

Abstract
Apoptosis is a finely regulated process that serves to determine the fate of cells in response to various stresses. One such stress is DNA damage, which not only can signal repair processes but is also intimately involved in regulating cell fate. In this review we examine the relationship between the DNA damage/repair response in cell survival and apoptosis following insults to the DNA. Elucidating these pathways and the crosstalk between them is of great importance, as they eventually contribute to the etiology of human disease such as cancer and may play key roles in determining therapeutic response. This article is part of a Special Issue entitled “Apoptosis: Four Decades Later”.
Keywords: poly(ADP-ribose) polymerase, EGFR, GSK3, BRCA1, apoptosis, p53, DNA damage and repair, balance between cell survival and death
Cell death is a fundamental cellular response that has a pivotal role in development as well as maintaining tissue homeostasis by eliminating unwanted cells. It is composed of both controlled and uncontrolled mechanisms, including apoptosis, autophagy, and necrosis. Apoptosis is a regulated cell death process that reflects the cellular decision to die in response to cues from the environment and is executed by intrinsic cellular machinery [1, 2]. In contrast, necrosis is uncontrolled cell death brought upon by overwhelming stress. Lastly, autophagy is characterized by self destruction starting with engulfment of cytoplasmic material by the phagophore and sequestration of material to the autophagic vacuoles, where they are eventually destroyed [3]. The type and strength of stimuli, tissue type, developmental stage of the tissue, and the physiologic cellular microenvironment determines which cell death process is undertaken [2].
The human body is continuously exposed to various external and internal stresses, such as hypoxia, toxins, oxidative stress, and many others [4–10]. The ability of individual cells to adapt to these stresses is crucial for their survival. Alternatively, if too much damage has been sustained, coordinated activation of cell death processes must occur to rid the body of cells that contain potential disease initiating mutations. Thus, complex adaptation strategies such as cell cycle checkpoints, DNA damage response pathways, and programmed cell death have evolved to combat these environmental and physiological threats [5]. In this review, we will focus on one of these stresses. DNA damage, as it relates to the cell death processes. Ultimately, imbalance between DNA damage/repair and activation/inactivation of these cell death processes leads to carcinogenesis and may even alter tumor response to therapy.
APOPTOSIS
Apoptosis is a vital process of programmed cell death characterized by distinct morphological characteristics and energy-dependent biochemical mechanisms [1, 2]. It is an integral component of various homeostatic and defense processes including normal cell turnover, aging, proper development and functioning of the immune system, hormone dependent atrophy, embryonic development, and chemical-induced cell death [2]. Either too much or too little apoptosis leads to various disease conditions including autoimmune and neurodegenerative disorders, ischemic damage, and cancer [2, 9–13]. Thus, the ability to modulate the life and death of a cell has immense therapeutic potential and has been the subject of intense research over the years.
Apoptosis ultimately leads to a series of coordinated and energy-dependent activation of a group of cysteine proteases — caspases [2, 10–18]. This leads to a cascade of events that link the initiating stimuli to cellular death (Fig. 1). Early apoptosis is characterized by cell shrinkage, dense cytoplasm, tightly packed organelles, and pyknosis due to chromatin condensation [2, 14, 18, 19]. This is followed by budding which involves extensive plasma membrane bleb bing, karyorrhexis and separation of cell fragments into apoptotic bodies [2, 19]. The apoptotic bodies are subsequently phagocytosed by macrophages, parenchymal cells, or neoplastic cells and degraded within phagolysosomes [2, 14, 19]. Since apoptotic cells do not release their cellular content into the interstitial tissue and there are no inflammatory cytokines produced, there are no inflammatory reactions associated with apoptosis [2, 14, 19].
Fig. 1.
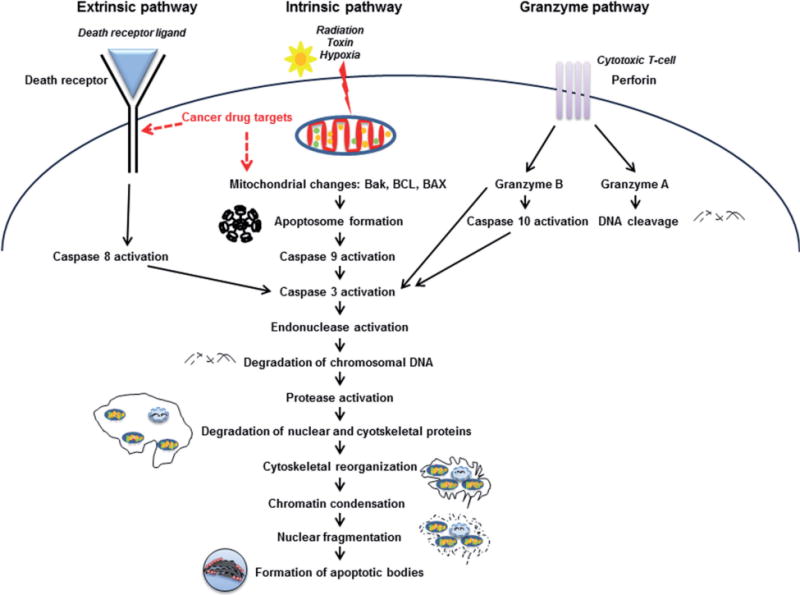
Signaling events characteristic of apoptosis
The major apoptotic pathways include the extrinsic or death receptor pathway, the intrinsic or mitochondrial pathway, and the perforin/granzyme pathway that involves T-cell mediated cytotoxicity (Fig. 1). For this review we will focus briefly on the extrinsic and intrinsic pathways. For a more in depth discussion, please refer to these excellent reviews [2, 16].
Extrinsic pathway
As mentioned above, the extrinsic apoptotic signaling is mediated by the activation of death receptors [2, 20, 21]. The death receptors are cell surface receptors that transmit apoptotic signals after binding with specific activating ligands. Death receptors belong to the tumor necrosis factor receptor (TNFR) gene superfamily, including TNFR-1, Fas/CD95, and the TRAIL receptors DR-4 and DR-5. They are characterized by cysteine rich extracellular subdomains which allow highly specific ligand recognition, subsequent trimerization, and activation of the death receptor [20]. Subsequent signaling is mediated by the cytoplasmic part of the death receptor which contains a conserved sequence termed the death domain (DD). Adapter molecules like Fas-associated death domain protein (FADD) or Tumor necrosis factor receptor type 1-associated DEATH domain (TRADD) possess the same sequence which allows them to form the death inducing signaling complex (DISC) and further propagate the signal [20, 22]. Another domain of the FADD, the death effector domain (DED), sequesters procaspase-8 to the DISC. Accumulation of procaspase-8 at the DISC leads to autocatalytic activation due to autopreoteolysis. This subsequently releases active caspase-8 which activates effector caspases resulting in cell death [2, 20, 22].
Intrinsic pathway
On the other hand, the intrinsic apoptosis pathways involve procaspase-9 which is activated downstream of mitochondrial proapoptotic events at the cytosolic death signaling protein complex, the apoptosome [2, 20]. Disruption of the inner mitochondrial transmembrane potential and permeability releases proapoptotic proteins from the mitochondrial intermembrane space into the cytoplasm. The released proteins include cytochrome c, which activates the apoptosome and therefore the caspase cascade [2, 20]. Dimerization of procaspase-9 molecules at the Apaf-1 scaffold induces caspase-9 activation and subsequent proteolytic activation of the effector procaspases-3, -6, and -7 [2, 20]. These cleave protein substrates, including procaspases, resulting in the mediation and amplification of the death signal and eventually in the execution of cell death [2, 20].
There is significant crosstalk between the pathways and molecules in one pathway can influence the other [2, 20]. The pathways converge on the same execution route which is initiated by the cleavage of caspase 3 and results in DNA fragmentation, degradation of cytoskeletal and nuclear proteins, cross-linking of various proteins, formation of apoptotic bodies, expression of ligands for phagocytic cell receptors and phagocytosis [2]. Ultimately, activation of caspases leads to an irreversible cascade of events progressing towards cell death.
As mentioned earlier, many cellular stresses can impact survival versus death pathways. In the next section, we focus on one particular cell stress, that is, DNA damage.
THE DNA DAMAGE RESPONSE
The human genome is under constant attack which leads to thousands of DNA lesions per day. The cellular response to DNA damage is critical for maintenance of genomic integrity [4, 5, 23]. Dysregulation of this DNA damage response leads to genomic instability which can result in the inactivation of pro-apoptotic pathways and the survival of cells that are polyploid, contain damaged DNA, and have dysregulated telomere maintenance [4, 5, 23, 24]. Suppression of the tightly regulated apoptotic process may thus play a critical role in the development of some cancers [4, 5, 8, 23].
Combating this malignant transformation process is the DNA damage response, a complex mechanism to detect the above mentioned lesions, signal their presence, and promote their repair. Additionally, mechanisms are in place such that if the damage is too great or repair is ineffective, activation of cell death pathways such as apoptosis or necrosis ensues (Fig. 2) [5, 8, 23]. These steps are in place to combat the threats posed by excess or unrepaired DNA damage [5, 8, 23]. In this next section, we briefly discuss the different types of DNA damage and the cellular responses to such damage to ultimately regulate cell fate.
Fig. 2.
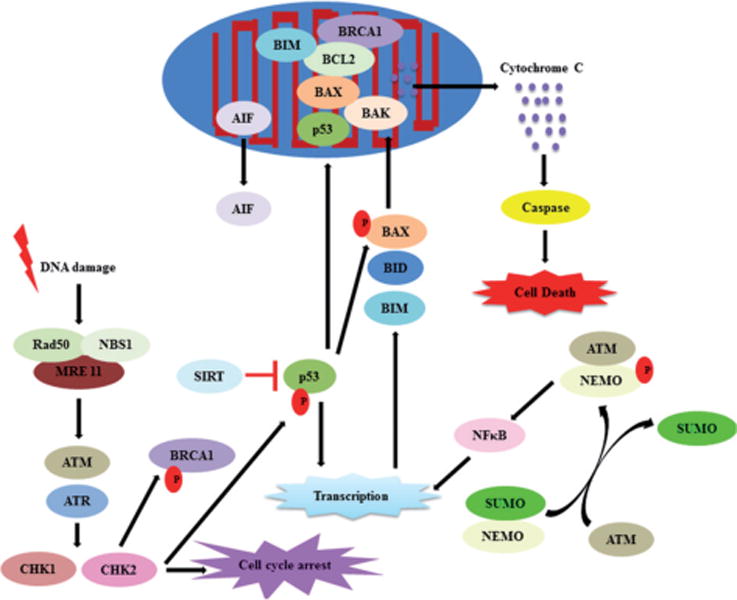
The complex relationship between DNA damage, repair and apoptosis. DNA damage triggers cellular responses such as cell cycle arrest, post-translational protein modifications, DNA repair, and transcription of pro/anti survival genes. The balance between these processes ultimately determines cell fate
DNA can be damaged by exogenous agents such as radiation, x-ray, UV, alkylating agents, as well as by the by-products from endogenous processes such as reactive oxygen and nitrogen species from metabolism and errors from DNA replication [5–8, 23]. The resultant DNA damage either involves one or both strands of the DNA (single strand vs. double strand breaks, respectively). While unresolved single strand breaks (SSBs) can be converted to double strand breaks (DSBs) and repaired, unrepaired DSBs can lead to severe consequences in cells. DSBs can be mutagenic, since they can potentially affect the expression of multiple genes. Most importantly, as little as one unrepaired DSB can be lethal to the cell [5–8, 23].
Exogenously, ultraviolet and other types of high energy radiation can induce SSBs and DSBs. UV-induced damage can also result in the production of pyrimidine dimers, where covalent cross-links occur in cytosine and thymine residues, disrupting DNA polymerases and preventing DNA replication [5–8]. Other agents can form DNA adducts and inter/intra strand crosslinks which, if left unrepaired, can lead to permanent mutations resulting in cell transformation and ultimately tumor development [5–8].
Endogenously, oxidative DNA damage can occur and involves oxidation of specific bases. 8-hydroxydeoxyguanosine (8-OHdG) is the most common marker for oxidative DNA damage [5–8]. Oxidative stress plays a central role in the pathophysiology of age-induced apoptosis via accumulated free radical-induced damage to the mitochondria. On the other hand, hydrolytic DNA damage involves deamination or the total removal of individual bases. Loss of DNA bases, known as AP (apurinic/apyrimidinic) sites, can be particularly mutagenic and if left unrepaired they can inhibit transcription [7, 25]. Interestingly, hydrolytic damage may result from the overabundance of reactive oxygen species, often a byproduct of respiration. Of course, the type and severity of damage to the DNA dictate the cellular response and ultimately cell fate [25].
Regardless of the type of DNA damage, the response to this insult involves sensing the damage, activating the checkpoints, and repairing/resolving the DNA lesions. These processes will be discussed below.
DNA damage sensors
In order to initiate the DNA damage response, DNA damage sensors must first detect the aberrant DNA lesions. The Mre11–Rad50–Nbs1 (MRN) complex acts as the sensor of DNA damage and maintains genomic stability by processing DNA ends and recruiting/bridging other members of the DNA damage response [5, 15, 23, 26–28]. Specifically, Rad50 recognizes the DNA, Nbs1 recruits other DNA repair proteins to DSB lesions, and Mre11 processes the DNA ends with its DNA nuclease activity [27]. This leads to activation of Ataxia Telangiectasia Mutated (ATM) or Ataxia Telangiectasia and Rad3 Related (ATR) depending on where the damage resides (ATM: DNA DSBs on chromatin vs. ATR: stalled replication forks) [26, 29, 30]. It is at this point where a cascade of signaling events is orchestrated to activate checkpoints and assemble the remaining members of the DNA repair complex.
Checkpoint activation
Upon sensing the DNA damage, a coordinated activation of DNA damage checkpoints as well as DNA repair proteins is required to arrest the cell cycle, thus allowing time for repair processes [24]. Checkpoints also induce changes in telomeric chromatin and recruitment of DNA repair proteins to sites of DNA damage, activation of transcription, telomere length, and induction of cell death by apoptosis [24]. Not surprisingly, several checkpoint genes are essential for cell and organism survival.
Chk1, a serine/threonine-protein kinase is required for checkpoint-mediated cell cycle arrest and activation of DNA repair in response to the presence of DNA damage or unreplicated DNA. Chk1 binds to and phosphorylates CDC25 which creates binding sites for 14-3-3 proteins and triggers degradation of CDC25 via ubiquitination pathway and proteosomal degradation [30, 31]. This leads to increased inhibitory tyrosine phosphorylation of CDK-cyclin complexes and blocks cell cycle progression. Chk1 also binds to Rad51 which promotes the release of Rad51 from BRCA2, increasing the chromatin association of Rad51 and subsequent HR-mediated DNA repair [32]. Chk1 also promotes repair of DNA cross-links by phosphorylating FANCE which is required for the nuclear accumulation of FANCC and provides a critical bridge between the FA complex and FANCD2 [33]. Chk1 also plays an essential role in maintenance of replication fork by regulating PCNA [33]. Besides, it also plays a role by modulating transcription of genes involved in cellcycle progression through phosphorylation of histones and subsequent epigenetic silencing of genes. Chk1 phosphorylates Rb1 to promote its interaction with the E2F family of transcription factors triggering subsequent cell cycle arrest and phosphorylates p53 activating the protein and promoting cell cycle arrest as well [31, 34].
Chk2 functions similar to Chk1, regulating cell cycle checkpoint arrest through phosphorylation of CDC25, inhibiting their activity [30, 32–37]. Inhibition of CDC25 phosphatase activity leads to increased inhibitory tyrosine phosphorylation of CDK-cyclin complexes and blocks cell cycle progression. Chk2 also phosphorylates NEK6 which is involved in G2/M cell cycle arrest [35]. Similar to Chk1. Chk2 regulates HR-mediated DNA repair through phosphorylation of BRCA2, enhancing the chromatin association of RAD51. Moreover, Chk2 promotes the transcription of genes involved in DNA repair (including BRCA2) through the phosphorylation and activation of the transcription factor FOXM1 [32]. Chk2 also regulates apoptosis through the phosphorylation of p53, MDM4 and PML [34, 36, 38]. Chk2 mediated phosphorylation of p53 reverses inhibition by MDM2, leading to accumulation of active p53. Chk2 dependent phosphorylation of MDM4 also functions to reduce degradation of p53. The kinase also controls the transcription of pro-apoptotic genes through phosphorylation of the transcription factor E2F1. Finally, Chk2 has a tumor suppressor role as well in that it functions in mitotic spindle assembly by phosphorylating BRCA1 and absence of Chk2 has been observed in some cancers [30, 33–38].
Cyclin dependent kinase (CDK) family of serine/threonine kinases regulate cell cycle progression through phosphorylation of proteins that function at specific phases of the cell cycle [26, 39]. Different CDKs act at different phases of the cell cycle and their activity is each dependent on association with a member of the cyclin family of proteins. Cdk1-cyclin B is important for the M phase transition while Cdk2-cyclin E association is critical for G1/S transition. Cdk2-cyclin E also functions in the S and G2 phases while CDK4-cyclin E and CDK6-cyclin D control progression through the G1 phase of the cell cycle by phosphorylation of the tumor suppressor protein. Rb [26, 39]. These proteins have thus been actively studied for cancer therapy.
DNA repair pathways
Once the DNA damage has been sensed and checkpoints activated, the process of repairing this damage is initiated. We will first focus on the DSB repair pathways, of which there are 2 major pathways: the homologous recombination (HR) and non-homologous end joining (NHEJ) [6–8, 25, 40]. HR relies on the presence of a sister chromatid and the cell cycle-regulated 5`-to-3` resection of DNA ends that generates stretches of single stranded DNA. This single stranded DNA is bound by replication protein A (RPA). BRCA2 subsequently binds Rad51 and promotes its loading onto RPA coated single stranded DNA to produce a RAD51-single stranded DNA nu-cleoprotein filament. RPA-coated ssDNA also leads to recruitment and activation of the checkpoint kinase ATR, which phosphorylates various targets, including the downstream checkpoint kinase CHK1. This cascade of events promotes DNA strand invasion and subsequent HR events. Because of the use of the homologous sister chromatid as template, HR is an error free repair pathway and the major mechanism utilized by cells for repairing DSBs and restarting stalled replication forks [8, 26].
On the contrary, the error-prone NHEJ involves connecting and resealing the two ends of DNA DSB without the need for sequence homology between the ends. Thus, this process is not dependent on the cell cycle and is, in fact, active throughout all phases of the cell cycle. It involves among others, Ku 70/80, DNA protein kinase family of proteins [8, 25, 40].
It is not yet clear what dictates the choice of repair pathway. Research suggests that the choice between NHEJ or HR pathways depends on cell cycle stage; NHEJ is active throughout the cell cycle, and its activity increases as cells progress from G1 to G2/M (G1 < S < G2/M). HR is nearly absent in G1, most active in the S phase, and declines in G2/M [7, 8, 25, 40]. The overall efficiency of NHEJ is higher than HR at all cell cycle stages. Cells usually utilize error-prone NHEJ as the major DSB repair pathway at all cell cycle stages, while HR is used primarily in the S phase. Reports also suggest key repair players such as CtIP, BRCA1, Ku, and others to impact the choice of DSB repair by controlling the initial events of DSB repair such as DSB end processing/resection [41–45].
For SSBs, different repair processes are utilized. Usually, the intact complementary strand can be used as a template to repair the damaged strand via a variety of repair mechanisms like base excision repair (BER) to repair damage to a single base caused by oxidation, alkylation, hydrolysis, or deamination, nucleotide excision repair (NER) to repair bulky, helix-distorting lesions such as pyrimidine dimers and photo-adducts, and mismatch repair (MMR) to corrects errors of DNA replication and recombination which may have resulted in mispaired (but undamaged) nucleotides [5, 7, 8, 23]. Formation of SSBs is closely linked to damaged bases and their attempted repair. It is worth noting that DSBs may form during the attempted repair of SSBs [5, 7, 8, 23]. Interestingly, the chromatin structure may be modulated to facilitate protein recruitment during repair [23]. Modifications of DNA-associated histone proteins maintain genomic stability. Upon the induction of DNA damage, phosphorylation of histones dictates if repair is justified or apoptosis is warranted [23, 46]. It is truly amazing how a cell decides whether to pursue repair and when to abort repair to favor apoptosis.
In this next section, we will discuss various key players in the DNA damage response who also play vital roles in regulating apoptosis.
INTERSECTION BETWEEN DNA DAMAGE RESPONSE AND APOPTOSIS
DNA damage sustained from normal DNA replication/cell processes, stress, mitotic catastrophe, agents such as radiation, toxins, hormones, growth factors, cytokines, and drugs, and reactive oxygen species can induce apoptosis if left unrepaired [4, 5, 7, 8, 23]. Additionally, current DNA damaging agents used in therapies act to overwhelm cellular DNA repair capacity to activate cell death processes. For example, irradiation, a standard treatment modality for a number of cancers such as brain, breast, and prostate, as well as a variety of chemotherapeutic agents induce DNA damage leading to apoptosis [4, 5, 7, 8, 23, 47]. Below, we will discuss several key players that intersect the DNA repair pathways with apoptotic pathways.
P53 and apoptosis
The tumor suppressor protein p53 has been shown to mediate cellular stress responses in that p53 can initiate DNA repair, cell-cycle arrest, senescence and, importantly, apoptosis [34, 36, 38, 40, 48–52]. These responses suppress tumor formation. Thus, it is not surprising that most tumors have p53 mutations [52]. p53 mediates DNA damage response by stimulating the nuclear release of histone H1. Phosphorylation is one of the primary post-translational modifications of p53 and this increases its stability. Various kinases such as ATM, Chk1 and Chk2 are responsible for the phosphorylation of p53. Other post-translational modifications such as acetylation, ubiquitination, methylation, sumoylation, and neddylation also regulates p53 protein stability and transcriptional activation [36, 38, 40, 48, 50, 52]. The E3 ubiquitin ligase MDM2 regulates p53 activity by binding to its N-terminal transactivation domain, thus preventing its interaction with other transcriptional factors. MDM2 also induces nuclear export of p53 and targets it for proteasomal degradation [36, 38, 40, 48, 50, 52]. In event of failed DNA repair p53 initiates apoptosis by transactivating pro-apoptotic proteins such as BAX, BID, PUMA and NOXA which permeabilizes the mitochondrial membrane and leaks the pro-apoptotic factors [36, 38, 40, 48, 50, 52]. p53 stimulates the extrinsic and/or the intrinsic pathway depending on the DNA damage. p53 has been reported to bind to the outer mitochondrial membrane and antagonize the anti-apoptotic function of BCL2 and BCL-XL. p53 represses the activity of BCL2, an anti-apoptotic protein involved in retaining the mitochondrial permeability, as well as survivin. Moreover, p53 also initiates apoptosis via proteins localized on the endoplasmic reticulum and plasma membrane such as DR5. p53 can also exert transcription independent effects on mitochondria membrane permeability by activating the pro-apoptotic protein BAX or by neutralizing the anti-apoptotic proteins BCL2 or BCL-XL [34, 36, 38, 40, 48, 50, 52].
Tumor cells possessing wild type p53 undergo apoptosis to a greater extent following DNA damage than cells that possess mutations in p53. DNA damage can also activate p53-independent apoptosis which may be called upon especially in cases in which p53 are mutated. The p53 homologs p63 and p73 are involved in this response. Unlike mutations in p53, mutations in p73 do not predispose to tumor formation but do have an impact on the DNA damage response. p73 is also often overexpressed in cancer. As mentioned above, in response to DNA lesions, ATM and/or ATR activate CHK1 and CHK2, which in turn activate E2F1 [38]. This in turn stimulates transcription of the p73 gene, increasing the levels of p73 protein. While p53 requires p63 and p73 to activate apoptosis, p73 is pro-apoptotic even in the absence of p53. p73-induced apoptosis is mediated by transcriptional upregulation of PUMA, which in turn induces mitochondrial translocation of BAX and cytochrome c release (discussed above). p73 also induces mitochondrial dysfunction via NOXA. On the other hand, p63 has the ability to suppress p73-mediated apoptosis [34, 36, 38, 40, 48, 50, 52].
Nuclear factor-κB (NF-κB) also has a role in p53-independent apoptosis. This transcription factor is generally anti-apoptotic and promotes survival. Activation of NF-κB in response to DNA damage is mediated by SUMOylation and ATM-dependent phosphorylation of NEMO (NF-κB essential modulator). Under some circumstances, however, NF-κB exhibits pro-apoptotic activity. For instance, presence of excess reactive oxygen species can induce NFκB-mediated transcription of the FAS ligand, thereby stimulating apoptosis [53]. NF-κB induces TNF-α production and subsequent receptor-interacting protein 1 (RIP1) autophosphorylation. In association with NEMO, RIP1 kinase promotes JNK3-mediated induction of IL-8 and recruits FADD to activate caspase 8 which then induces apoptosis [54]. p53-independent apoptosis can also be triggered by BCL-2 degradation and GSK3 which is detailed in a later section [55].
BRCA1 and apoptosis
The tumor suppressor BRCA1 plays an integral role in the maintenance of genomic stability and modulates cellular response to DNA damage [56–58]. It is involved in both the major DNA double strand break repair pathways — HR and NHEJ [56]. BRCA1 functions in a variety of cellular processes including chromatin remodeling, protein ubiquitination, DNA replication, DNA repair, regulation of transcription, cell cycle checkpoint control, and apoptosis [56–58]. BRCA1 function is regulated through diverse mechanisms including transcription control, protein-protein interaction, and post-translational modification [49, 56, 58, 59]. BRCA1 is a nuclear-cytoplasmic shuttling protein and its function may be regulated via active shuttling between the cellular compartments [49, 56, 58, 59]. When nuclear, BRCA1 controls high fidelity repair of damaged DNA. In contrast, BRCA1 has been shown to enhance p53-independent apoptosis when cytoplasmic [49, 56, 58, 59]. BRCA1 contains two nuclear localization signals which target it to the nucleus via importin and two nuclear export sequences which transport it to the cytoplasm via the CRM1/exportin pathway [49, 56, 58–60].
As mentioned above, BRCA1 shuttling can also be regulated via protein-protein interaction. The BRCA1-associated RING domain protein (BARD1) has been shown to prevent CRM1 dependent nuclear export of BRCA1 by binding to and masking the BRCA1 NES located at the N-terminal RING domain. On the contrary, the BRCA1 C-terminus (BRCT) domain has been shown to play a crucial role in DNA damage-induced nuclear import of BRCA1 through association with numerous other proteins, including p53, CtIP and BACH [45, 49, 56, 60]. p53 seems to be an important player in DNA damage induced BRCA1 nuclear export since human breast cancer cells with deficiency in p53 function exhibit aberrant BRCA1 shuttling [49]. Mutations that target the BRCT region of BRCA1 have been shown to exclude BRCA1 from the nucleus by blocking nuclear import. Thus, the critical region responsible for regulating the location of BRCA1 appears to reside in the BRCT domain. Nuclear exclusion of BRCA1 can be therapeutically exploited with poly(ADP-ribose) polymerase (PARP) inhibitors as well as other DNA damaging agents such as cisplatin [49].
In addition to the repair of damaged DNA. BRCA1 plays a role in apoptosis [61–64]. Specifically overexpression of BRCA1 induces apoptosis and the process has been linked to DNA damage-induced BRCA1 nuclear export and the c-Jun N-terminal kinase pathway [65]. BARD1, which binds and masks the BRCA1 nuclear export sequence to prevent BRCA1 nuclear export, inhibits this BRCA1-mediated apoptosis. Moreover, the apoptotic pathway stimulated by BRCA1 is independent of p53 [56, 59].
BRCA1 has also been reported to be present in the mitochondria where it promotes BCL2 mediated apoptosis. BCL2-mediated targeting of BRCA1 to the endomembranes depletes BRCA1 from the nucleus resulting in decreased HR-mediated repair [66, 67]. In addition, BCL2 expression is low in BRCA1-associated tumors [68].
DNA-dependent protein kinase (DNA PK) and apoptosis
The DNA-dependent protein kinase (DNA PK) plays a critical role in DSB repair and V(D)J recombination [69]. DNA PK plays a central role in the NHEJ pathway for DSB repair in mammalian cells via autophosphorylation events as well as its association with other DNA repair proteins such as BRCA1 and Ku. Ku binds to the DNA end and recruits DNA PK, stabilizing its binding to DNA. This is followed by bridging of the broken ends by DNA PK to facilitate rejoining. DNA-PK also recruits and activates proteins involved in DNA end-processing and ligation. The coordinated assembly of Ku and DNA-PKcs on DNA ends is followed by recruitment of the DNA ligase IV–XRCC4 complex that is responsible for the rejoining step [5, 7, 8, 23, 69].
DNA PK is present at the telomere and cap chromosome ends, protecting them telomere and preventing chromosome end-to-end fusions. This interaction with telomerase helps to maintain telomere length as well [70]. Recently, it was reported that poly(ADP-ribose) polymerase 1 (PARP1) interacts genetically with the DNA PK catalytic subunit to prevent cancer (lymphoma) by suppressing p53 mutation and telomere fusions [71]. The role of PARP in DNA repair and apoptosis is discussed in a subsequent section.
DNA PK also plays a crucial role in triggering apoptosis in response to severe DNA damage or critically shortened telomeres [70, 72, 73]. The ability to trigger apoptosis in the presence of unresolved DNA damage is critical for preventing progression to cancer. In response to DNA damage, DNA PK phosphorylates p53 and triggers p53-dependent apoptosis. Conversely, DNA PK undergoes proteasomal degradation later on in the apoptotic process which aids to suppress pro-survival signals [70]. The Ku70 subunit of DNA-PK has been shown to suppress apoptosis by sequestering Bax from mitochondria [74]. Increased acetylation of cytoplasmic Ku70 disrupts the Ku70-Bax interaction augmenting apoptosis [75].
PARP and apoptosis
PARP is a family of proteins involved in a number of cellular processes including DNA repair and apoptosis [76]. PARP is predominantly located in the nucleus where it promotes BER-mediated DNA single strand break repair by binding to the DNA and inducing a structural modification [77]. It also induces the synthesis of poly(ADP-ribose) (PAR) chains which acts as a signal for other DNA repair proteins. An early transient burst of poly(ADP-ribosyl)ation (PARylation) of nuclear proteins followed by caspase-3 mediated cleavage of PARP is required for apoptosis to proceed [8, 76–81]. This inactivation of PARP prevents depletion of NAD (a PARP substrate) and ATP, which are required for later events in apoptosis. PARylation plays diverse roles in many molecular and cellular processes, including DNA damage detection and repair, chromatin modification, transcription, cell death pathways, and mitotic apparatus function [76, 77, 79–81]. These processes are critical for genome maintenance, carcinogenesis, aging, inflammation, and neuronal function [8, 76–82].
PARP-1 interacts physically and functionally with various proteins involved in these DNA repair pathways, and recruits the repair proteins to sites of DNA damage such as XRCC-1 in BER and DNA PK in NHEJ-mediated repair. PAR, as covalent attachment of automodified PARP-1 and PARP-2, acts to recruit repair proteins to sites of DNA damage [8, 71, 76–78, 81, 82].
PARP dependent SSB repair and BRCA1- and BRCA2-dependent DSB repair has been exploited for cancer therapy [6–8, 78, 83]. As mentioned above. BRCA1 and BRCA2 are tumor-suppressor proteins important for DSB repair by HR, and mutation of the genes encoding these proteins causes predisposition to breast and ovarian cancers. PARP inhibitors have shown promising results in BRCA deficient tumors and other DNA repair deficient tumors in clinical trials when combined with other cytotoxic agents [83–87].
We and others have recently reported that the PARP inhibition induces apoptosis in a variety of cell types. We have shown that in conjunction with EGFR inhibitors, the PARP inhibitor ABT-888 activates the intrinsic apoptotic pathway as evidenced by cleavage of caspase 3 and 9 [78]. PARP inhibitor treatment also induces phosphorylation of DNA PK and stimulates error-prone NHEJ-mediated repair in HR-deficient cells, resulting in cell death. PARP1 catalytic activity possibly regulates NHEJ in the absence of HR and thus, deregulated NHEJ may explain the exquisite cytotoxicity of HR deficient cells to PARP inhibitors [78, 88].
PARP1 inhibitor induces caspase-independent cell death as well. It causes mitochondrial depolarization, mitochondrial permeability transition and mitochondrial release of AIF which, upon release from the mitochondria, translocates into the nucleus, where it triggers nuclear DNA fragmentation [89, 90]. Thus, PARP inhibitors tilt cell death from necrosis to apoptosis in cancer cells [91 ].
ATM/ATR and apoptosis
Defects in ATM are associated with cancers such as T-cell pro-lymphocytic leukemia, and B-cell chronic lymphocytic leukemia [6–8, 92]. Defective ATM also predisposes to sporadic colon cancer in tumors with microsatellite instability [92]. Loss of ATM results in hypersensitivity to radiation and defect in cell cycle arrest [92]. ATM is also involved in p73 mediated apoptosis [93]. Radiation induces ATM-dependent c-Abl phosphorylation which then activates p73. Upon commitment to apoptosis, caspases (cysteine aspartic acid proteases) are activated in a proteolytic cascade and ATM is cleaved by a caspase-3-like apoptotic protease. This generates a truncated ATM protein devoid of kinase activity but still retaining its DNA binding ability. This functions to prevent further DNA repair and propagation of DNA damage signaling [94, 95].
As mentioned before, ATR activates p53 in response to DNA damage by phosphorylating p53 [29, 30, 36, 50]. In response to DNA damage, ATR also phosphorylates and activates Chk1, which in turn phosphorylates p53 and regulates cell cycle progression. ATR also mediates phosphorylation of BRCA1 in response to UV [29, 30, 36, 50]. Thus, these signaling pathways can converge on both p53 and BRCA1 mediated apoptosis.
ATM/ATR also mediates BID phosphorylation, which is required for DNA damage-induced intra-S phase checkpoint [96]. An intact BH3 domain is required for apoptosis but not BID-mediated S phase effects. Thus the two functions may be distinct. BID accumulates in both the nucleus and mitochondria following stress suggesting a possible role in DNA damage response. Furthermore, like ATM and ATR. BID localizes to the chromatin fraction of the nucleus following treatment with DNA-damaging agents [96].
Sirtuins (SIRT) and apoptosis
Sirtuins (SIRT) are a family of histone deacetylases which also has a role in the maintenance of genomic stability [97]. They also possess mono-ribosyltransferase activity [97, 98]. SIRT have been implicated in influencing aging and regulating transcription, apoptosis and stress resistance [99]. For instance, SIRT1 can deacetylate various factors linked to the repair of DNA damage, including the Werner helicase and NBS1 [100–102]. Absence of SIRT results in increased chromosomal aberrations and impaired DNA repair. In addition SIRTs are recruited to sites of DNA breaks following DNA damage to avoid genomic instability [103]. Thus, SIRTs regulate epigenetic silencing and chromatin modification.
SIRT1, the human Sir2 homolog, acts as a negative regulator of the transactivation function of p53 by binding and deacetylating p53, thereby repressing apoptosis induced by DNA damage [104, 105]. Persistent lesions keep PARP in an activated state and the nicotinamide produced by the process inhibits SIRT1. This leads to hyperacetylation and enhanced transactivation of p53 which in turn leads to increase in the transcription of pro-apoptotic genes and subsequent apoptosis. Besides p53, SIRT1 can regulate other targets linked to cell death, including Ku70, E2F1 and TGF-β signaling [106, 107].
Another SIRT-mediated pro-survival pathway involves the Forkhead box class O (FOXO) transcription factors which control the expression of genes involved in apoptosis such as Fas ligand, Bim, TRAIL, cyclin D. Gadd45, p27/Kip1, Gadd45, MnSOD. Akt phosphorylates FOXO factors in the presence of growth factors [108, 109]. This prevents their nuclear translocation. However, when the growth factor signaling is switched off, FOXOs are located in the nuclei and act as transcription factors. Acetyltransferases, PCAF and p300/CBP mediated acetylation silences the transcription factors. The transcriptional activity of FOXO3 is restored by deacetylation carried out by SIRT1 which leads to resumption of gene transcription including DNA damage checkpoint genes. This increases the ability of FOXO to induce cell cycle arrest and resist oxidative stress [97–99, 106–109]. Thus SIRT promotes cell survival via transcriptional regulation. Moreover, the concerted action of SIRT3 and SIRT4 appear to inhibit cell death by maintaining mitochondrial NAD levels following stress [98, 110]. It should be noted that although SIRT predominantly antagonize stressinduced cell death pathways, SIRT1 can also deacety late components of the NFκB complex, leading to increased cell death, primarily senescence [98, 111].
Epidermal growth factor receptor (EGFR) and apoptosis
The epidermal growth factor receptor (EGFR) plays an important role in the development and progression of solid tumors. In addition, EGFR activation also mediates resistance to chemotherapy and radiation therapy [78, 88]. EGFR inhibition down-modulates survival pathways and shifts towards the proapoptotic Bcl-2 expression and/or activation [112].
There are multiple inhibitors of EGFR that are currently either used as a standard of care or are in clinical trials. Inhibitors of the tyrosine kinase activity of EGFR compete with ATP for binding to the tyrosine kinase pocket of the receptor. They have significant antitumor activity since the EGFR-TKIs block signaling by both ERK and AKT pathways and induce apoptosis [113]. EGFR mediated apoptosis requires an active kinase but not EGFR autophosphorylation sites, meaning the truncated receptor can generate the apoptotic signal. EGFR can activate Ras and the induction in apoptosis is due to impaired Akt activation. EGFR inhibition induces BIM expression via inhibition of the MEK-ERK pathway and BIM induction plays a key role in EGFR-TKI-induced apoptosis. Research suggests that both the PI3K-AKT-survivin and MEK-ERK-BIM pathways contribute independently to EGFR inhibitor-induced apoptosis [114–116].
The BH3-only protein PUMA (p53 upregulated modulator of apoptosis) plays an essential role in p53-dependent and -independent apoptosis. PUMA mediates apoptosis through the Bcl-2 family proteins Bax/Bak and the mitochondrial pathway [117]. PUMA is also induced by EGFR inhibitors independent of p53. EGFR inhibitors block phosphorylation of EGFR and inhibit the PI3K/AKT pathway, which leads to increased expression of p73 and its binding to the PUMA promoter and subsequent transactivation [118]. Thus, PUMA functions as a critical mediator of EGFR inhibitor-induced apoptosis, especially in head and neck cancer cells where EGFR inhibitors are widely used. Moreover, p73, p63, and the PI3K/AKT pathway serve as key regulators of PUMA induction after EGFR inhibition [119].
Recent evidence also suggests a key role of EGFR in both major DNA DSB repair pathways. Specifically EGFR binds and activates DNA PK for NHEJ [120, 121]. Additionally, EGFR inhibitors have been shown to attenuate DNA repair pathways. Interestingly, we recently reported that the EGFR inhibitor cetuximab reduced both NHEJ and HR in head and neck cancer cells and subsequently induced a synthetic lethality with the PARP inhibitor ABT-888 [78]. This enhanced cytotoxicity was associated with activation of the intrinsic apoptotic pathway [78]. This brings forth the possibility that other DNA repair proteins may be involved in the apoptotic response. We are actively investigating this avenue.
Glycogen synthase kinase 3 (GSK3) and apoptosis
Another important player in linking extracellular signals to DNA damage/repair and ultimately apoptosis is the glycogen synthase kinase 3 (GSK3). Phosphorylation of substrates by GSK3 allows it to modulate key processes including cell structure, metabolism, gene expression and apoptosis [122]. GSK3 has the unique capacity to either increase or decrease the apoptotic threshold due to its opposing regulation of the two major apoptotic signaling pathways. GSK3 promotes cell death caused by the mitochondrial intrinsic apoptotic pathway, but inhibits the death receptormediated extrinsic apoptotic signaling pathway [123]. GSK3 is involved in the apoptotic response following growth factor withdrawal, inhibition of the PI3K/Akt signaling pathway, DNA damage, ER stress, hypoxia/ischemia, and oxidative stress [123]. Intrinsic apoptotic signaling which is activated by cell damage is promoted by GSK3 by facilitation of signals that cause disruption of mitochondria and by regulation of transcription factors that control the expression of anti- or pro-apoptotic proteins. These transcription factors include p53 which was discussed above and cyclic AMP response element binding protein (CREB) [123, 124]. GSK3β activity in the nucleus promotes p53-induced expression of Bax in response to DNA damage and inhibits CREB, which can block the CREB-dependent expression of the anti-apoptotic protein Bcl-2 [123]. GSK3 can regulate p53 levels through the phosphorylation of the p53-regulating protein MDM2 as well as by directly interacting with p53 [125]. GSK3 promotes p53-mediated transcription of specific genes and regulates the intracellular localization of p53 [125]. p53 is also able to activate apoptosis independently of its transcription function by acting directly on mitochondrial proteins, and GSK3β binds p53 in the mitochondria, which may contribute to p53-induced apoptosis [126]. In the canonical Wnt signaling pathway, the transcriptional co-activator β-catenin promotes growth and survival, but phosphorylation of β-catenin by GSK3 targets it for proteosomal degradation thereby promoting apoptosis [122]. Activation of Wnt signaling inhibits GSK3 selectively in the Wnt signaling protein complex, causing accumulation of β-catenin and its translocation to the nucleus where it interacts with the TCF/LEF transcription factors to induce expression of pro-survival genes. GSK3 phosphorylates heat shock factor-1 (HSF-1), a pro-survival transcription factor to inhibit its activity, thereby reducing expression of heat shock proteins, an action that can facilitate apoptosis [127]. Since GSK3 is present in the mitochondria as well and there is a spike in GSK3 levels following DNA damage. GSK3 is uniquely positioned to regulate apoptosis.
Pro-apoptotic members of the Bcl-2 family of proteins such as Bax transmit the apoptotic signal to the mitochondria following phosphorylation by GSK3. Stress such as DNA damage induces a conformational change in Bax that promotes its translocation from the cytoplasm to the mitochondria where it can both sequester anti-apoptotic Bcl-2 family proteins and oligomerize within the mitochondrial membrane. This as well as phosphorylation of the voltage-dependent anion channels by GSK3 disrupts the mitochondrial membrane potential and releases apoptotic proteins such as cytochrome c from the mitochondrial intermembrane space into the cytoplasm. Cytochrome c in turn binds to the protein apoptotic protease activating factor-1 (APAF-1), ATP/dATP, and procaspase-9 to form the apoptosome in the cytoplasm. This causes the activation of caspase-9, thereby triggering the activation of the caspase cascade as discussed above [122, 123].
The extrinsic apoptotic pathway entails extracellular ligands stimulating cell-surface death receptors that initiate apoptosis by activating caspase-8, and this early step in extrinsic apoptotic signaling is inhibited by GSK3. Examples of death receptors include p55. Fas, DR4 and DR5. Cellular insults induce receptor homo-trimerization followed by the recruitment of cytoplasmic adaptor and effector proteins which activates the receptor. This complex subsequently binds to the cytoplasmic proteins FADD and procaspase-8 (or procaspase-10) to form the DISC. DISC formation can allow autoactivation of caspase-8, which then leads to the activation of effector caspases, primarily caspases-3, -6, and -7 [122, 123]. This pathway is discussed in detail above. Thus, GSK3 modulates key steps in each of the two major pathways of apoptosis, but in opposite directions.
GSK3β knockout mice are embryonically lethal due to massive hepatocyte apoptosis, which demonstrates that GSK3β is an important inhibitor of apoptosis [47, 122, 128]. GSK3 inhibitors promote apoptosis induced by stimulation of DD-containing receptors but provide protection from many other insults that induce apoptosis. For instance, we have previously reported that inhibition of GSK3 using lithium and other chemical inhibitors selectively kill tumor cells such as gliomas and leukemia but protect normal tissues from radiation-induced toxicity. The mechanism involved enhanced NHEJ mediated DSB repair following IR in normal tissues but not cancer. Since radiation cannot be selectively delivered to cancer cells, it leads to many destructive cellular processes including apoptosis, genomic instability, and autophagy. Cranial irradiation therapy is a standard method for treatment of brain cancer but results in long term neurocognitive deficits, especially in children. Thus, treatment with GSK3 inhibitors may potentially improve the quality of life of cancer patients undergoing radiation treatment. Several clinical trials have been initiated to test the efficacy of lithium in neuroprotection during the treatment of brain tumors but the trials are still at their infancy [47, 122, 128].
It is perplexing that inhibition of GSK3 upregulates DNA repair exclusively in normal cells. A possible explanation may be that GSK3 is already maximally inhibited in the majority of cancer. p53 status may also play a role in determining cellular response to GSK3 inhibition but further research is warranted in this avenue. Our lab is currently investigating the role of GSK3 in DNA damage/repair and how this relates to GSK3-induced neuroprotection.
CONCLUSION
A wide array of key players in the DNA damage response also is also involved in the interplay between cell survival and apoptosis. Further research is necessary to decipher the mechanisms by which cell fate is determined. In this complex network, uncovering these mechanisms may allow for the understanding of certain diseases and the generation of more effective therapies.
Acknowledgments
Some of the work described in this review was supported by the IMPACT Award from the Department of Radiation Oncology, University of Alabama-Birmingham Comprehensive Cancer Center, a translational scholar award from the Sidney Kimmel Foundation for cancer research, a pilot award from the Center for Clinical and Translational Science (CCTS) and the Council of Center Directors (COCD) Translational Research Intramural Pilot Grant Program (grant number 5UL1 RR025777-04) from the NIH National Center for Research Resources, and a medical research award from the Gabrielle’s Angel Foundation (to ESY). We recognize that we were unable to cover all aspects of DNA damage response and cell death pathways in this review. We apologize to those whom we have been unable to cite owing to space constraints.
Abbreviations used
- 8-OHdG
8-hydroxydeoxyguanosine
- AP
apurinic/apyrimidinic
- APAF-1
apoptotic protease activating factor-1
- ATM
ataxia telangiectasia mutated
- ATR
ataxia telangiectasia and Rad3 related
- BARD
BRCA1-associated RING domain protein
- BER
base excision repair
- BRCT
BRCA1 C-terminus
- CDK
cy clin dependent kinase
- CREB
cyclic AMP response element binding protein
- DD
death domain
- DED
death effector domain
- DISC
death inducing signaling complex
- DNA PK
DNA-dependent protein kinase
- DSB
double strand break
- EGFR
epidermal growth factor receptor
- FADD
Fas-associated death domain protein
- FOXO
Forkhead box class O
- GSK3
glycogen synthase kinase 3
- HR
homologous recombination
- HSF-1
heat shock factor-1
- MMR
mismatch repair
- MRN
Mre11–Rad50–Nbs1
- NEMO
NF-κB essential modulator
- NER
nucleotide excision repair
- NF-κB
nuclear factor-κB
- NHEJ
non-homologous end joining
- PAR
poly(ADP-ribose)
- PARP
poly(ADP-ribose) polymerase
- PARylation
poly(ADP-ribosyl)ation
- PUMA
p53 upregulated modulator of apoptosis
- RIP1
receptor-interacting protein 1
- RPA
replication protein A
- SIRT
sirtuins
- SSB
single strand break
- SUMO
small ubiquitin like modifier
- TNFR
tumor necrosis factor receptor
- TRADD
tumor necrosis factor receptor type 1-associated DEATH domain
References
- 1.Chittenden T, Harrington EA, O’Connor R, et al. Induction of apoptosis by the Bcl-2 homologue Bak. Nature. 1995;374:733–6. doi: 10.1038/374733a0. [DOI] [PubMed] [Google Scholar]
- 2.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- 5.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aziz K, Nowsheen S, Georgakilas AG. Nanotechnology in cancer therapy: targeting the inhibition of key DNA repair pathways. Curr Mol Med. 2010;10:626–39. doi: 10.2174/156652410792630599. [DOI] [PubMed] [Google Scholar]
- 7.Aziz K, Nowsheen S, Pantelias G, et al. Targeting DNA damage and repair: Embracing the pharmacological era for successful cancer therapy. Pharmacol Ther. 2011;133:334–50. doi: 10.1016/j.pharmthera.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 8.Nowsheen S, Whitley AC, Yang ES. Biomarkers to assess the targeting of DNA repair pathways to augment tumor response to therapy. Curr Mol Med. 2012;12:788–803. doi: 10.2174/156652412800792615. [DOI] [PubMed] [Google Scholar]
- 9.Carson DA, Ribeiro JM. Apoptosis and disease. Lancet. 1993;341:1251–4. doi: 10.1016/0140-6736(93)91154-e. [DOI] [PubMed] [Google Scholar]
- 10.Savill J. Apoptosis in disease. Eur J Clin Invest. 1994;24:715–23. doi: 10.1111/j.1365-2362.1994.tb01067.x. [DOI] [PubMed] [Google Scholar]
- 11.Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer. 2005;5:231–7. doi: 10.1038/nrc1560. [DOI] [PubMed] [Google Scholar]
- 12.Kerr JFR, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reed JC, Green DR. Apoptosis: Physiology and Pathology. Cambridge: Cambridge University Press; 2011. [Google Scholar]
- 14.Kroemer G, Martin SJ. Caspase-independent cell death. Nat Med. 2005;11:725–30. doi: 10.1038/nm1263. [DOI] [PubMed] [Google Scholar]
- 15.Roos WP, Kaina B. DNA damage-induced apoptosis: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2012 doi: 10.1016/j.canlet.2012.01.007. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 16.Zhivotovsky B, Kroemer G. Apoptosis and genomic instability. Nat Rev Mol Cell Biol. 2004;5:752–62. doi: 10.1038/nrm1443. [DOI] [PubMed] [Google Scholar]
- 17.Fan TJ, Han LH, Cong RS, et al. Caspase family proteases and apoptosis. Acta Biochim Biophys Sin (Shanghai) 2005;37:719–27. doi: 10.1111/j.1745-7270.2005.00108.x. [DOI] [PubMed] [Google Scholar]
- 18.Trisciuoglio L, Bianchi ME. Several nuclear events during apoptosis depend on caspase-3 activation but do not constitute a common pathway. PloS One. 2009;4:e6234. doi: 10.1371/journal.pone.0006234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Ann Rev Physiol. 1998;60:619–42. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- 20.Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25:4798–811. doi: 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- 21.Wajant H. The Fas signaling pathway: more than a paradigm. Science. 2002;296:1635–6. doi: 10.1126/science.1071553. [DOI] [PubMed] [Google Scholar]
- 22.Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22:8628–33. doi: 10.1038/sj.onc.1207232. [DOI] [PubMed] [Google Scholar]
- 23.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–33. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou BBS, Elledge SJ. The DNA damage respons e: putting checkpoints in perspective. Nature. 2000;408:433–9. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 25.Robertson A, Klungland A, Rognes T, et al. DNA repair in mammalian cells. Cell Mol Life Sci. 2009;66:981–93. doi: 10.1007/s00018-009-8736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Falck J, Forment JV, Coates J, et al. CDK targeting of NBS1 promotes DNA-end resection, replication restart and homologous recombination. EMBO Rep. 2012;13:561–8. doi: 10.1038/embor.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JH, Paull TT. ATM activation by DNA doublestrand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–4. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 28.Uziel T, Lerenthal Y, Moyal L, et al. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 200322:5612–2. 1. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flynn RL, Zou L. ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem Sci. 2011;36:133–40. doi: 10.1016/j.tibs.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith J, Tho LM, Xu N, et al. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 31.Vermeulen K, van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36:131–49. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bahassi EM, Ovesen JL, Riesenberg AL, et al. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene. 2008;27:3977–85. doi: 10.1038/onc.2008.17. [DOI] [PubMed] [Google Scholar]
- 33.Stracker TH, Usui T, Petrini JHJ. Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair (Amst) 2009;8:1047–54. doi: 10.1016/j.dnarep.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shieh SY, Ahn J, Tamai K, et al. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000;14:289–300. [PMC free article] [PubMed] [Google Scholar]
- 35.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–9. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 36.Pabla N, Huang S, Mi QS, et al. ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis. J Biol Chem. 2008;283:6572–83. doi: 10.1074/jbc.M707568200. [DOI] [PubMed] [Google Scholar]
- 37.Stracker TH, Couto SS, Cordon-Cardo C, et al. Chk2 suppresses the oncogenic potential of DNA replication associated DNA damage. Mol Cell. 2008;31:21–32. doi: 10.1016/j.molcel.2008.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Polager S, Ginsberg D. p53 and E2f: partners in life and death. Nat Rev Cancer. 2009;9:738–48. doi: 10.1038/nrc2718. [DOI] [PubMed] [Google Scholar]
- 39.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–66. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 40.Bernstein C, Bernstein H, Payne CM, et al. DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis. Mutat Res. 2002;511:145–78. doi: 10.1016/s1383-5742(02)00009-1. [DOI] [PubMed] [Google Scholar]
- 41.Fattah F, Lee EH, Weisensel N, et al. Ku regulates the non-homologous end joining pathway choice of DNA doublestrand break repair in human somatic cells. PLoS Genet. 2010;6:e1000855. doi: 10.1371/journal.pgen.1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–71. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 43.Shibata A, Conrad S, Birraux J, et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011;30:1079–92. doi: 10.1038/emboj.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459:460–3. doi: 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakamura K, Kogame T, Oshiumi H, et al. Collaborative Action of Brca1 and CtIP in elimination of covalent modifications from double-strand breaks to facilitate subsequent break repair. PLoS Genet. 2010;6:e1000828. doi: 10.1371/journal.pgen.1000828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ikura T, Ogryzko VV, Grigoriev M, et al. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000;102:463–73. doi: 10.1016/s0092-8674(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 47.Yang ES, Wang H, Jiang G, et al. Lithium-mediated protection of hippocampal cells involves enhancement of DNA-PKdependent repair in mice. J Clin Invest. 2009;119:1124–35. doi: 10.1172/JCI34051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dai C, Tang Y, Jung SY, et al. Differential effects on p53-mediated cell cycle arrest vs. apoptosis by p90. Proc Natl Acad Sci USA. 2011;108:18937–42. doi: 10.1073/pnas.1110988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang J, Yang ES, Jiang G, et al. p53-dependent BRCA1 nuclear export controls cellular susceptibility to DNA damage. Cancer Res. 2011;71:5546–57. doi: 10.1158/0008-5472.CAN-10-3423. [DOI] [PubMed] [Google Scholar]
- 50.Pauklin S, Kristjuhan A, Maimets T, et al. ARF and ATM/ATR cooperate in p53-mediated apoptosis upon oncogenic stress. Biochem Biophys Res Commun. 2005;334:386–94. doi: 10.1016/j.bbrc.2005.06.097. [DOI] [PubMed] [Google Scholar]
- 51.Thangaraju M, Kaufmann SH, Couch FJ. BRCA1 facilitates stress-induced apoptosis in breast and ovarian cancer cell lines. J Biol Chem. 2000;275:33487–96. doi: 10.1074/jbc.M005824200. [DOI] [PubMed] [Google Scholar]
- 52.Levine AJ. p53, the cellular gatekeeper review for growth and division. Cell. 1997;88:323–31. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 53.Ryan KM, Ernst MK, Rice NR, et al. Role of NF-kB in p53-mediated programmed cell death. Nature. 2000;404:892–6. doi: 10.1038/35009130. [DOI] [PubMed] [Google Scholar]
- 54.Biton S, Ashkenazi A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-α feedforward signaling. Cell. 2011;145:92–103. doi: 10.1016/j.cell.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 55.Bock FJ, Villunger A. GSK3 TIPping off p53 to unleash PUMA. Mol Cell. 2011;42:555–6. doi: 10.1016/j.molcel.2011.05.022. [DOI] [PubMed] [Google Scholar]
- 56.Yang ES, Xia F. BRCA1 16 years later: DNA damage-induced BRCA1 shuttling. FEBS J. 2010;277:3079–85. doi: 10.1111/j.1742-4658.2010.07734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nowsheen S, Aziz K, Tran PT, et al. Epigenetic inactivation of DNA repair in breast cancer. Cancer Lett. 2012 doi: 10.1016/j.canlet.2012.05.015. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 58.Zhang J, Powell SN. The role of the BRCA1 tumor suppressor in DNA double-strand break repair. Mol Cancer Res. 2005;3:531–9. doi: 10.1158/1541-7786.MCR-05-0192. [DOI] [PubMed] [Google Scholar]
- 59.Wang H, Yang ES, Jiang J, et al. DNA damage-induced cytotoxicity is dissociated from BRCA1’s DNA repair function but is dependent on its cytosolic accumulation. Cancer Res. 2010;70:6258–267. doi: 10.1158/0008-5472.CAN-09-4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Feng Z, Kachnic L, Zhang J, et al. DNA damage induces p53-dependent BRCA1 nuclear export. J Biol Chem. 2004;279:28574–84. doi: 10.1074/jbc.M404137200. [DOI] [PubMed] [Google Scholar]
- 61.Fitzgerald LD, Bailey CK, Brandt SJ, et al. BRCA1 accumulates in the nucleus in response to hypoxia and TRAIL and enhances TRAIL-induced apoptosis in breast cancer cells. FEBS J. 2007;274:5137–46. doi: 10.1111/j.1742-4658.2007.06033.x. [DOI] [PubMed] [Google Scholar]
- 62.Gu M, Li H, Shen C, et al. Cloning and characterization of a new BRCA1 variant: A role for BRCT domains in apoptosis. Cancer Lett. 2010;295:205–13. doi: 10.1016/j.canlet.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 63.Martin SA, Ouchi T. BRCA1 phosphorylation regulates caspase-3 activation in UV-induced apoptosis. Cancer Res. 2005;65:10657–62. doi: 10.1158/0008-5472.CAN-05-2087. [DOI] [PubMed] [Google Scholar]
- 64.Shao N, Chai YL, Shyam E, et al. Induction of apop-tosis by the tumor suppressor protein BRCA1. Oncogene. 1996;13:1–7. [PubMed] [Google Scholar]
- 65.Harkin DP, Bean JM, Miklos D, et al. Induction of GADD45 and JNK/SAPK-dependent apoptosis following inducible expression of BRCA1. Cell. 1999;97:575–86. doi: 10.1016/s0092-8674(00)80769-2. [DOI] [PubMed] [Google Scholar]
- 66.Wang YL, Wang B, Zhang H, et al. BRCA1 involved in regulation of Bcl-2 expression and apoptosis susceptibility to ionizing radiation. Sci China Phys Mech Astron. 2011;54:916–22. [Google Scholar]
- 67.Laulier C, Barascu A, Guirouilh-Barbat J, et al. Bcl-2 inhibits nuclear homologous recombination by localizing BRCA1 to the endomembranes. Cancer Res. 2011;71:3590–602. doi: 10.1158/0008-5472.CAN-10-3119. [DOI] [PubMed] [Google Scholar]
- 68.Freneaux P, Stoppa-Lyonnet D, Mouret E, et al. Low expression of bcl-2 in Brca1-associated breast cancers. Br J Cancer. 2000;83:1318–22. doi: 10.1054/bjoc.2000.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mandal PK, Blanpain C, Rossi DJ. DNA damage response in adult stem cells: pathways and consequences. Nat Rev Mol Cell Biol. 2011;12:198–202. doi: 10.1038/nrm3060. [DOI] [PubMed] [Google Scholar]
- 70.Burma S, Chen DJ. Role of DNA PK in the cellular response to DNA double-strand breaks. DNA Repair (Amst) 2004;3:909–18. doi: 10.1016/j.dnarep.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 71.Rybanska I, Ishaq O, Chou J, et al. PARP1 and DNA-PKcs synergize to suppress p53 mutation and telomere fusions during T-lineage lymphomagenesis. Oncogene. 2012 doi: 10.1038/onc.2012.199. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Espejel S, Franco S, Sgura A, et al. Functional interaction between DNA-PKcs and telomerase in telomere length maintenance. EMBO J. 2002;21:6275–87. doi: 10.1093/emboj/cdf593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Espejel S, Martín M, Klatt P, et al. Shorter telomeres, accelerated ageing and increased lymphoma in DNA-PKcs deficient mice. EMBO Rep. 2004;5:503–9. doi: 10.1038/sj.embor.7400127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sawada M, Sun W, Hayes P, et al. Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat Cell Biol. 2003;5:320–9. doi: 10.1038/ncb950. [DOI] [PubMed] [Google Scholar]
- 75.Cohen HY, Lavu S, Bitterman KJ, et al. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Baxmediated apoptosis. Mol Cell. 2004;13:627–38. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- 76.Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly (ADP-ribose) and PARP-1. Genes Dev. 2012;26:417–32. doi: 10.1101/gad.183509.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39:8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nowsheen S, Bonner JA, LoBuglio AF, et al. Cetuximab augments cytotoxicity with poly (ADP-ribose) polymerase inhibition in head and neck cancer. PloS One. 2011;6:e24148. doi: 10.1371/journal.pone.0024148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yu SW, Andrabi SA, Wang H, et al. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci USA. 2006;103:18314–9. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Andrabi SA, Kim NS, Yu SW, et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci USA. 2006;103:18308–13. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim MY, Zhang T, Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: “PAR-laying” NAD+ into a nuclear signal. Genes Dev. 2005;19:1951–67. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- 82.Kraus WL, Lis JT. PARP goes transcription. Cell. 2003;113:677–83. doi: 10.1016/s0092-8674(03)00433-1. [DOI] [PubMed] [Google Scholar]
- 83.O’Shaughnessy J, Osborne C, Pippen JE, et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N Engl J Med. 2011;364:205–14. doi: 10.1056/NEJMoa1011418. [DOI] [PubMed] [Google Scholar]
- 84.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–94. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 85.Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235–44. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 86.Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376:245–51. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- 87.Fong PC, Yap TA, Boss DS, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28:2512–9. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 88.Nowsheen S, Bonner JA, Yang ES. The poly(ADP ribose) polymerase inhibitor ABT-888 reduces radiation induced nuclear EGFR and augments head and neck tumor response to radiotherapy. Radiother Oncol. 2011;99:331–8. doi: 10.1016/j.radonc.2011.05.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yu SW, Wang H, Poitras MF, et al. Mediation of poly (ADP-ribose) polymerase-1-dependent cell death by apopto-sis-inducing factor. Science. 2002;297:259. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 90.Cregan SP, Dawson VL, Slack RS. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene. 2004;23:2785–96. doi: 10.1038/sj.onc.1207517. [DOI] [PubMed] [Google Scholar]
- 91.Ye K. PARP inhibitor tilts cell death from necrosis to apoptosis in cancer cells. Cancer Biol Ther. 2008;7:942–4. doi: 10.4161/cbt.7.6.6198. [DOI] [PubMed] [Google Scholar]
- 92.Derheimer FA, Kastan MB. Multiple roles of ATM in monitoring and maintaining DNA integrity. FEBS Lett. 2010;584:3675–81. doi: 10.1016/j.febslet.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yoshida K, Ozaki T, Furuya K, et al. ATM-dependent nuclear accumulation of IKK-alpha plays an important role in the regulation of p73-mediated apoptosis in response to cisplatin. Oncogene. 2007;27:1183–8. doi: 10.1038/sj.onc.1210722. [DOI] [PubMed] [Google Scholar]
- 94.Smith GCM, d’Adda di Fagagna F, Lakin ND, et al. Cleavage and inactivation of ATM during apoptosis. Mol Cell Biol. 1999;19:6076–84. doi: 10.1128/mcb.19.9.6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang J, Pabla N, Wang C-Y, et al. Caspase-mediated cleavage of ATM during cisplatin-induced tubular cell apoptosis: inactivation of its kinase activity toward p53. Am J Physiol Renal Physiol. 2006;291:F1300–7. doi: 10.1152/ajprenal.00509.2005. [DOI] [PubMed] [Google Scholar]
- 96.Zinkel SS, Hurov KE, Ong C, et al. A role for proapoptotic BID in the DNA-damage response. Cell. 2005;122:579–91. doi: 10.1016/j.cell.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 97.Kelly GS. A review of the sirtuin system, its clinical implications, and the potential role of dietary activators like resveratrol: part 2. Altern Med Rev. 2010;15:313–28. [PubMed] [Google Scholar]
- 98.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–91. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Longo VD, Kennedy BK. Sirtuins in aging and age related disease. Cell. 2006;126:257–68. doi: 10.1016/j.cell.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 100.Yuan Z, Zhang X, Sengupta N, et al. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol Cell. 2007;27:149–62. doi: 10.1016/j.molcel.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vaitiekunaite R, Butkiewicz D, Krzesniak M, et al. Expression and localization of Werner syndrome protein is modulated by SIRT1 and PML. Mech Ageing Dev. 2007;128:650–61. doi: 10.1016/j.mad.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 102.Uhl M, Csernok A, Aydin S, et al. Role of SIRT1 in homologous recombination. DNA Repair (Amst) 2009;9:383–93. doi: 10.1016/j.dnarep.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 103.Wang RH, Sengupta K, Li C, et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312–23. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen WY, Wang DH, Yen RWC, et al. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123:437–48. doi: 10.1016/j.cell.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 105.Kim EJ, Kho JH, Kang MR, et al. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell. 2007;28:277–90. doi: 10.1016/j.molcel.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 106.Brooks CL, Gu W. How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer. 2008;9:123–8. doi: 10.1038/nrc2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Giannakou ME, Partridge L. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol. 2004;14:408–12. doi: 10.1016/j.tcb.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 109.Yang Y, Hou H, Haller EM, et al. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 2005;24:1021–32. doi: 10.1038/sj.emboj.7600570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ahn B-H, Kim H-S, Song S, et al. A role for the mito-chondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA. 2008;105:14447–52. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kawahara TLA, Michishita E, Adler AS, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kB dependent gene expression and organismal life span. Cell. 2009;136:62–74. doi: 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huang SM, Bock JM, Harari PM. Epidermal growth factor receptor blockade with C225 modulates proliferation, apoptosis, and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Res. 1999;59:1935–40. [PubMed] [Google Scholar]
- 113.Sarkar S, Mazumdar A, Dash R, et al. ZD6474, a dual tyrosine kinase inhibitor of EGFR and VEGFR-2, inhibits MAPK/ERK and AKT/PI3-K and induces apoptosis in breast cancer cells. Cancer Biol Ther. 2010;9:592–602. doi: 10.4161/cbt.9.8.11103. [DOI] [PubMed] [Google Scholar]
- 114.Okamoto K, Okamoto I, Okamoto W, et al. Role of survivin in EGFR inhibitor induced apoptosis in non-small cell lung cancers positive for EGFR mutations. Cancer Res. 2010;70:10402–10. doi: 10.1158/0008-5472.CAN-10-2438. [DOI] [PubMed] [Google Scholar]
- 115.Costa DB, Halmos B, Kumar A, et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007;4:e315. doi: 10.1371/journal.pmed.0040315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gong Y, Somwar R, Politi K, et al. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 2007;4:e294. doi: 10.1371/journal.pmed.0040294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 118.Allocati N, Di Ilio C, De Laurenzi V. p63/p73 in the control of cell cycle and cell death. Exp Cell Res. 2012;318:1285–90. doi: 10.1016/j.yexcr.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 119.Sun Q, Ming L, Thomas SM, et al. PUMA mediates EGFR tyrosine kinase inhibitor-induced apoptosis in head and neck cancer cells. Oncogene. 2009;28:2348–57. doi: 10.1038/onc.2009.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dittmann K, Mayer C, Kehlbach R, Rodemann HP. Radiation-induced caveolin-1 associated EGFR internalization is linked with nuclear EGFR transport and activation of DNA-PK. Mol Cancer. 2008;7:69. doi: 10.1186/1476-4598-7-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bandyopadhyay D, Mandal M, Adam L, et al. Physical interaction between epidermal growth factor receptor and DNA-dependent protein kinase in mammalian cells. J Biol Chem. 1998;273:1568–73. doi: 10.1074/jbc.273.3.1568. [DOI] [PubMed] [Google Scholar]
- 122.Mills CN, Nowsheen S, Bonner JA, et al. Emerging roles of glycogen synthase kinase 3 in the treatment of brain tumors. Front Mol Neurosci. 2011;4:47. doi: 10.3389/fnmol.2011.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Beurel E, Jope RS. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol. 2006;79:173–89. doi: 10.1016/j.pneurobio.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gotschel F, Kern C, Lang S, et al. Inhibition of GSK3 differentially modulates NF-kB, CREB, AP-1 and B-catenin signaling in hepatocytes, but fails to promote TNF alpha-induced apoptosis. Exp Cell Res. 2008;314:1351–66. doi: 10.1016/j.yexcr.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 125.Charvet C, Wissler M, Brauns-Schubert P, et al. Phosphorylation of Tip60 by GSK-3 determines the induction of PUMA and apoptosis by p53. Mol Cell. 2011;42:584–96. doi: 10.1016/j.molcel.2011.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Watcharasit P, Bijur GN, Song L, et al. Glycogen synthase kinase-3B (GSK3B) binds to and promotes the actions of p53. J Biol Chem. 2003;278:48872–9. doi: 10.1074/jbc.M305870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bijur GN, Jope RS. Opposing actions of phosphati dylinositol 3′kinase and glycogen synthase kinase 3B in the regulation of HSF1 activity. J Neurochem. 2000;75:2401–8. doi: 10.1046/j.1471-4159.2000.0752401.x. [DOI] [PubMed] [Google Scholar]
- 128.Yang ES, Nowsheen S, Wang T, et al. Glycogen synthase kinase 3B inhibition enhances repair of DNA doublestrand breaks in irradiated hippocampal neurons. Neuro-Oncology. 2011;13:459–70. doi: 10.1093/neuonc/nor016. [DOI] [PMC free article] [PubMed] [Google Scholar]