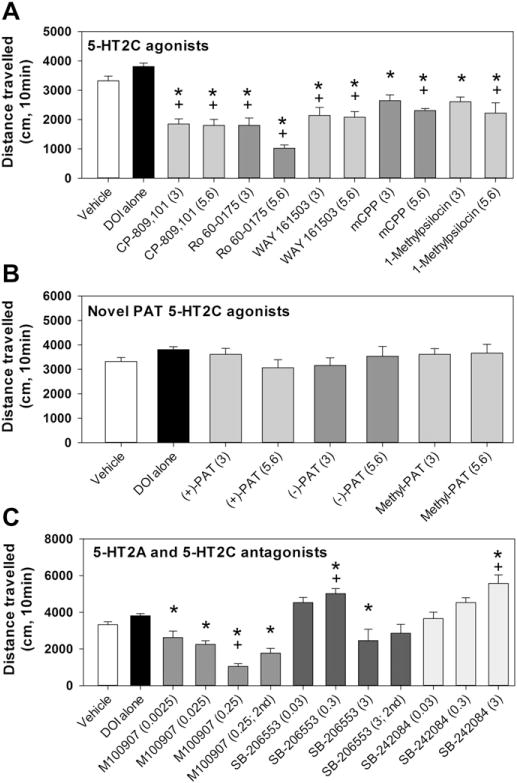
The effects of commercially available 5-HT2C agonists (A), 4-phenyl-2-aminotetralin 5-HT2C agonists (B), and selective 5-HT2 antagonists (C) on locomotion following administration of saline, DOI (1 mg/kg), or DOI in combination with the various compounds. The dose (mg/kg) is indicated in parentheses. “2nd” refers to test sessions in which the antagonist was administered after DOI. *Indicates a statistically significant difference from DOI alone. +Indicates a statistically significant difference from vehicle alone.
3.4. Experiment 4: effect of 5-HT2A and 5-HT2C antagonists on locomotion
Fig. 2c shows the effects of DOI in combination with selective 5-HT2 antagonists. The highest dose combination of DOI and M100907 (0.25 mg/kg) resulted in decreased activity relative to vehicle administration, whereas the 5-HT2C antagonists SB-206553 (0.3 mg/kg) and SB-242084 (3 mg/kg) resulted in increased levels of activity (F11,59= 13.4; p < 0.001). Compared to DOI administration (F11,94= 14.7; p < 0.001), all doses of M100907 decreased locomotion. When DOI was administered 10 min prior to M100907 (0.25 mg/kg, 2nd), the resulting locomotion remained suppressed. Compared to DOI alone, SB-206553 (0.3 mg/kg) and SB-242084 (3 mg/kg) increased activity. A higher dose of SB-206553 (3 mg/kg) in combination with DOI resulted in decreased activity relative to DOI alone. Two-way ANOVA demonstrated that 3.0 mg/kg of SB-206553 resulted in lower activity compared to 0.03 and 0.3 mg/kg SB-206553, and 3.0 mg/kg of SB-242084 (F2,36= 15.0; p < 0.001).
3.5. Experiment 5: ligand affinity and function at mouse and human 5-HT2A and 5-HT2C receptors
To provide a molecular pharmacological context for interpretation of the in vivo behavioral results obtained in mice (above), affinity and function of ligands at mouse and human 5-HT2A and 5-HT2C receptors were determined (Table 1 and Table 2). There are no published affinity values of the selective 5-HT2 antagonists SB-206553, SB-242084, or M100907 at mouse 5-HT2A and 5-HT2C receptors. Thus, it was important to assess whether ligand receptor selectivity remains at the mouse receptors. Similarly, affinity values were assessed for the PAT compounds at mouse receptors, as these values have also not been reported. First, the Kd values of [3H]-ketanserin and [3H]-mesulergine at C57Bl/6 mouse 5-HT2A and 5-HT2C receptors were determined using membranes prepared from transiently transfected HEK cellsdresults were similar to values obtained using the native receptors (Canal et al., 2010; Dougherty and Aloyo, 2011). Mean (±S.E.M.) Kd values of [3H]-ketanserin and [3H]-mesulergine were 0.92 (0.18) nM and 2.2 (0.15) nM, respectively. Regarding comparison of affinity of 5-HT2 ligands at human vs. mouse 5-HT2A and 5-HT2C receptors, no general trend emerged (Table 1). For example, the 5-HT2 agonists 5-HTand DOI, as well as, the novel 5-HT2C agonists (−)-PATand (+)-PAT, showed no differences in affinity at mouse vs. human 5-HT2A receptors (p > 0.05), but showed greater affinity at human compared to mouse 5-HT2C receptors (p < 0.05). In contrast to the other PAT-type 5-HT2C agonists, methyl-PAT showed greater affinity at both the human 5-HT2A and 5-HT2C receptors compared to the mouse homologs. The 5-HT2C antagonist SB-242084 demonstrated higher affinity at mouse vs. human 5-HT2A and 5-HT2C receptors, but SB-206553 had greater affinity at only mouse relative to human 5-HT2A receptors (p < 0.05), with no species difference at 5-HT2C receptors. The 5-HT2A antagonist M100907 showed greater affinity at human compared to mouse 5-HT2A and 5-HT2C receptors (p < 0.05). (Notably ligand Ki values did not significantly differ across different membrane receptor preparations as is illustrated in the low variance associated with the means.)
Table 2.
Approximate potency (EC50) and efficacy values of novel 4-phenyl-2-aminotetralin ligands relative to 5-HTand (±)-DOI for activating mouse and human 5-HT2A and 5-HT2C receptor-mediated PLC signaling. EC50 data are expressed in nM (±SEM). Efficacy data represent percent of maximum 5-HT response (±SEM). m5-HT2A = C57Bl/6 mouse 5-HT2A receptor; m5-HT2C = C57Bl/6 mouse 5-HT2C-VNV receptor; h5-HT2A = human 5-HT2A receptor; h5-HT2C = human 5-HT2C-INI receptor; N/A = not applicable.*Pilot test only; #results from Booth et al., 2009 using CHO-K1 cells.
| Ligand | m5-HT2A
|
h5-HT2A
|
m5-HT2C
|
h5-HT2C
|
||||
|---|---|---|---|---|---|---|---|---|
| EC50 | Efficacy | EC50 | Efficacy | EC50 | Efficacy | EC50 | Efficacy | |
| 5-HT | 71 (2.6) | 100 | 118 (7.50) | 100 | 16 (2.5) | 100 | 4.2 (1.3) | 100 |
| (±)-DOI | 2.6 (0.1) | 98 (4.6) | 1.8 (0.7) | 97 (3.5) | 3.4 (0.9) | 77 (2.6) | 5.5* | 106* |
| (−)-PAT | Inactive | N/A | Inactive | N/A | 39 (1.7) | 72 (2.9) | 20# | 100# |
| (+)-PAT | 352 (18.5) | 31 (1.2) | 233(20.7) | 18 (0.5) | 2115 (87.50) | 69 (1.2) | 171 (37.0) | 88 (9.9) |
| Methyl-PAT | Inactive | N/A | Inactive | N/A | 808 (24.3) | 67 (2.6) | 1120 (145.5) | 96 (3.0) |
Table 2 shows the functional activity (EC50 and percent of maximum 5-HT response (efficacy)) of the 5-HT2 agonists 5-HTand DOI compared to the novel PATs, at mouse vs. human 5-HT2A and 5-HT2C receptors. Functional data for other 5-HT2 ligands at both human and rodent receptors has been reported in the literature, so these experiments were not performed. In contrast to affinity differences noted above at mouse vs. human 5-HT2A and 5-HT2C receptors, the potency (EC50) and efficacy for most of the 5-HT2 agonists tested was similar at mouse and human 5-HT2A and 5-HT2C receptors. One notable difference was (+)-PAT. This compound showed agonist activity at both mouse and human 5-HT2A and 5-HT2C receptors (unlike (−)-PAT which activated only 5-HT2C receptors), yet showed over 10× greater potency for activating human compared to mouse 5-HT2C receptors. (Notably the EC50 values for ligands did not significantly differ across individual experiments, as are illustrated in the low variance associated with the means.)
3.6. Experiment 6: functional activity of 5-HT2C agonists alone and in combination with DOI
In line with the functional selectivity hypothesis, experiments were performed to determine whether selective 5-HT2C agonists alter 5-HT2C receptor signaling stimulated by DOI (Fig. 3). HEK cells expressing the mouse 5-HT2C receptor were treated with 500 nM (−)-PAT, mCPP, or 1-methylpsilocin simultaneously with increasing concentrations of DOI (0.1 nM−10 μM), and PLC signaling was assessed. The 500 nM concentration of (−)-PAT, mCPP, and 1-methylpsilocin was chosen based on competitive antagonist studies showing that doses approximately 10× the Ki’s of antagonists reliably alter agonist signaling yet in a surmountable manner. Individual t-tests revealed that each of the three 5-HT2C agonists, which were most potent for attenuating the DOI-elicited-HTR, significantly reduced the maximum efficacy of DOI to stimulate mouse 5-HT2C receptor-mediated PLC signaling (p < 0.05). Notably, each selective 5-HT2C agonist stimulated 5-HT2C receptor PLC signaling by itself, yet no agonist led to additive or synergistic effects with DOI at the 500 nM dose tested.
Fig. 3.
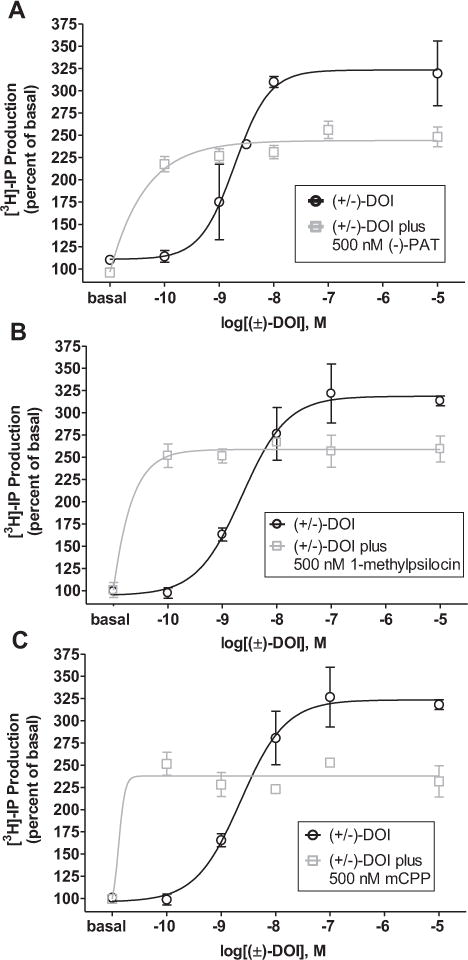
Representative mouse 5-HT2C-VNV-mediated PLC functional data showing the effects of co-administration of 500 nM (−)-PAT (A), 1-methylpsilocin (B), or mCPP (C) with DOI on the ability of DOI to stimulate 5-HT2C receptor-mediated inositol phosphate (IP) production in HEK cells. Note that each of the selective 5-HT2C agonists significantly suppressed the maximal IP response to DOI.
4. Discussion
Using the common inbred strain of mice, C57Bl/6J, the present experiments show that both 5-HT2C-preferring agonists and antagonists, administered at doses commonly used in vivo to explore 5-HT2C receptor function, significantly attenuate the DOI-elicited-HTR. These data suggest that seemingly contradictory literature reports of both 5-HT2C agonists and antagonists suppressing the DOI-elicited-HTR are not contradictory, and demand a new explanation.
Since the recognition of the phenomenon of G-protein-coupled receptor (GPCR) functional selectivity, it has become clear that describing a ligand as an agonist or antagonist does not have comprehensive explanatory value unless a specific context, e.g. receptor density, cell type, cellular location of the GPCR, in vitro versus in vivo, or signaling pathway, is described. For example, lisuride is a 5-HT2C partial agonist in vitro using recombinant CHO cells (Cussac et al., 2008), but an antagonist, lacking no agonist effects on its own, in vivo or ex vivo using choroid plexus cells that express high densities of 5-HT2C receptors (Burris et al., 1991). Similar observations have been reported recently for ligands acting on the dopamine D2 receptor (Koener et al., 2012) and the β2-adrenergic receptor (Kaya et al., 2012). It is conceivable that these findings are related mechanistically to ligand functional selectivity across receptor contexts: a particular ligand can more favorably stabilize one of many possible GPCR conformations that determine intracellular signaling outcomes (Mary et al., 2012), and the specific conformation(s) that is stabilized may also depend on the receptor’s context, e.g. the presence of unique sets of scaffolding proteins (Abbas et al., 2009; Becamel et al., 2004). Thus, every ligand interacting with a GPCR may produce its own unique signaling signature in unique cellular contexts (Flordellis, 2012; Fowler et al., 2012).
The fact that both selective agonists and antagonists of the traditional 5-HT2C receptor signaling pathway (5-HT2C-Gq-PLC) attenuated the DOI-elicited-HTR in the same strain of mouse, from the same vendor, under identical experimental conditions suggests a few explanations: 1) the 5-HT2C agonist ligands tested are functionally selective, orthosterically stabilizing a 5-HT2C receptor conformation(s) that is unique from the conformation(s) that DOI stabilizes, and thus prevent (similar to 5-HT2C selective antagonists) DOI from binding and activating 5-HT2C receptors to positively affect the HTR; 5-HT2C receptor knockout mice show a reduction in, but not an elimination of the HTR elicited by DOI (Canal et al., 2010), suggesting they contribute positively to the DOI-elicited-HTR. Similarly, 5-HT2C selective agonists, at the doses tested here, suppress but do not eliminate the DOI-elicited-HTR. Indeed, this functional antagonist explanation can be extended to 5-HT2A receptors, as all reported 5-HT2C selective agonists, besides (−)-PAT, activate 5-HT2A receptors at higher concentrations (discussed below); 2) 5-HT2C selective agonists stabilize a 5-HT2C receptor conformation(s) that leads to signaling outcomes that antagonize 5-HT2A signaling activated by DOI that produces the HTR, i.e., mutually opposing signaling antagonism (cross-talk); or 3) selective 5-HT2C ligands are in fact not selective in vivo, and other receptors that bind these ligands negatively modulate the DOI-elicited-HTR.
Each of these outcomes, either on their own, or in combination could explain the behavioral data, and the data reported have limitations that preclude specifically defining which outcome accounts for the most variance. Nevertheless, although the third explanation cannot be ruled out without further tests (e.g. whether the behavioral effects of 5-HT2C ligands observed are eliminated in 5-HT2C receptor knock-out mice), the data support the former explanations, which are in accordance with functional selectivity. First, the 5-HT2C agonists most potent for attenuating the DOI HTR in vivo also suppressed the efficacy of DOI to stimulate mouse 5-HT2C receptor-mediated Gq/PLC signaling in vitro; Gq signaling is known to be involved in the production of the DOI-elicited-HTR (Garcia et al., 2007). Notably, there were no additive or synergistic effects of selective 5-HT2C agonists with DOI in vitro, but suppressive effects consistent with the in vivo results. It should be mentioned, however, that exclusive signaling through Gq does not mediate the DOI-elicited-HTR; Gq knock-out mice show only about a 40% reduction in the number of DOI-elicited-HTRs (Garcia et al., 2007). Also, in the present studies, irrespective of the direction of intrinsic efficacy in terms of 5-HT2C Gq-PLC signaling, i.e. inverse agonist/antagonist/agonist, all 5-HT2C-targeting compounds tested suppressed the HTR elicited by DOI, strongly suggesting that another signaling pathway(s) influences the suppressive effects of 5-HT2C selective ligands on the DOI-elicited-HTR Notably, β-arrestin-2 knock-out mice did not show changes in the DOI-elicited-HTR, suggesting alternative signaling via β-arrestin-2 can be ruled out (Schmid et al., 2008). Although the present experiments do not reveal novel signaling pathways that regulate the DOI-elicited-HTR, they encourage further testing in this area.
Surprisingly, the 5-HT2C agonists WAY 161503, Ro 60-0175, mCPP, and CP 809101 showed no difference in potency or efficacy for attenuating the DOI HTR, suggesting that potential pharmacokinetic differences between these compounds (see CLogP values in Table 1) did not affect their behavioral potencies. More attention to pharmacokinetics issues would be in order had one or more compounds within a class had opposite or no effects on the DOI HTR. Also, it is acknowledged that the negative modulation of the DOI elicited HTR by each compound could be due to a metabolite(s) and/or the parent compound, yet the similarities in behavioral effects within classes of compounds imply that the effects are related to 5-HT2 receptor modulation, and not off-targets. It seems unlikely, with what is presently known about the pharmacology of the structurally diverse compounds utilized, that they and/or their metabolites interact with a common target(s) other than 5-HT2C receptors to produce their comparable effects on the HTR (and locomotion, noted below), albeit, 5-HT2A and/or 5-HT2B receptor activation elicited by selective 5-HT2C agonists could play a role in the negative modulation of the HTR observed, as described below. These considerations, nevertheless, illustrate that there are a host of factors intervening between in vitro cellular pharmacological assays and behavioral pharmacology phenotypes.
Interestingly, 1-methylpsilocin was more potent for attenuating the DOI-elicited-HTR than all other 5-HT2C-preferring agonists that also activate 5-HT2A receptors (Sard et al., 2005). Like psilocin, 1-methylpsilocin may also activate 5-HT1 receptors that are known to negatively modulate the HTR (Darmani et al., 1990), perhaps accounting for the superior potency of 1-methylpsilocin here. Finally, the 4-phenyl-2-aminotetralin compound, (−)-PAT, that is a 5-HT2C agonist and 5-HT2A/2B antagonist in vitro and lacks appreciable affinity for other GPCRs known to modulate the DOI HTR (Booth et al., 2009), was as potent and efficacious as 1-methylpsilocin in suppressing the DOI HTR, likely due to its unique 5-HT2 pharmacology (5-HT2A/2B antagonism together with 5-HT2C agonism).
Regarding the 5-HT2 antagonists studied, the results are comparable to previous findings from the C57Bl/6J strain of mice (Canal et al., 2010), with the exception that the 5-HT2C/2B inverse agonist SB-206553 was more efficacious in suppressing the DOI-elicited-HTR than the 5-HT2C antagonist, SB-242084. Also, SB-206553 led to a bi-phasic effect on locomotion after DOI treatment, increasing activity at lower doses, and decreasing activity at higher doses. These data suggest that 5-HT2B receptors may be involved in the behaviors tested here, and/or that, at doses above 0.3 mg/kg, SB-206553 may be interacting with off-targets, including alpha-7 nicotinic receptors (Dunlop et al., 2009), that affect behavior.
The present data show clearly that 5-HT2C agonists that also activate 5-HT2A and/or 5-HT2B receptors (e.g. Ro 60-0175, mCPP, CP 809101, WAY 161503, 1-methylpsilocin) are effective at attenuating a 5-HT2A receptor-dependent behavior, the DOI-elicited-HTR Ro 60-0175 is considered a selective 5-HT2C agonist and is commonly chosen for studying in vivo functions of 5-HT2C receptors. This compound, however, also stimulates human 5-HT2A and 5-HT2B receptor-mediated Ca++ signaling in vitro, and among the 5-HT2 receptor subtypes is most potent at activating 5-HT2B receptors (Porter et al., 1999). Also, in a pilot experiment measuring human 5-HT2A receptor-mediated PLC signaling in vitro, we observed Ro 60-0175 to be a potent agonist with an EC50 value and efficacy nearly equivalent to 5-HT (data not shown). Finally, in vivo, Ro 60-0175 is known to possess activity at receptors other than 5-HT2C (Damjanoska et al., 2003). Thus, suppression of the DOI-elicited-HTR by selective 5-HT2C agonists may not be mediated solely by activation of 5-HT2C receptors, but may also involve activation of 5-HT2A and/or 5-HT2B receptors. Indeed, the non-hallucinogenic and non-HTR-eliciting compound lisuride has 5-HT2A agonist properties, but attenuates the HTR in mice elicited by the structurally and pharmacologically similar lysergic acid diethylamide (LSD) (Gonzalez-Maeso et al., 2007). Similarly, hallucinogenic 5-HT2 agonist compounds attenuate the HTR in mice elicited by LSD (Corne and Pickering, 1967). Moreover, although to a lesser extent than DOI, 1-methylpsilocin produces an HTR on its own (Halberstadt et al., 2011), yet, as shown here, 1-methylpsilocin also attenuates the DOI-elicited-HTR These observations may be accounted for by the functional selectivity hypothesis, i.e., different 5-HT2 ligands stabilize particular 5-HT2A, 5-HT2B, and/or 5-HT2C receptor conformations leading to different signaling outcomes that have unique influences on behavior. Molecular modeling studies docking DOI and other 5-HT2 ligands in the 5-HT2A, 5-HT2B, and/or 5-HT2C receptor binding pockets will help resolve whether there are specific amino acid interactions that lead to DOI’s HTR-eliciting effect. Alternative explanations for the results obtained include the possibility that different 5-HT2C ligands interact uniquely with known homodimerized 5-HT2C receptors (Herrick-Davis et al., 2012, 2004) which could impact signaling associated with the HTR. Also, the 5-HT2C receptor may heterodimerize with 5-HT2A and/or other receptors that might impact the pharmacology of 5-HT2C ligands (Gonzalez-Maeso et al., 2008). Finally, DOI may act directly at receptor systems separate from 5-HT (Ray, 2010) that could influence the HTR, complicating the picture.
Numerous studies have demonstrated that compounds interacting with 5-HT2 receptor subtypes can influence locomotion in mice. Results from the present study are generally consistent with the literature (Fletcher et al., 2009; Gleason et al., 2001 ; Halberstadt et al., 2009; Heisler and Tecott, 2000). Each of the commercially-available 5-HT2C agonists given prior to DOI produced decreases in activity, whereas the antagonists produced dose-dependent increases in activity. Notably, 5-HT2C agonist and antagonist effects on locomotion are abolished in 5-HT2C receptor knock-out mice (Fletcher et al., 2009). The 5-HT2A antagonist given prior to DOI also suppressed locomotion, as would be expected (Halberstadt et al., 2009). In contrast, the novel 4-phenyl-2-aminotetralin compounds (+)-PAT, (−)-PAT, and methyl-PAT did not alter activity relative to vehicle or DOI only administration. Based on the pharmacological profile of these compounds (5-HT2C agonism and 5-HT2A antagonism/inverse agonism), this failure to alter locomotion is somewhat surprising, however from a medication development perspective this may represent a promising and desirable side effect profile (e.g. no sedation or hyperlocomotion). The dissociation of behavioral effects (all classes of compounds decreased the DOI-elicited-HTR, while different classes of compounds showed differential effects on locomotion) demonstrates that the suppression of the DOI-elicited-HTR was not a direct consequence of an alteration in locomotor activity.
Few studies have compared directly the potencies of 5-HT2 ligands for mouse versus human 5-HT2 receptors (Dougherty and Aloyo, 2011). The current study revealed subtle differences in binding affinities of 4-phenyl-2-aminotetralin ligands and commercially available 5-HT2 antagonists for mouse versus human 5-HT2A and 5-HT2C receptors, yet the selectivity of ligands for 5-HT2A and/or 5-HT2C receptors did not change across the receptor species, suggesting that the behavioral pharmacology data cannot be explained by a loss of ligand receptor selectivity in mice. Though ligands show unique potencies for 5-HT2C receptors depending on the specific edited 5-HT2C isoform analyzed, 5-HT was shown to have equivalent potency for activating the human 5-HT2C-VNV and 5-HT2C-INI isoforms (Burns et al., 1997), suggesting the difference between mouse (5-HT2C-VNV) and human (5-HT2C-INI) receptors studied herein did not contribute greatly to the variability in binding potency. The potencies of 4-phenyl-2-aminotetralin ligands for activating mouse 5-HT2C receptor-mediated PLC signaling matched closely with their binding affinities, and each ligand was a partial 5-HT2C agonist. Interestingly, although (−)-PAT and methyl-PAT were inactive at the mouse 5-HT2A receptor, (+)-PAT displayed consistent, yet weak, partial agonist activity at this receptor. Given (+)-PAT’s higher potency for activating mouse 5-HT2A receptors, relative to mouse 5-HT2C receptors, it is notable that (+)-PAT retained efficacy at attenuating the DOI HTR, bolstering previous findings that agonists of 5-HT2A receptors unlike DOI may effectively attenuate DOI’s HTR-eliciting effects.
Results from this study suggest the possibility that 5-HT2C ligands, whether agonists or antagonists of the 5-HT2-Gq receptor signaling pathway, block the DOI-elicited-HTR by functionally attenuating or altering 5-HT2C receptor signaling activated by DOI and/or by re-directing 5-HT2C receptor signaling that subsequently alters 5-HT2A receptor signaling initiated by DOI (cross-talk). Selective 5-HT2C agonists, therefore, may behave as biased agonists for the receptor, stimulating 5-HT2C receptor signaling that is unique from DOI. The DOI-elicited-HTR may thus be an in vivo behavioral model to study ligand-specific 5-HT2C (and/or 5-HT2A or 5-HT2B) receptor conformations, and could assist in drug development of 5-HT2 receptor ligands, like the 4-phenyl-2-aminotetralins, that may have antipsychotic properties, and/or provide other therapeutic outcomes, such as weight reduction, without locomotor or sedative side effects.
Acknowledgments
The authors thank Drs. Myong Sang Kim, Rajeev Sakhuja, and Zhuming Sun at the University of Florida Department of Medicinal Chemistry for synthesizing PAT compounds. The authors also wish to thank the referees of the manuscript, who provided thoughtful comments and suggestions. These studies were funded by grants from the National Institutes of Health (NIH R01 DA023928, DA030989, and MH081193). The NIH did not have a role in the study design, in the collection, analysis, and interpretation of the data, in the writing of the manuscript, or in the decision to submit the paper for publication.
Footnotes
Conflict of interest
All authors declare that they have no financial conflicts of interest.
References
- Abbas A, Roth BL. Pimavanserin tartrate: a 5-HT2A inverse agonist with potential for treating various neuropsychiatric disorders. Expert Opin Pharmacother. 2008;9:3251–3259. doi: 10.1517/14656560802532707. [DOI] [PubMed] [Google Scholar]
- Abbas AI, Yadav PN, Yao WD, Arbuckle MI, Grant SG, Caron MG, Roth BL. PSD-95 is essential for hallucinogen and atypical antipsychotic drug actions at serotonin receptors. J Neurosci. 2009;29:7124–7136. doi: 10.1523/JNEUROSCI.1090-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arena Pharmaceuticals Inc. Arena Pharmaceuticals and Eisai Announce FDA Approval of BELVIQ® (Lorcaserin HCl) for Chronic Weight Management in Adults Who are Overweight with a Comorbidity or Obese. 2012 http://invest.arenapharm.com/releasedetail.cfm?ReleaseID=687182.
- Becamel C, Gavarini S, Chanrion B, Alonso G, Galeotti N, Dumuis A, Bockaert J, Marin P. The serotonin 5-HT2A and 5-HT2C receptors interact with specific sets of PDZ proteins. J Biol Chem. 2004;279:20257–20266. doi: 10.1074/jbc.M312106200. [DOI] [PubMed] [Google Scholar]
- Bentley JM, Adams DR, Bebbington D, Benwell KR, Bickerdike MJ, Davidson JE, Dawson CE, Dourish CT, Duncton MA, Gaur S, George AR, Giles PR, Hamlyn RJ, Kennett GA, Knight AR, Malcolm CS, Mansell HL, Misra A, Monck NJ, Pratt RM, Quirk K, Roffey JR, Vickers SP, Cliffe IA. Indoline derivatives as 5-HT(2C) receptor agonists. Bioorg Med Chem Lett. 2004;14:2367–2370. doi: 10.1016/j.bmcl.2003.05.001. [DOI] [PubMed] [Google Scholar]
- Booth RG, Fang L, Huang Y, Wilczynski A, Sivendran S. (1R, 3S)-(-)-trans-PAT: a novel full-efficacy serotonin 5-HT2C receptor agonist with 5-HT2A and 5-HT2B receptor inverse agonist/antagonist activity. Eur J Pharmacol. 2009;615:1–9. doi: 10.1016/j.ejphar.2009.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucholtz EC, Brown RL, Tropsha A, Booth RG, Wyrick SD. Synthesis, evaluation, and comparative molecular field analysis of 1-phenyl-3-amino-1,2,3,4-tetrahydronaphthalenes as ligands for histamine H(1) receptors. J Med Chem. 1999;42:3041–3054. doi: 10.1021/jm980428x. [DOI] [PubMed] [Google Scholar]
- Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997;387:303–308. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- Burris KD, Breeding M, Sanders-Bush E. (+)Lysergic acid diethylamide, but not its nonhallucinogenic congeners, is a potent serotonin 5HT1C receptor agonist. J Pharmacol Exp Ther. 1991;258:891–896. [PubMed] [Google Scholar]
- Canal CE, Cordova-Sintjago TC, Villa NY, Fang LJ, Booth RG. Drug discovery targeting human 5-HT(2C) receptors: residues S3.36 and Y7.43 impact ligand-binding pocket structure via hydrogen bond formation. Eur J Pharmacol. 2011;673:1–12. doi: 10.1016/j.ejphar.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canal CE, Morgan D. Head-twitch response in rodents induced by the hallucinogen 2,5-dimethoxy-4-iodoamphetamine: a comprehensive history, a re-evaluation of mechanisms, and its utility as a model. Drug Test Anal. 2012;4:556–576. doi: 10.1002/dta.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canal CE, Olaghere da Silva UB, Gresch PJ, Watt EE, Sanders-Bush E, Airey DC. The serotonin 2C receptor potently modulates the head-twitch response in mice induced by a phenethylamine hallucinogen. Psychopharmacology (Berl) 2010;209:163–174. doi: 10.1007/s00213-010-1784-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Corne SJ, Pickering RW. A possible correlation between drug-induced hallucinations in man and a behavioural response in mice. Psychopharmacologia. 1967;11:65–78. doi: 10.1007/BF00401509. [DOI] [PubMed] [Google Scholar]
- Cunningham KA, Fox RG, Anastasio NC, Bubar MJ, Stutz SJ, Moeller FG, Gilbertson SR, Rosenzweig-Lipson S. Selective serotonin 5-HT(2C) receptor activation suppresses the reinforcing efficacy of cocaine and sucrose but differentially affects the incentive-salience value of cocaine- vs. sucrose-associated cues. Neuropharmacology. 2011;61:513–523. doi: 10.1016/j.neuropharm.2011.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cussac D, Boutet-Robinet E, Ailhaud MC, Newman-Tancredi A, Martel JC, Danty N, Rauly-Lestienne I. Agonist-directed trafficking of signalling at serotonin 5-HT2A, 5-HT2B and 5-HT2C-VSV receptors mediated Gq/11 activation and calcium mobilisation in CHO cells. Eur J Pharmacol. 2008;594:32–38. doi: 10.1016/j.ejphar.2008.07.040. [DOI] [PubMed] [Google Scholar]
- Cussac D, Newman-Tancredi A, Quentric Y, Carpentier N, Poissonnet G, Parmentier JG, Goldstein S, Millan MJ. Characterization of phospholipase C activity at h5-HT2C compared with h5-HT2B receptors: influence of novel ligands upon membrane-bound levels of [3H]phosphatidylinositols. Naunyn Schmiedebergs Arch Pharmacol. 2002;365:242–252. doi: 10.1007/s00210-001-0505-y. [DOI] [PubMed] [Google Scholar]
- Damjanoska KJ, Muma NA, Zhang Y, D’Souza DN, Garcia F, Carrasco GA, Kindel GH, Haskins KA, Shankaran M, Petersen BR, Van De Kar LD. Neuroendocrine evidence that (S)-2-(chloro-5-fluoro-indol-l-yl)-1-methylethylamine fumarate (Ro 60-0175) is not a selective 5-hydroxytryptamine(2C) receptor agonist. J Pharmacol Exp Ther. 2003;304:1209–1216. doi: 10.1124/jpet.102.043489. [DOI] [PubMed] [Google Scholar]
- Darmani NA, Martin BR, Pandey U, Glennon RA. Do functional relationships exist between 5-HT1A and 5-HT2 receptors? Pharmacol Biochem Behav. 1990;36:901–906. doi: 10.1016/0091-3057(90)90098-3. [DOI] [PubMed] [Google Scholar]
- Dougherty JP, Aloyo VJ. Pharmacological and behavioral characterization of the 5-HT2A receptor in C57BL/6N mice. Psychopharmacology (Berl) 2011;215:581–593. doi: 10.1007/s00213-011-2207-6. [DOI] [PubMed] [Google Scholar]
- Dunlop J, Lock T, Jow B, Sitzia F, Grauer S, Jow F, Kramer A, Bowlby MR, Randall A, Kowal D, Gilbert A, Comery TA, Larocque J, Soloveva V, Brown J, Roncarati R. Old and new pharmacology: positive allosteric modulation of the alpha7 nicotinic acetylcholine receptor by the 5-hydroxytryptamine(2B/C) receptor antagonist SB-206553 (3,5-dihydro-5-methyl-N-3-pyridinylbenzo[1,2-b:4,5-b’]di pyrrole-1(2H)-carboxamide) J Pharmacol Exp Ther. 2009;328:766–776. doi: 10.1124/jpet.108.146514. [DOI] [PubMed] [Google Scholar]
- Ebdrup BH, Rasmussen H, Arnt J, Glenthoj B. Serotonin 2A receptor antagonists for treatment of schizophrenia. Expert Opin Investig Drugs. 2011;20:1211–1223. doi: 10.1517/13543784.2011.601738. [DOI] [PubMed] [Google Scholar]
- Fantegrossi WE, Simoneau J, Cohen MS, Zimmerman SM, Henson CM, Rice KC, Woods JH. Interaction of 5-HT2A and 5-HT2C receptors in R(−)-2,5-dimethoxy-4-iodoamphetamine-elicited head twitch behavior in mice. J Pharmacol Exp Ther. 2010;335:728–734. doi: 10.1124/jpet.110.172247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher PJ, Tampakeras M, Sinyard J, Slassi A, Isaac M, Higgins GA. Characterizing the effects of 5-HT(2C) receptor ligands on motor activity and feeding behaviour in 5-HT(2C) receptor knockout mice. Neuropharmacology. 2009;57:259–267. doi: 10.1016/j.neuropharm.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Flordellis CS. The plasticity of the 7TMR signaling machinery and the search for pharmacological selectivity. Curr Pharm Des. 2012;18:145–160. doi: 10.2174/138161212799040556. [DOI] [PubMed] [Google Scholar]
- Fowler JC, Bhattacharya S, Urban JD, Vaidehi N, Mailman RB. Receptor conformations involved in dopamine D2L receptor functional selectivity induced by selected transmembrane 5 serine mutations. Mol Pharmacol. 2012;81:820–831. doi: 10.1124/mol.111.075457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox M, French H, LaPorte J, Blackler A, Murphy D. The serotonin 5-HT2A receptor agonist TCB-2: a behavioral and neurophysiological analysis. Psychopharmacology. 2010;212:13–23. doi: 10.1007/s00213-009-1694-1. [DOI] [PubMed] [Google Scholar]
- Garcia EE, Smith RL, Sanders-Bush E. Role of G(q) protein in behavioral effects of the hallucinogenic drug 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane. Neuropharmacology. 2007;52:1671–1677. doi: 10.1016/j.neuropharm.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason SD, Lucaites VL, Shannon HE, Nelson DL, Leander JD. m-CPP hypolocomotion is selectively antagonized by compounds with high affinity for 5-HT(2C) receptors but not 5-HT(2A) or 5-HT(2B) receptors. Behav Pharmacol. 2001;12:613–620. doi: 10.1097/00008877-200112000-00005. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Maeso J, Ang RL, Yuen T, Chan P, Weisstaub NV, Lopez-Gimenez JF, Zhou M, Okawa Y, Callado LF, Milligan G, Gingrich JA, Filizola M, Meana JJ, Sealfon SC. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature. 2008;452:93–97. doi: 10.1038/nature06612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Maeso J, Weisstaub NV, Zhou M, Chan P, Ivic L, Ang R, Lira A, Bradley-Moore M, Ge Y, Zhou Q, Sealfon SC, Gingrich JA. Hallucinogens recruit specific cortical 5-HT(2A) receptor-mediated signaling pathways to affect behavior. Neuron. 2007;53:439–452. doi: 10.1016/j.neuron.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Maeso J, Yuen T, Ebersole BJ, Wurmbach E, Lira A, Zhou M, Weisstaub N, Hen R, Gingrich JA, Sealfon SC. Transcriptome fingerprints distinguish hallucinogenic and nonhallucinogenic 5-hydroxytryptamine 2A receptor agonist effects in mouse somatosensory cortex. J Neurosci. 2003;23:8836–8843. doi: 10.1523/JNEUROSCI.23-26-08836.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halberstadt AL, Koedood L, Powell SB, Geyer MA. Differential contributions of serotonin receptors to the behavioral effects of indoleamine hallucinogens in mice. J Psychopharmacol. 2011;25:1548–1561. doi: 10.1177/0269881110388326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halberstadt AL, van der Heijden I, Ruderman MA, Risbrough VB, Gingrich JA, Geyer MA, Powell SB. 5-HT(2A) and 5-HT(2C) receptors exert opposing effects on locomotor activity in mice. Neuropsychopharmacology. 2009;34:1958–1967. doi: 10.1038/npp.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisler LK, Tecott LH. A paradoxical locomotor response in serotonin 5-HT(2C) receptor mutant mice. J Neurosci. 2000;20:RC71. doi: 10.1523/JNEUROSCI.20-08-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick-Davis K, Grinde E, Lindsley T, Cowan A, Mazurkiewicz JE. Oligomer size of the serotonin 5-hydroxytryptamine 2C (5-HT2C) receptor revealed by fluorescence correlation spectroscopy with photon counting histogram analysis: evidence for homodimers without monomers or tetramers. J Biol Chem. 2012;287:23604–23614. doi: 10.1074/jbc.M112.350249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick-Davis K, Grinde E, Mazurkiewicz JE. Biochemical and biophysical characterization of serotonin 5-HT2C receptor homodimers on the plasma membrane of living cells. Biochemistry. 2004;43:13963–13971. doi: 10.1021/bi048398p. [DOI] [PubMed] [Google Scholar]
- Hutcheson JD, Setola V, Roth BL, Merryman WD. Serotonin receptors and heart valve diseaseeit was meant 2B. Pharmacol Ther. 2011;132:146–157. doi: 10.1016/j.pharmthera.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen NH, Cremers TI, Sotty F. Therapeutic potential of 5-HT2C receptor ligands. ScientificWorldJournal. 2010;10:1870–1885. doi: 10.1100/tsw.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya AI, Onaran HO, Ozcan G, Ambrosio C, Costa T, Balli S, Ugur O. Cell contact-dependent functional selectivity of beta2-adrenergic receptor ligands in stimulating cAMP accumulation and extracellular signal-regulated kinase phosphorylation. J Biol Chem. 2012;287:6362–6374. doi: 10.1074/jbc.M111.301820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight AR, Misra A, Quirk K, Benwell K, Revell D, Kennett G, Bickerdike M. Pharmacological characterisation of the agonist radioligand binding site of 5-HT(2A), 5-HT(2B) and 5-HT(2C) receptors. Naunyn Schmiedebergs Arch Pharmacol. 2004;370:114–123. doi: 10.1007/s00210-004-0951-4. [DOI] [PubMed] [Google Scholar]
- Koener B, Focant MC, Bosier B, Maloteaux JM, Hermans E. Increasing the density of the D2L receptor and manipulating the receptor environment are required to evidence the partial agonist properties of aripiprazole. Prog Neuropsychopharmacol Biol Psychiatry. 2012;36:60–70. doi: 10.1016/j.pnpbp.2011.08.007. [DOI] [PubMed] [Google Scholar]
- Landolt HP, Wehrle R. Antagonism of serotonergic 5-HT2A/2C receptors: mutualimprovementofsleep,cognitionandmood? Eur J Neurosci. 2009:29, 1795–1809. doi: 10.1111/j.1460-9568.2009.06718.x. [DOI] [PubMed] [Google Scholar]
- Mailman RB. GPCR functional selectivity has therapeutic impact. Trends Pharmacol Sci. 2007;28:390–396. doi: 10.1016/j.tips.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mary S, Damian M, Louet M, Floquet N, Fehrentz JA, Marie J, Martinez J, Baneres JL. Ligands and signaling proteins govern the conformational landscape explored by a G protein-coupled receptor. Proc Natl Acad Sci USA. 2012;109:8304–8309. doi: 10.1073/pnas.1119881109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ. Serotonin 5-HT2C receptors as a target for the treatment of depressive and anxious states: focus on novel therapeutic strategies. Therapie. 2005;60:441–460. doi: 10.2515/therapie:2005065. [DOI] [PubMed] [Google Scholar]
- Milligan G. Mechanisms of multifunctional signalling by G protein-linked receptors. Trends Pharmacol Sci. 1993;14:239–244. doi: 10.1016/0165-6147(93)90019-g. [DOI] [PubMed] [Google Scholar]
- Monti JM. Serotonin 5-HT(2A) receptor antagonists in the treatment of insomnia: present status and future prospects. Drugs Today (Barc) 2010;46:183–193. doi: 10.1358/dot.2010.46.3.1437247. [DOI] [PubMed] [Google Scholar]
- Motulsky HJ, Christopoulos A. A Practical Guide to Curve-fitting. GraphPad Software, Inc.; San Diego, CA: 2003. Fitting Models to Biological Data Using Linear and Non-linear Regression. [Google Scholar]
- Moya PR, Berg KA, Gutierrez-Hernandez MA, Saez-Briones P, Reyes-Parada M, Cassels BK, Clarke WP. Functional selectivity of hallucinogenic phenethylamine and phenylisopropylamine derivatives at human 5-hydroxytryptamine (5-HT)2A and 5-HT2C receptors. J Pharmacol Exp Ther. 2007;321:1054–1061. doi: 10.1124/jpet.106.117507. [DOI] [PubMed] [Google Scholar]
- Nichols DE. Hallucinogens. Pharmacol Ther. 2004;101:131–181. doi: 10.1016/j.pharmthera.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Porter RH, Benwell KR, Lamb H, Malcolm CS, Allen NH, Revell DF, Adams DR, Sheardown MJ. Functional characterization of agonists at recombinant human 5-HT2A, 5-HT2B and 5-HT2C receptors in CHO-K1 cells. Br J Pharmacol. 1999;128:13–20. doi: 10.1038/sj.bjp.0702751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray TS. Psychedelics and the human receptorome. PLoS One. 2010;5:e9019. doi: 10.1371/journal.pone.0009019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig-Lipson S, Zhang J, Mazandarani H, Harrison BL, Sabb A, Sabalski J, Stack G, Welmaker G, Barrett JE, Dunlop J. Antiobesity-like effects of the 5-HT2C receptor agonist WAY-161503. Brain Res. 2006:1073–1074. doi: 10.1016/j.brainres.2005.12.052. [DOI] [PubMed] [Google Scholar]
- Rosenzweig-Lipson S, Dunlop J, Marquis KL. 5-HT2C receptor agonists as an innovative approach for psychiatric disorders. Drug News Perspect. 2007;20:565–571. doi: 10.1358/dnp.2007.20.9.1162244. [DOI] [PubMed] [Google Scholar]
- Sard H, Kumaran G, Morency C, Roth BL, Toth BA, He P, Shuster L. SAR of psilocybin analogs: discovery of a selective 5-HT 2C agonist. Bioorg Med Chem Lett. 2005;15:4555–4559. doi: 10.1016/j.bmcl.2005.06.104. [DOI] [PubMed] [Google Scholar]
- Schmid CL, Bohn LM. Serotonin, but not N-methyltryptamines, activates the serotonin 2A receptor via a ss-arrestin2/Src/Akt signaling complex in vivo. J Neurosci. 2010;30:13513–13524. doi: 10.1523/JNEUROSCI.1665-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid CL, Raehal KM, Bohn LM. Agonist-directed signaling of the serotonin 2A receptor depends on beta-arrestin-2 interactions in vivo. Proc Natl Acad Sci USA. 2008;105:1079–1084. doi: 10.1073/pnas.0708862105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuciak JA, Chapin DS, McCarthy SA, Guanowsky V, Brown J, Chiang P, Marala R, Patterson T, Seymour PA, Swick A, Iredale PA. CP-809,101, a selective 5-HT2C agonist, shows activity in animal models of antipsychotic activity. Neuropharmacology. 2007;52:279–290. doi: 10.1016/j.neuropharm.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Teegarden BR, Al Shamma H, Xiong Y. 5-HT(2A) inverse-agonists for the treatment of insomnia. Curr Top Med Chem. 2008;8:969–976. doi: 10.2174/156802608784936700. [DOI] [PubMed] [Google Scholar]
- Thomsen WJ, Grottick AJ, Menzaghi F, Reyes-Saldana H, Espitia S, Yuskin D, Whelan K, Martin M, Morgan M, Chen W, Al-Shamma H, Smith B, Chalmers D, Behan D. Lorcaserin, a novel selective human 5-hydroxytryptamine2C agonist: in vitro and in vivo pharmacological characterization. J Pharmacol Exp Ther. 2008;325:577–587. doi: 10.1124/jpet.107.133348. [DOI] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Vincek AS, Booth RG. New Approach to 4-Phenyl-beta-aminotetralin from 4-(3-Halophenyl)tetralen-2-ol Phenylacetate. Tetrahedron Lett. 2009;50:5107–5109. doi: 10.1016/j.tetlet.2009.06.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welmaker GS, Nelson JA, Sabalski JE, Sabb AL, Potoski JR, Graziano D, Kagan M, Coupet J, Dunlop J, Mazandarani H, Rosenzweig-Lipson S, Sukoff S, Zhang Y. Synthesis and 5-hydroxytryptamine (5-HT) activity of 2,3,4,4a-tetrahydro-1H-pyrazino[1,2-a]quinoxalin-5-(6H)ones and 2,3,4,4a,5,6-hexahydro-1H-pyrazino[1,2-a]quinoxalines. Bioorg Med Chem Lett. 2000;10:1991–1994. doi: 10.1016/s0960-894x(00)00400-5. [DOI] [PubMed] [Google Scholar]