

Abstract
Although various cell encapsulation materials are available commercially for a wide range of potential therapeutic cells, their combined clinical impact remains inconsistent. Synthetic materials such as poly(ethylene glycol) (PEG) hydrogels are mechanically robust and have been extensively explored but lack natural biofunctionality. Naturally derived materials including collagen, fibrin and alginate-chitosan are often labile and mechanically weak. In this paper we report the development of a hybrid biomatrix based on the thiol-ene reaction of PEG diacrylate (PEGdA) and cysteine/PEG-modified gelatin (gel-PEG-Cys). We hypothesized that covalent crosslinking decreases gelatin dissolution thus increasing gelatin resident time within the matrix and the duration of its biofunctionality; at the same time the relative ratio of PEGdA to gel-PEG-Cys in the matrix formulation directly affects hydrogel bulk and local microenvironment properties. Bulk viscoelastic properties were highly dependent on PEGdA concentration and total water content, while gel-PEG-Cys concentration was more critical to swelling profiles. Microviscoelastic properties were related to polymer concentration. The covalently crosslinked gel-PEG-Cys with PEGdA decreased gelatin dissolution out of the matrix and collagenase-mediated degradation. Fibroblasts and keratinocyte increased adhesion density and formed intercellular connections on stiffer hydrogel surfaces, while cells exhibited more cytoplasmic spreading and proliferation when entrapped within softer hydrogels. Hence, this material system contains multiparametric factors that can easily be controlled to modulate the chemical, physical and biological properties of the biomatrix for soft tissue scaffolding and cell presentation to reconstruct lost tissue architecture and physical functionality.
Keywords: Bioactivity, Cell encapsulation, Biomatrix, Gelatin, Poly(ethylene glycol)
1. Introduction
Hydrogel scaffolds are extensively used for cell encapsulation and drug delivery. An ideal scaffold should have well-matched physicochemical properties for a specific microenvironment and should interact with surrounding cells [1–3]. Several kinds of materials have been developed for hydrogel scaffolds. Hydrogels based on synthetic polymers, such as poly(ethylene glycol) (PEG), poly(vinyl alcohol) and poly(acrylic acid), are compositionally consistent, nontoxic, hydrophilic and possess controllable chemical and mechanical properties [4,5]. However, these synthetic materials lack biologically active sites. Therefore, extensive conjugation chemistry is required to introduce bioactive molecules such as RGD peptides (arginine–glycine–aspartic acid) to promote cell response [6]. Scaffolds derived from natural biomacromolecules such as collagen, gelatin, chitosan and hyaluronic acid could actively support cell proliferation, migration and differentiation [6]. However, these materials are mechanically less robust and hard to process, and it is also difficult to maintain product consistency [7]. To overcome these limitations, the combination of naturally derived and synthetic materials to create a biomatrix for 3-D cell culture represents a new strategy in scaffold design [8,9].
We have reported a tunable hydrogel network based on the combination of gelatin and PEG derivatives as a cell- and drug-delivery platform [9]. Gelatin contains cell-binding motifs, such as RGD oligopeptides, which support cell adhesion and proliferation. However, unmodified gelatin is not suitable as a 3-D culture platform due to the sol–gel transition property and its rapid dissolution at physiological temperature [10,11]. Several strategies have been employed to crosslink gelatin macromolecules. The most commonly used method involves gluteraldehyde which is well known to be cytotoxic and thus cannot be used to encapsulate living cells [12]. Methacrylate-modified gelatin, which can be photopolymerized under physiological conditions, has been successfully applied for the encapsulation of NIH3T3 cells and valvular interstitial cells [13,14]. However, the preparation of methacrylate-modified gelatin requires a processing temperature higher than physiological temperature to avoid the sol–gel transition. In this study, a new method has been adopted to modify gelatin with l-cysteine, which is absent in the primary structure of gelatin macromolecule, via a homobifunctional PEG-bis-NHS linker (gel-PEG-Cys, Fig. 1A) [9]. This thiolated gelatin derivative can be photopolymerized with PEGdA via the thiol–ene reaction under physiological conditions to achieve cell encapsulation.
Fig. 1.
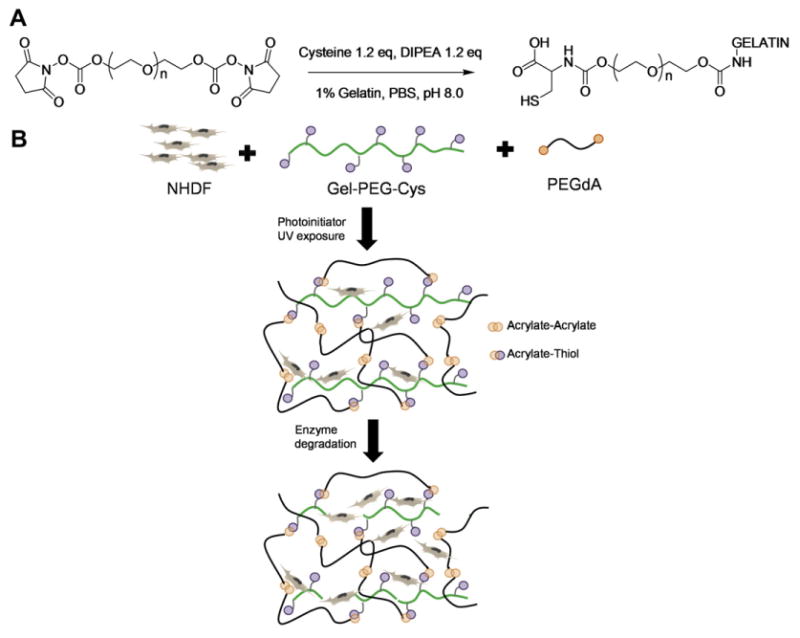
(A) Synthesis of PEG/cysteine-modified gelatin (gel-PEG-Cys). Gelatin was modified to introduce free thiols on the backbone via grafting with PEG-bis-NHS and l-cysteine. (B) Scaffold structure of gelatin-PEG matrix for living cell encapsulation. Two possible reactions, acrylate–acrylate and acrylate–thiol, can occur simultaneously. The gelatin-PEG hydrogels can undergo degradation with exogenous enzyme.
The physicochemical characteristics of 3-D living culture matrices not only affect cell behavior but also impact its application in reconstructing lost tissue architecture [15]. The network structure and swelling characteristics impact the penetration of essential nutrients and gases as well as the removal of waste products that are critical for the survival of encapsulated cells [16]. The viscoelastic properties of the hydrogels are crucial for cell migration, proliferation and differentiation. For example, human bone marrow stromal cell differentiation is directly related with the matrix modulus [17]. Additionally, a controllable degradation profile of the hydrogel is also important in tissue regeneration applications where temporary scaffolding is desired. Recently, hydrogels formed with PEG and natural proteins have been extensively studied [18–20]. In hydrogels formed with PEGdA and fibrinoon, the molecular weight of PEG linker and the polymer concentration directly contributed to the network structure, physical properties and cellular morphology [18,21]. In this study, gelatin was used as a model protein for other biomacromolcules. Gel-PEG-Cys fabricated from type A or type B gelatin was used to form hydrogel scaffolds with PEGdA. Modifications to the hydrogel network structure were achieved by varying the concentration of gel-PEG-Cys to PEGdA. Hydroonl bulk mechanical and microviscoelastic properties, swelling profiles and gelatin dissolution were quantified to determine their combined impact on encapsulated cells.
2. Materials and Methods
2.1. Materials
PEG-diol (average molecular weight 2 kDa and 3.4 kDa), acryloyl chloride, triethylamine (TEA), N,N′-disuccinimidyl carbonate (DSC), 4-(dimethylamino) pyridine (DMAP), N,N′-diisopropylethylamine (DIPEA), sodium azide, l-cysteine, gelatin type A (300 bloom, from porcine skin), gelatin type B (75 bloom, from bovine skin), collagenase type I (from Clostridium histolyticum) and dimethyformamide (DMF) were purchased from Sigma-Aldrich (St Louis, MO, USA). Irgacure® 2959 was obtained from Ciba Specialty Chemicals (New York, USA). Collagen (Type I, from rat tail) was purchased from BD Bioscience (Franklin Lakes, NJ). Polystyrene beads with carboxyl surface groups (diameter 1.87 μm) and beads without specific surface groups (diameter 1.09 μm) were purchased from Spherotech Inc. (Lake Forest, IL, USA).
2.2. Synthesis of PEGdA and thiolation of gelatin
PEGdA was synthesized following previously reported procedures with minor modifications [22]. Acrylation of PEG-diol (MW 3.4 kDa) was carried out by reacting a dichloromethane (DCM) solution of PEG-diol with acryloyl chloride and TEA at a relative molar ratio of 1:4:4. The raw product was filtered with aluminum oxide and washed three times with 0.5 N NaOH. The final product was precipitated in cold diethyl ether and dried under vacuum for at least 2 days.
PEGylated cysteine-grafted gelatin (gel-PEG-Cys) was synthesized according to a previously established method [23]. Briefly, N-hydroxysuccinimide-functionalized PEG (PEG-bis-NHS) was first synthesized via carbonate linkage between PEG-diol (MW 2 kDa) and DSC with DMAP as catalyst. To modify gelatin with l-cysteine, PEG-bis-NHS (1.0 g, 0.43 mmol) was dissolved in 5 ml dry DMF. l-cysteine (0.08 g, 0.66 mmol) was then added to PEG-bis-NHS solution followed by additional 110 μl DIPEA. The reaction was kept under argon protection for 20 min, after which 1% gelatin in PBS solution was added (60 ml, type A or type B). The reaction was further stirred for 1 h at room temperature under argon protection and the pH was maintained at 8.0. The products were dialyzed (6–8 kDa cutoff) for 2 days against double-distilled (dd) H2O. The gel-PEG-Cys solution was then filtered through a 0.22 μm filter, snap frozen and lyophilized. The amount of lysine groups modified on the gelatin macromolecule was estimated by the trinitrobenzene sulfonic acid (TNBS) method and the relative free thiol concentration in the gel-PEG-Cys solution was calculated from the Ellman test result.
2.3. Hydrogel preparation
Gelatin-PEG hydrogels were formed by crosslinking PEGdA and gel-PEG-Cys via photopolymerization (Fig. 1B). 20% (w/w) gel-PEG-Cys (type A or type B) and 20% (w/w) PEGdA solution were prepared with 0.5% Irgacure 2959® in DPBS at 37 °C. Gel-PEG-Cys solution was then mixed with PEGdA solution at various concentrations (Table 1). The precursor solution was transferred to a 10 mm diameter glass-bottomed Petri dish (sample thickness ∼1 mm) and photocrosslinked with long-wavelength UV (λmax = 365 nm, 10 mW cm−2) for 90 s. The hydrogel nomenclature used in this study is defined as “GAXPY” or “GBXPY”, where “A” represents type A gel-PEG-Cys and “B” represents type B gel-PEG-Cys. “X” is the gel-PEG-Cys weight percentage and “Y” is the PEGdA weight percentage. For the preparation of collagen hydrogel, rat tail type I collagen was mixed with 10 × PBS, 1 N NaOH and ice-cold ddH2O at 4 °C according to manufacturer's protocol. The final concentration of the collagen was 5 mg ml−1. The mixed collagen solution was then allowed to gel at 37 °C for 30 min.
Table 1.
Formulation table of gelatin-PEG hydrogels.
| Sample formula | PEGdA (wt.%) | Gelatin-PEG-Cys (type A, 300 bloom, wt.%) | Gelatin-PEG-Cys (type B, 75 bloom, wt.%) | Water (wt.%) |
|---|---|---|---|---|
| GA10P5 | 5 | 10 | 0 | 85 |
| GA10P7.5 | 7.5 | 10 | 0 | 82.5 |
| GA10P10 | 10 | 10 | 0 | 80 |
| GA5P5 | 5 | 5 | 0 | 90 |
| GA15P5 | 5 | 15 | 0 | 80 |
| GB10P5 | 5 | 0 | 10 | 85 |
| GB10P7.5 | 7.5 | 0 | 10 | 82.5 |
| GB10P10 | 10 | 0 | 10 | 80 |
| GB5P5 | 5 | 0 | 5 | 90 |
| GB15P5 | 5 | 0 | 15 | 80 |
2.4. Hydrogel bulk physical characterizations
To perform swelling analysis, hydrogel disks 10 mm in diameter and 1 mm thick were fabricated, and then incubated in DPBS (3 ml) with 0.1% sodium azide at 37 °C. At predetermined time points, samples were carefully removed from DPBS and lightly blotted dry, and the swollen weight (Wt) was recorded. After 96 h, samples were washed with ddH2O, lyophilized and the dried weight (Wdry) was measured. The equilibrium weight swelling ratio (Qs) was calculated as: Qs = (Wt − Wdry)/Wdry. The volume swelling profiles of hydrogel disks were measured according to their surface area change. Hydrogels were allowed to reach equilibrium via incubation in DPBS for 3 days. The surface area of each hydrogel was then recorded and compared with the initial area (10 mm diameter plate). Since the thickness of each sample was not observably changed throughout the swelling experiment, the change in the thickness was not considered.
To evaluate FITC-dextran release profiles, type B gelatin-PEG hydrogels were incorporated with 1 mg ml−1 FITC-dextran (MW 4, 70 and 500 kDa; hydrodynamic radii, 4, 21 and 51 nm, respectively) immediately before gelation [24]. The samples were then incubated in DPBS containing 0.1% sodium azide. The supernatant was collected and replaced with fresh DPBS at each time point. The fluorescence intensity of FITC-dextran in the supernatant was detected by a microplate reader with excitation at 485 nm and emission at 520 nm (FluoStar Omega, BMG Labtech, Germany). The cumulative dextran amount in the supernatant was calculated and compared with the dextran amount in the initial hydrogel.
To evaluate gelatin dissolution profiles, samples were incubated in DPBS containing 0.1% sodium azide at 37 °C. At each time point, the entire volume of supernatant was collected and replaced with fresh DPBS. The gelatin concentration in the supernatant was determined using a BCA™ protein assay kit (Thermo Scientific, USA). The gelatin dissolution percentage was calculated as the cumulative gelatin weight in the supernatant (Ws) compared with the gelatin weight in the initial hydrogel (Wh), i.e. percentage of gelatin dissolution = (Ws)/(Wh) × 100%.
To determine the possible effect of collagenase on hydrogel degradation, samples were prepared as described above, placed in glass vials with DPBS containing 0.4 mg ml−1 type I collagenase and 0.1% sodium azide [25]. Samples were then placed on a platform shaker at 37 °C. The collagenase solution was changed every 2 days to maintain enzyme activity. After 1, 3 and 7 days, samples were washed with ddH2O and lyophilized to obtain the dried weight. The percentage of degradation was calculated by the dried weight of each time point divided by the dried weight of the initial hydrogel. Samples incubated in DPBS without collagenase were used as a baseline comparison.
Bulk rheological characterization of the hydrogel was performed with an ARES-LS2 2000ex rheometer (TA Instruments, USA) equipped with 8 mm parallel disk geometry. The frequency-sweep was applied with a range of 0.1–10 Hz with 5% strain at 25 ± 0.1 °C. Hydrogel samples ∼1 mm thick were prepared and covered with silicone oil to minimize water evaporation. Hydrogel samples were unswollen, swollen (incubated with DPBS for 7 days) or encapsulated with fibroblasts (106 cell ml−1 hydrogel, culture for 7 days). The magnitude of the complex shear modulus (G*) was calculated as G* = G′ + iG″.
2.5. Hydrogel microrheological properties
The microrheological properties of hydrogels were determined by measuring the Brownian motion of polystyrene beads embedded in gelatin-PEG hydrogels and trapped by optical tweezers. The hydrogel precursor solutions were prepared according to Sec. 2.3, and a 20% v/v bead solution (0.005% w/v in PBS) was then added. The solution was then injected into a glass microchamber with a coverglass bottom and a glass slide top (Fig. 7A). The entire assembly was subsequently sealed and exposed to UVC (∼4 mW cm−2) for 60 s to set the gel. The structure of the optical tweezers system and detailed experiment procedures were described in our previous publication [26]. In short, a single beam from a Nd:YVO4 laser (1064 nm, Spectra-Physics Lasers) was used as trapping laser and another wavelength (830 nm, Point Source) was used as the detection laser. The bead position was recorded by a QPD sensor with nanometer resolution at 20 kHz. The time domain data was then Fourier transformed and analyzed in the frequency domain. Eq. (1) describes the position power spectrum, Sx(f), of a trapped bead, where [kB] is Boltzmann's constant; [T] is absolute temperature; [γ] is the drag coefficient, which relates to the local viscosity and the radius of bead as shown in Eq. (2); [fc] is the corner frequency, which relates to the drag coefficient and the trapping stiffness of the optical tweezers as shown in Eq. (3) [27]. The local viscosity, [η], of the hydrogel was obtained from the power spectrum of the bead (Fig. 7B and C). Due to the short working distance of the high NA oil immersion lens used for optical trapping and imaging, only the layer within 10 μm of the coverglass was clearly visible. Beads that moved on the camera feed from a camera attached to the microscope were deemed to have Brownian motion (Fig. 7B and Movie 1 in Supplementary Materials). Those that did not move were considered as stuck (Fig. 7C and Movie 2 in Supplementary Materials). Experiments for each formula were repeated at least once, and 10 beads from various locations in each microchamber were observed. Only samples with beads showing Brownian motion consistently throughout the observable region were used to determine local viscosity. All measurements were done at a temperature of 23.5 ± 0.5 °C, in the central region of the gel slab 5 μm above the coverglass,
Fig. 7.
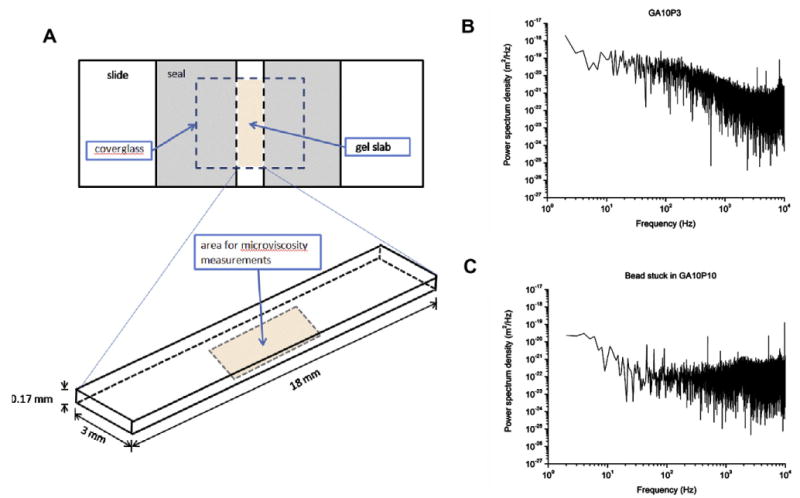
(A) Set-up of the glass microchamber. All beads used to measure microviscosity were located in the central bottom area of the gel slab, 5 μm above the coverglass and at least 0.2 mm from the sides. (B) The sample power spectrum of 1.87 μm diameter beads with visible Brownian motion in GA10P3 hydrogels. This particular spectrum is typical of a microparticle showing Brownian motion in a liquid: it is flat in the low-frequency region until the corner frequency (∼100 Hz) is reached, at which point it drops at a constant rate. (C) The sample power spectrum of the stuck bead in GA10P10 hydrogels: it is flat in the high-frequency region.
| (1) |
| (2) |
| (3) |
2.6. In vitro cell culture
Neonatal human dermal fibroblasts (NHDFs) and neonatal human dermal keratinocytes (NHEKs) were purchased from Lonza (NJ, USA). NHDFs were cultured in fibroblast basic medium (FBM) supplemented with 10% fetal bovine serum (FBS). Fibroblasts were passaged every 3 days and cells between passages 5 and 10 were used for all experiments. Human keratinocytes were cultured in KGM-Gold® keratinocyte growth medium (Lonza). Keratinocytes between passages 2 and 5 were used. 2-D cell adhesion was performed on hydrogel samples prepared as described above. Samples were transferred into a 24 well culture plate and incubated with respective culture medium for 2 h at 37 °C to reach equilibrium. NHDFs or NHEKs were trypsinized and resuspended to single cell suspension at 1 × 105 cells ml−1. 1 ml of cell suspension was added on each hydrogel sample and, after 3 days, live/dead staining (Invitrogen, NJ, USA) was used to evaluate cell viability and adhesion on the hydrogel surfaces.
For 3-D cell encapsulation, precursor solutions of various PEGdA and gel-PEG-Cys concentrations were prepared. The NHDFs were trypsinized, resuspended and mixed with hydrogel precursor solutions at a final concentration of 2 × 106 cells ml−1. Mixed precursor solutions were gently agitated and transferred to glass-bottomed Petri dishes and subjected to UV exposure for 90 s (10 mWcm−2). Subsequently, cell-encapsulated hydrogels were detached from the mold and incubated in culture medium. After 14 days, cell-entrapped hydrogels were washed with DPBS and fixed in 4% paraformaldehyde for 4 h at room temperature followed by incubation in 0.25% Triton X-100 plus 1% BSA in PBS solution for 2 h. Cytoskeletal F-actin was stained with 1 U ml−1 Alexfluo 488-conjugated phalloidin (Invitrogen) at 4 °C overnight. Fluorescence images of the F-actin distribution were acquired by confocal microscopy (F1000, Olympus, Japan).
2.7. Statistical analysis
All analyses were performed using standard Student's t-test and represented as a mean ± standard deviation. Statistical significance was considered for P < 0.05. At least three independent experiments were performed for each study.
3. Results and discussion
3.1. Gelatin modification and hydrogel fabrication
Proton NMR spectrum of gel-PEG-cys in D2O showed: δ 1.3, d, 2Hs, −CH2SH; δ 2.90, t, 1H, −CHCH2SH; δ 3.65, m, −CH2– from PEG backbone; broad peaks composed of many overlapping small peaks at 1.7, 1.75, 1.92, 3.18 and 4.2 ppm were characteristic gelatin peaks [28]. The introduction of PEG molecule to the gelatin backbone increased the solubility of gel-PEG-Cys and minimized phase separation with PEGdA. The reaction of PEG-bis-NHS, l-cysteine and gelatin resulted in an approximate modification ratio of 70% with type A gelatin and 53% with type B gelatin (Table 2). The average molecular mass of different gelatin products indicated that approximately 17 lysyl residues of each type A gelatin chain and 4 lysyl residues of each type B gelatin chain were modified. Although type A gelatin had a greater degree of lysyl residue modification than type B gelatin, there is no significant difference with the reactive free thiol concentration between two types of gel-PEG-cys solutions (Table 2). Both types of gelatin derivatives were further used in gelatin-PEG hydrogel fabrication via thiol–ene photopolymerization with PEGdA. Two concurrent reactions were possible in the mixture containing PEGdA and gel-PEG-Cys (Fig. 1B). The acrylic radical could either react with another acrylate group or with a thiol functional group via hydrogen abstraction/chain transfer [29]. Changing the polymer concentration and the molar ratio of thiol to acrylate monomers can greatly influence the reaction kinetics, the network structures and physicochemical properties of hydrogel [30,31].
Table 2.
Gelatin modification ratio and free thiol concentration in gel-PEG-Cys solution.
| Lysyl modification ratio (%) | Free thiol concentration in 20% (w/w) gel-PEG-cys solution (mmol) | |
|---|---|---|
| Gel-PEG-Cys, type A, bloom 300, average molecular mass: 50–100 kDa | 70.5 ±4.5 | 11.3 ±5.3 |
| Gel-PEG-Cys, type B, bloom 75, average molecular mass: 20–25 kDa | 53.3 ±5.0 | 12.5 ± 1.2 |
3.2. Hydrogel bulk properties: stiffness, swelling and enzymatic degradation
Increasing either PEGdA or gel-PEG-Cys concentration significantly increased the G* value in unswollen samples (Fig. 2A, black bars). With constant PEGdA concentration (5%), the G* value increased from 2610 ± 400 to 6102 ± 399 Pa with increasing gel-PEG-Cys content (P < 0.01). PEGdA concentration also showed a positive correlation with the hydrogel stiffness. G* values increased from 4073 ± 192 Pa for GA10P5 to 5140 ± 551 Pa for GA10P7.5 and 7627 ± 1200 Pa for GA10P10 (P < 0.01). These phenomena, however, were quite different when using fully hydrated samples (Fig. 2A, white bars). All five swollen samples had significantly lower G* values than respective unswollen samples, especially those containing 5% PEGdA. When completely swollen, hydrogels containing different gelatin concentrations exhibited similar G* values (Fig. 2A, white bars). The PEGdA concentration in the swollen hydrogels remained positively correlated with G* (i.e. 294 ± 47 Pa for GA10P5 to 1203 ± 129 Pa for GA10P7.5 and 1959 ± 469 Pa for GA10P10, Fig. 2A, white bars, P < 0.01). The original G′ and G″ data from the frequency sweep are shown in Fig. 1S in the Supplementary Materials. Similar trends were also observed in hydrogels fabricated with type B gel-PEG-Cys (Fig. 2B, black bars). When compared with hydrogels formed with type A gel-PEG-cys, hydrogels containing type B gel-PEG-Cys generally had higher G* values (Fig. 2B). With the same polymer composition under unswollen status, the storage modulus was directly dependent on the crosslink density of the network [32]. Higher PEGdA or gel-PEG-Cys concentration increased the crosslink density and thus resulted in a higher G* value. Swollen gels had significantly lower G* values than unswollen gels; this is mainly due to the higher water content and more relaxed state of the polymer chains. The bulk property of the hydrogels could also be influenced by the presence of encapsulated cells [33]. However, there were no statistical differences between cell-containing and cell-free PEG-gelatin hydrogel samples (Fig. 2C). Our results showed that water content and PEGdA concentration played a significant role in the bulk viscoelasticity of the hydrogels.
Fig. 2.
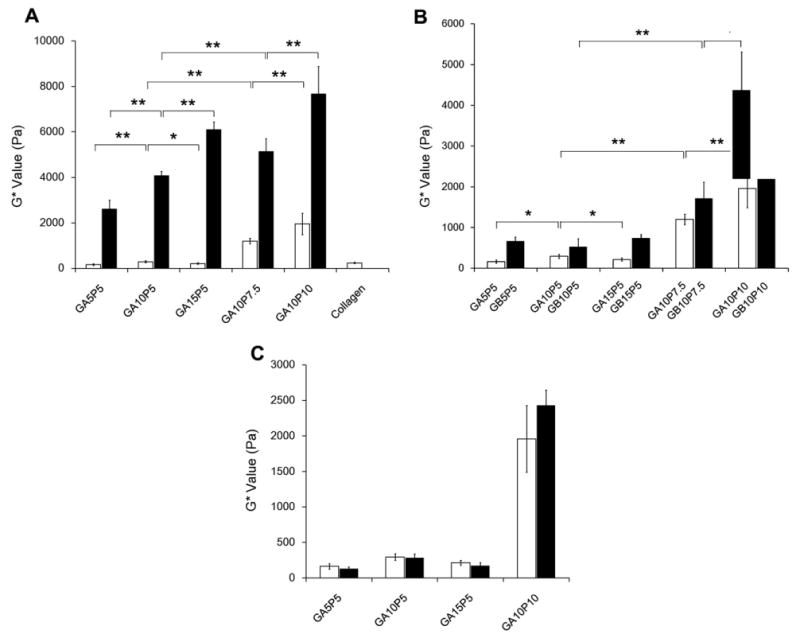
Bulk rheology properties of gelatin-PEG hydrogels. (A) Complex shear modulus (G*) value of type A gelatin-PEG hydrogels with unswollen status (black bars) vs. swollen status (white bars). (B) Complex shear modulus of type A gelatin-PEG hydrogels (white bars) and type B gelatin-PEG hydrogel (black bars) with completely swollen status. (C) Complex shear modulus of cell encapsulated type A gelatin-PEG hydrogels after 14 days culture (black bars) vs. cell free samples (white bars). **P < 0.01, *P < 0.05.
Both PEGdA and gel-PEG-Cys concentrations significantly affected the mass swelling characteristics of the hydrogel (Fig. 3A). Increasing the PEGdA concentration resulted in a lower mass swelling ratio. GA10P5 (Qmax = 50.4 ± 8.3 at 2 h) displayed about a 2-fold larger Qmax value than GA10P7.5 (Qmax = 28.4 ± 4.3 at 2 h) and GA10P10 (Qmax = 21.9 ± 2.4 at 1 h). The negative effect of PEG concentration on the mass swelling profiles of hydrogels had been well studied and characterized [9,34,35]. Increasing the PEG-dA concentration resulted in a higher crosslink density and led to a lower mass swelling ratio. Gelatin concentration had the opposite effect on the hydrogel mass swelling profile (Fig. 3A). Hydrogels containing 5% PEGdA with 5%, 10% and 15% gel-PEG-cys had Qmax values of 39.1 ± 3.4, 50.4 ± 8.3 and 61.2 ± 7.6, respectively (P < 0.01). Hydrogels composed of type B gel-PEG-cys also showed similar mass swelling trends as hydrogels with type A gel-PEG-Cys (Fig. 3B). Only the gel-PEG-Cys concentration had a positive correlation with the area change profile of the hydrogels (Fig. 3C). The swelling area increased significantly with increasing gel-PEG-Cys concentration. Since gel-PEG-Cys could react with PEGdA via the thiol–acrylate reaction and compete with the acrylate–acrylate reaction, increasing gel-PEG-Cys concentration increased the probability of thiol–acrylate reaction while decreasing the acrylate–acrylate reaction. Since the thiol–acrylate crosslinker involves larger macromolecules that are more flexible than acrylate–acrylate crosslinker, hydrogels containing more gel-PEG-Cys would be expected to have less chain rigidity and higher water content, mass swelling and larger final area.
Fig. 3.
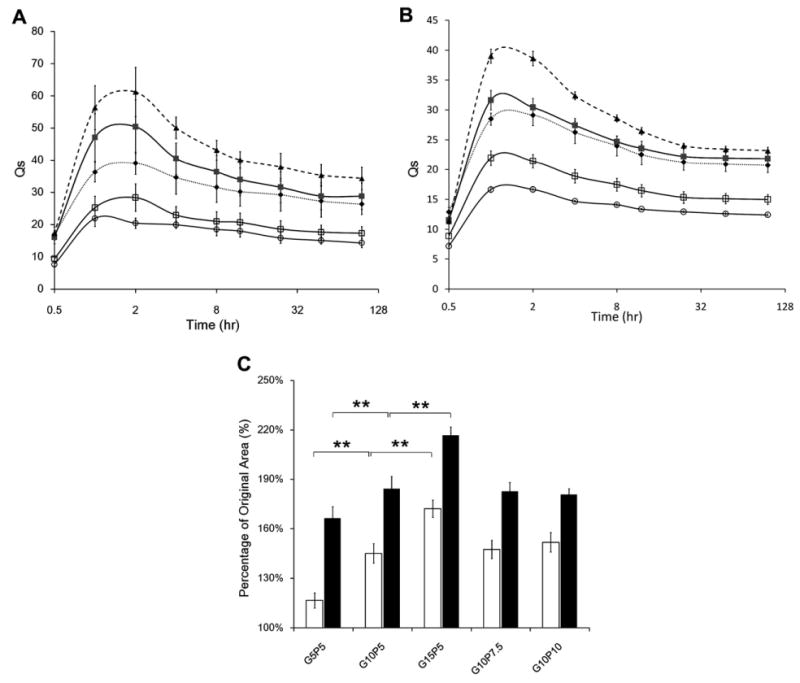
(A) Swelling profiles of gelatin-PEG hydrogels with type A gel-PEG-Cys (◆, GA5P5; ■, GA10P5; ▲, GA15P5; □, GA10P7.5; ○, GA10P10). (B) Swelling profiles of gelatin-PEG hydrogels with type B gel-PEG-Cys (◆, GB5P5; ■, GB10P5; ▲, GB15P5; □, GB10P7.5; ○, GB10P10). (C) Relative surface area change of type A gelatin-PEG hydrogels (white bars) and type B gelatin-PEG hydrogels (black bars). **P < 0.01.
Gelatin-PEG hydrogels showed a rapid initial gelatin dissolution that equilibrated within 24 h (Fig. 4), indicating most of the gel-PEG-Cys was stably crosslinked within the hydrogel network. There was no significant difference in the gelatin dissolution profile in all five formulations with either type A or type B gelatins (Fig. 4A and B). Different to the gelatin dissolution, non-covalently incorporated FITC-dextran displayed significantly different release profiles. As shown in Fig. 5, FITC-dextran with lower molecular weight (4 kDa, Fig. 5A) rapidly diffused out of the hydrogel within 24 h while FITC-dextran with higher molecular weight (70 kDa, Fig. 5B; 500 kDa, Fig. 5C) diffused much more slowly. The variation of gelatin concentration has no effect on the FITC-dextran release profile, while the higher PEG concentration significantly retarded the release of dextran. These results indicated that the hydrogels with higher PEG concentration had a tighter structure which impeded the diffusion of large molecules. Hydrogels incubated with collagenase displayed larger mass loss than those incubated in the DPBS at 3 and 7 days (Fig. 6), indicating that the modified gelatin could be recognized and digested by collagenase I. The mass loss was more pronounced in hydrogels with higher gelatin concentration (Fig. 6A and C) and lower PEG concentration (Fig. 6B and D). Contrasting with unmodified gelatin or collagen hydrogels, which could be completely degraded by collagenase, all gelatin-PEG hybrid hydrogels maintained structural integrity after 7 days. This could be due to the polymerization of PEGdA-PEGdA formulated the robust PEG network which would not be affected by collagenase, thus maintained the overall structure. Similar observations were reported when PEGdA was introduced into methacrylated gelatin hydrogels [20].
Fig. 4.
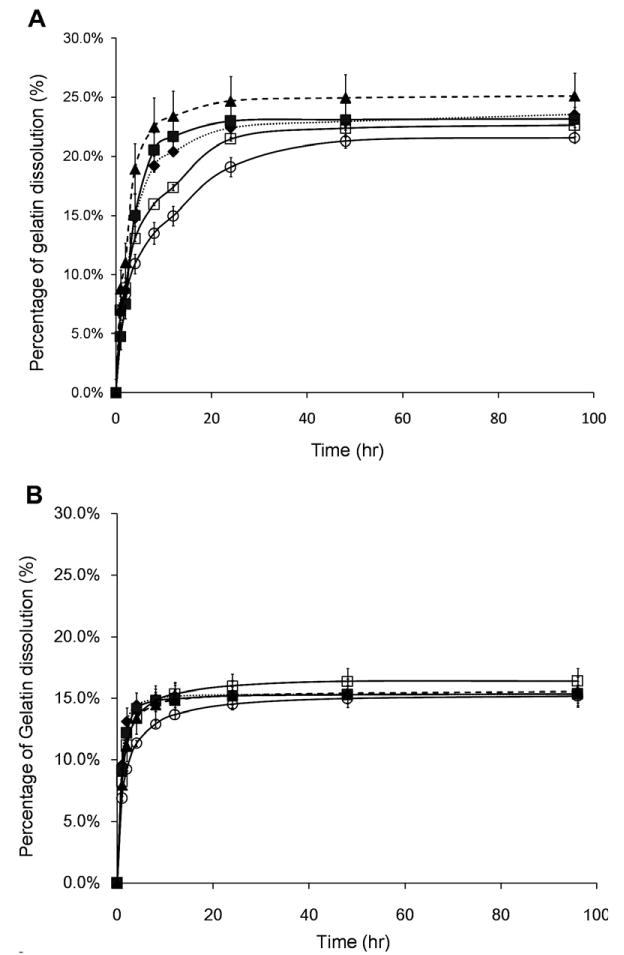
(A) Gelatin dissolution behavior of type A gelatin-PEG hydrogels (◆, GA5P5; ■, GA10P5; ▲, GA15P5; □, GA10P7.5; ○, GA10P10). (B) Gelatin dissolution behavior of type B gelatin-PEG hydrogels (◆, GB5P5; ■, GB10P5; ▲, GB15P5; □, GB10P7.5; ○, GB10P10).
Fig. 5.
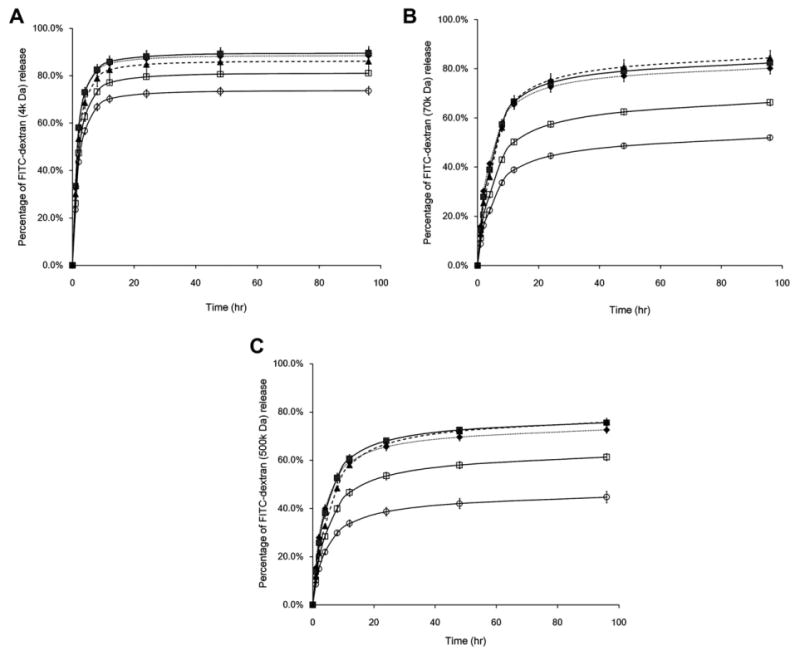
The release kinetics of FITC-dextran with different molecular weights (A, 4 kDa; B, 70 kDa; C, 500 kDa) from type B gelatin-PEG hydrogels (◆, GB5P5; ■, GB10P5; ▲, GB15P5; □, GB10P7.5; ○, GB10P10).
Fig. 6.
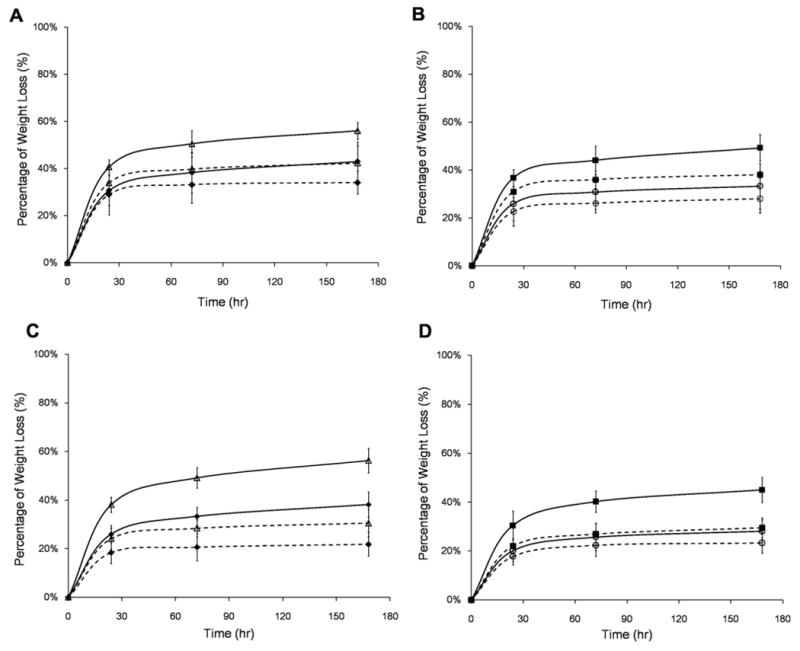
Enzymatic degradation profiles of type A gelatin-PEG hydrogels (A, GA5P5 (◆) vs. GA15P5 (△); B, GA10P5 (■) vs. GA10P10 (○) and type B gelatin-PEG hydrogels (C, GB5P5 (◆) vs. GB15P5 (△); D, GB10P5 (■) vs. GB10P10 (○)). The degradation profile of each formulation incubated with type I collagenase (solid line) was compared with hydrogel incubated in PBS (dashed line).
Covalently crosslinked gelatin molecules significantly increased their resident time within the matrix and rendered the system biodegradable. Incorporating PEG component provided a tight polymeric network to maintain the hydrogel structure and increased the mechanical rigidity of the system.
3.3. Hydrogel microviscoelastic properties
The diameters of the polystyrene beads used in this experiment were 1.09 and 1.87 μm, i.e. much smaller than mammalian cells. Thus, the local viscosity represented the viscoelastic properties of hydrogels on a subcellular scale. The surface modification of the polystyrene beads did not affect their Brownian motion within the hydrogel. Both types of beads behaved similarly in all formulae. Visible Brownian motion of the beads was observed in GA10P3 (Fig. 7B and Movie 1) with different viscosity values at different locations (2.71 ± 0.18, 1.05 ± 0.08, 2.78 ± 0.05 and 2.29 ± 0.36 mPa s at four locations, the local viscosity of each trapped bead was measured three times), which indicated that the microviscosity in PEG-gelatin hydrogel was not homogeneous. When the PEG-dA concentration was raised to 5% and 7.5% in the precursor solutions (GA5P5, GA10P5, GA15P5 and GA10P7.5), the Brownian motion of the beads was only obtained in the boundary area (60–100 μm from the sides of the gel slabs) and was not measurable in the middle of the hydrogels (Fig. 7C and Movie 2). All the beads appeared stuck when the PEGdA concentration was raised to 10% (GA10P10). Different to the PEGdA concentration, the gel-PEG-Cys concentration did not have any significant effect on the behavior of the beads. The local viscosity properties are mainly dependent on the close proximity of the cavity walls formed by PEGdA and gel-PEG-Cys fiber networks and the extra drag from the long, flexible gelatin-PEG polymers present inside the cavity. Individual pores in one hydrogel may have different sizes and different distributions of free polymer ends, thus producing different local viscosities for each bead. Increasing the concentration of PEG-dA in the hydrogels will increase the density of crosslinking and decrease pore size, thus directly restricting the movement of the beads.
3.4. 2-D cell adhesion and 3-D cell encapsulation
G5P5, G10P5 and G15P5 were selected for 2-D cell culture since these formulations exhibited similar bulk rheological properties but different degrees of biofunctionality from various gelatin concentrations. G10P10 had the highest gel stiffness and was employed as a comparison. NHDF adhered to all surfaces but with different cell morphologies by 3 days (Fig. 8A). On hydrogels containing 5% PEG, most cells exhibited elongated morphology with few interconnections. More adherent cells were observed on GA15P5 and GB15P5 (Fig. 8A). Fibroblasts tended to display more spindle-like shapes on the rigid surface (G10P10) than on softer samples. On GA10P10 and GB10P10, fibroblasts covered most of the hydrogel surface after 3 days, and had a morphology similar to those on TCPS. Most of the hydrogels only supported minimal keratinocyte adhesion with round morphology (Fig. 8B). Although higher adherent density could be observed on GB15P5, keratinocytes formed extensive cell clusters which indicated insufficient cell spreading and the likelihood of apoptosis. On GB10P10, however, keratinocytes spread out, exhibited a flattened morphology and formed intercellular networks. Cell interaction with the extracellular matrix (ECM) depends on ligation-induced and traction-induced signaling pathways. Higher gelatin concentration provided more cell-binding motifs, thus promoting cell adhesion. Similarly, smooth muscle cells formed more focal adhesion and stretched F-actin fibers on substrates with increasing stiffness [36]. Ghosh et al. reported that on hyaluronan and fibronectin hydrogels, fibroblasts showed higher proliferation on stiffer hydrogel surfaces [37]. Thus, the increased cell density on GA10P10 and GB10P10 is due to the higher initial adherent density and the higher rate of cell proliferation.
Fig. 8.
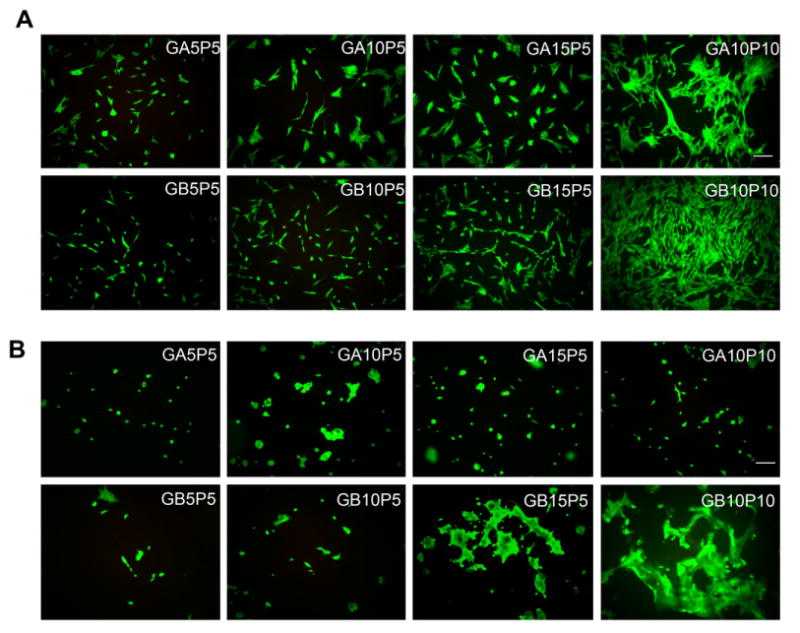
Cell adhesion on the surface of gelatin-PEG hydrogels. Fibroblasts (A) or keratinocytes (B) were applied on hydrogel surface at the same seeding density and cultured for 3 days. Cells were stained via a live/dead staining kit. Scale bar = 100 μm.
When entrapped in the hydrogel, cell behavior was markedly different from that on the 2-D surface. After culture for 2 weeks, fibroblasts started to spread and formed extensions in softer type A gelatin-PEG hydroge (Fig. 9A, upper panel). Although elongated cell phenotype could be found in GA5P5, GA10P5 and GA15P5 (all have similar G* values), extensive cell spreading and intracellular network formation were only observed in GA15P5, which had the highest gelatin concentration. The higher gelatin concentration likely provided more cell-binding sites to support cell spreading and proliferation. With the same gelatin concentration, GA10P5 and GA10P10, well-spread cells were found within the softer hydrogel. GA15P5 and GA10P10 were then imaged at four different depths. As shown in Fig. 9B, cell spreading and network formation could be observed at all four depths in G15P5. In contrast, fibro-blasts were found in three depths within the bulk of G10P10. The extensively spread cells in the first layer of G10P10 are believed to be cells on the hydrogel surface and thus behave similarly as those in the 2-D culture. No cell spreading was observed in hydrogels fabricated with type B gel-PEG-Cys of the same formulation as type A gel-PEG-Cys (Fig. 9A, lower panel). Since hydrogels fabricated with type B gel-PEG-Cys had higher G* values than those with type A (Fig. 2B), these hydrogels might be too rigid for cells to spread. To test this hypothesis, we fabricated hydrogel with 15% type B gel-PEG-Cys and 3% PEGdA (GB15P3) which had lower stiffness and higher swelling profile. After 7 days, fibroblasts exhibited a well-spread morphology and formed intercellular connections, indicating that GB15P3 had the optimal mechanical stiffness to facilitate this type of cell behavior (Fig. 9C).
Fig. 9.
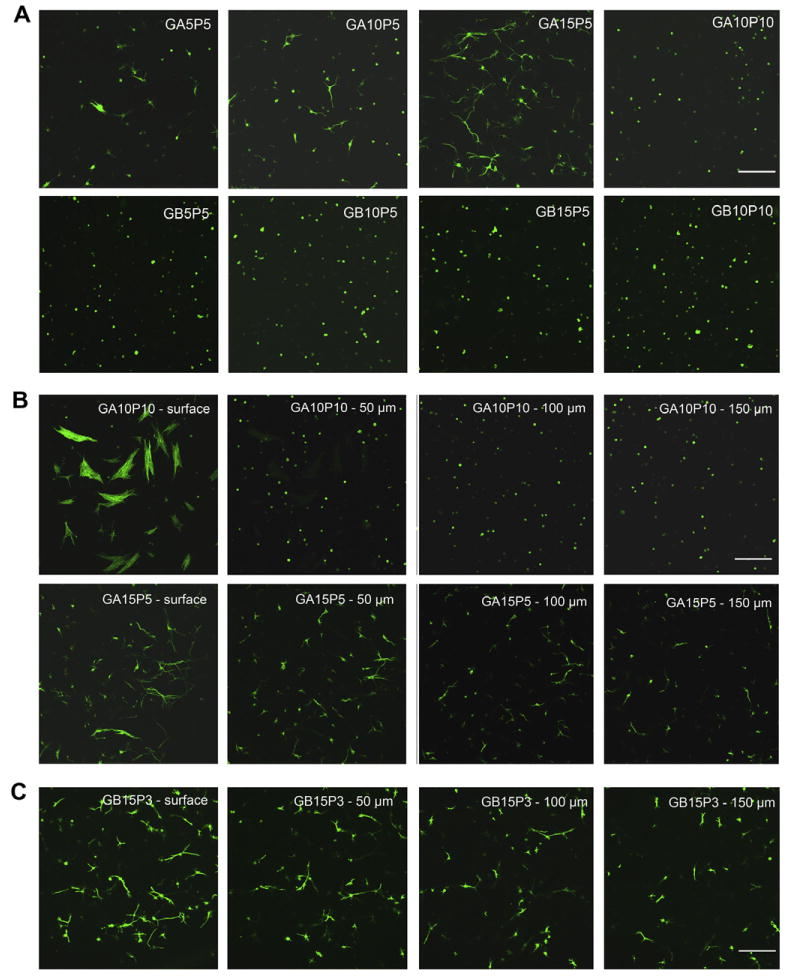
Three-dimensional cell encapsulation in gelatin-PEG hydrogels. Fibroblasts were photoencapsulated in gelatin-PEG hydrogels and cultured for 14 days. Cell cytoskeleton was stained with Alex-488 conjugated phalloidin. (A) F-actin morphology of cells entrapped in type A or type B gelatin-PEG hydrogels. (B) Cell morphology in GA10P10 and GA15P5 hydrogel. Images were taken from four different depths of the hydrogel with 50 μm intervals between each depth. (C) Fibroblasts spread inside the GB15P3 hydrogel. Scale bar = 200 μm.
Hydrogel based on gelatin or collagen has long been used for cell encapsulation. Two major disadvantages of collagen-based scaffolds are their limited physical integrity to match soft tissue structure and extensive contraction when encapsulated with contracting cells [38]. In this study, fibroblasts were encapsulated in 5 mg ml−1 collagen hydrogel and compared with GA15P5. These two hydrogels had similar complex shear moduli and both supported cell adhesion and spreading in a 3-D environment. As shown in Fig. 10, unmodified collagen hydrogel underwent rapid contraction and curling. In contrast, the bulk volume of GA15P5 was comparable to the initial state after 14 days of culture. The significant difference in contraction might be due to the different extent of cell proliferation as well as the enhanced hydrogel rigidity from the covalently crosslinked gelatin-PEG matrix.
Fig. 10.
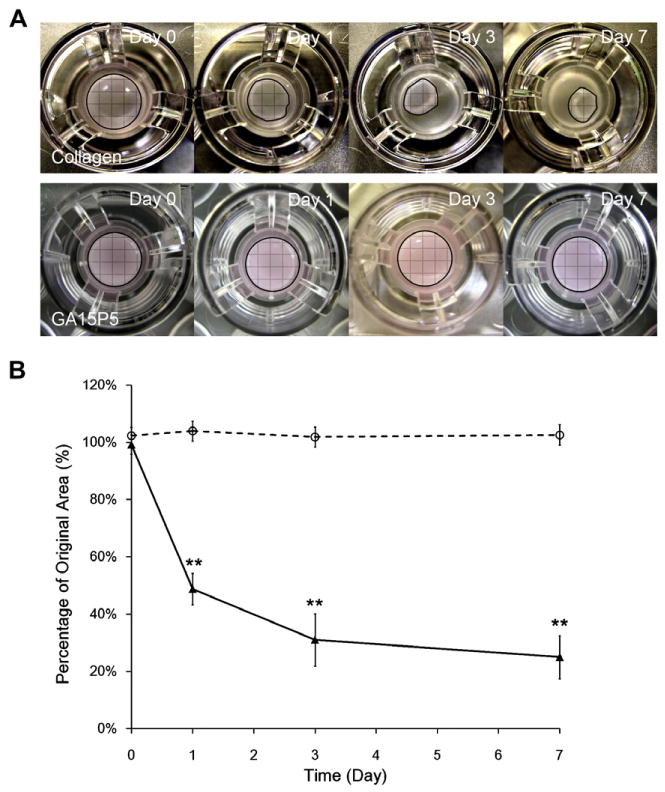
Contraction of PEG-gelatin hydrogel and collagen hydrogel. Fibroblasts were encapsulated in 5 mg ml−1 collagen gel (upper panel in A and solid line in B) and GA15P5 hydrogel (lower panel in A and dashed line in B). Contraction images were taken at predetermined time points (A) and resistance to gel contraction was calculated by the percentage of original gel area at day 0. **P < 0.01, compared with GA15P5.
The in vitro cell culture results illustrated that the covalently crosslinked gelatin molecules in hydrogel promoted cell growth, proliferation and migration. The physiochemical characteristics can be readily adjusted to modulate the cellular behavior, indicating that the current PEG-gelatin hydrogel is a potential substitute for collagen-based matrix.
4. Conclusion
We demonstrated a thiol–ene-based biological/synthetic hybrid biomatrix for living cell encapsulation. Covalently crosslinking thiolated gelatin (gel-PEG-Cys) with PEGdA provided a long-term bio-active 3-D environment for cell growth and migration. The incorporation of gelatin molecule provided a facile platform to control bulk and microenvironment viscoelasticity and physicochemical properties including swelling and degradation. Retaining gelatin molecules within the biomatrix and the controllable physicochemical properties rendered PEG/gelatin hydrogel a suitable scaffold for soft tissue regeneration and cell presentation to reconstruct lost tissue architecture and physical functionality.
Supplementary Material
{kind=link}
Acknowledgments
We gratefully acknowledge Dr. Jeffrey A. Giacomin for assistance with bulk rheology characterization and Whitney Johnson for the assistance of dextran release experiment. We thank the National Institute of Health (NIH Grant R01EB6613) for funding support.
Footnotes
Appendix A. Figures with essential colour discrimination
Certain figures in this article, particularly Figs. 1 and 7–10, are difficult to interpret in black and white. The full colour images can be found in the on-line version, at http://dx.doi.org/10.1016/j.actbio.2012.03.049.
Appendix B. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.actbio.2012.03.049.
References
- 1.Lutolf MP, Hubbell JA. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat Biotechnol. 2005;23(1):47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 2.Drury JL, Mooney DJ. Hydrogels for tissue engineering: scaffold design variables and applications. Biomaterials. 2003;24(24):4337–51. doi: 10.1016/s0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 3.Griffith LG, Naughton G. Tissue engineering – current challenges and expanding opportunities. Science. 2002;295(5557):1009–14. doi: 10.1126/science.1069210. [DOI] [PubMed] [Google Scholar]
- 4.Nuttelman CR, Rice MA, Rydholm AE, Salinas CN, Shah DN, Anseth KS. Macromolecular monomers for the synthesis of hydrogel niches and their application in cell encapsulation and tissue engineering. Prog Polym Sci. 2008;33(2):167–79. doi: 10.1016/j.progpolymsci.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ifkovits JL, Burdick JA. Review: photopolymerizable and degradable biomaterials for tissue engineering applications. Tissue Eng. 2007;13(10):2369–85. doi: 10.1089/ten.2007.0093. [DOI] [PubMed] [Google Scholar]
- 6.Cushing MC, Anseth KS Materials science. Hydrogel cell cultures. Science. 2007;316(5828):1133–4. doi: 10.1126/science.1140171. [DOI] [PubMed] [Google Scholar]
- 7.Dawson E, Mapili G, Erickson K, Taqvi S, Roy K. Biomaterials for stem cell differentiation. Adv Drug Deliv Rev. 2008;60(2):215–28. doi: 10.1016/j.addr.2007.08.037. [DOI] [PubMed] [Google Scholar]
- 8.Burmania JA, Stevens KR, Kao WJ. Cell interaction with protein-loaded interpenetrating networks containing modified gelatin and poly(ethylene glycol) diacrylate. Biomaterials. 2003;24(22):3921–30. doi: 10.1016/s0142-9612(03)00270-9. [DOI] [PubMed] [Google Scholar]
- 9.Fu Y, Xu K, Zheng X, Giacomin AJ, Mix AW, Kao WJ. 3D cell entrapment in crosslinked thiolated gelatin-poly(ethylene glycol) diacrylate hydrogels. Biomaterials. 2011;33(1):48–58. doi: 10.1016/j.biomaterials.2011.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu SH, Jamieson AM. Viscoelastic behaviour at the thermal sol–gel transition of gelatin. Polymer. 1993;34(12):2602–8. [Google Scholar]
- 11.Waldeck H, Kao WJ. Effect of the addition of a labile gelatin component on the degradation and solute release kinetics of a stable PEG hydrogel. J Biomater Sci Polym Ed. 2011 doi: 10.1163/092050611X587547. http://dx.doi.org/10.1163/092050611X587547. [DOI] [PMC free article] [PubMed]
- 12.Mwangi JW, Ofner CM., IIIrd Crosslinked gelatin matrices: release of a random coil macromolecular solute. Int J Pharm. 2004;278(2):319–27. doi: 10.1016/j.ijpharm.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 13.Benton JA, DeForest CA, Vivekanandan V, Anseth KS. Photocrosslinking of gelatin macromers to synthesize porous hydrogels that promote valvular interstitial cell function. Tissue Eng Part A. 2009;15(11):3221–30. doi: 10.1089/ten.tea.2008.0545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlar S, Khademhosseini A. Cell-laden microengineered gelatin methacrylate hydrogels. Biomaterials. 2010;31(21):5536–44. doi: 10.1016/j.biomaterials.2010.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brandl F, Sommer F, Goepferich A. Rational design of hydrogels for tissue engineering: impact of physical factors on cell behavior. Biomaterials. 2007;28(2):134–46. doi: 10.1016/j.biomaterials.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 16.Li RH, Altreuter DH, Gentile FT. Transport characterization of hydrogel matrices for cell encapsulation. Biotechnol Bioeng. 1996;50(4):365–73. doi: 10.1002/(SICI)1097-0290(19960520)50:4<365::AID-BIT3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 17.Parekh SH, Chatterjee K, Lin-Gibson S, Moore NM, Cicerone MT, Young MF, et al. Modulus-driven differentiation of marrow stromal cells in 3D scaffolds that is independent of myosin-based cytoskeletal tension. Biomaterials. 2011;32(9):2256–64. doi: 10.1016/j.biomaterials.2010.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dikovsky D, Bianco-Peled H, Seliktar D. The effect of structural alterations of PEG-fibrinogen hydrogel scaffolds on 3-D cellular morphology and cellular migration. Biomaterials. 2006;27(8):1496–506. doi: 10.1016/j.biomaterials.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 19.Almany L, Seliktar D. Biosynthetic hydrogel scaffolds made from fibrinogen and polyethylene glycol for 3D cell cultures. Biomaterials. 2005;26(15):2467–77. doi: 10.1016/j.biomaterials.2004.06.047. [DOI] [PubMed] [Google Scholar]
- 20.Hutson CB, Nichol JW, Aubin H, Bae H, Yamanlar S, Al-Haque S, et al. Synthesis and characterization of tunable poly(ethylene glycol): gelatin methacrylate composite hydrogels. Tissue Eng Part A. 2011;17(13–14):1713–23. doi: 10.1089/ten.tea.2010.0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gonen-Wadmany M, Goldshmid R, Seliktar D. Biological and mechanical implications of PEGylating proteins into hydrogel biomaterials. Biomaterials. 2011;32(26):6025–33. doi: 10.1016/j.biomaterials.2011.04.055. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt DR, Kao WJ. Monocyte activation in response to polyethylene glycol hydrogels grafted with RGD and PHSRN separated by interpositional spacers of various lengths. J Biomed Mater Res A. 2007;83(3):617–25. doi: 10.1002/jbm.a.31270. [DOI] [PubMed] [Google Scholar]
- 23.Miron T, Wilchek M. A simplified method for the preparation of succinimidyl carbonate polyethylene glycol for coupling to proteins. Bioconjug Chem. 1993;4(6):568–9. doi: 10.1021/bc00024a022. [DOI] [PubMed] [Google Scholar]
- 24.Armstrong JK, Wenby RB, Meiselman HJ, Fisher TC. The hydrodynamic radii of macromolecules and their effect on red blood cell aggregation. Biophys J. 2004;87:4259–70. doi: 10.1529/biophysj.104.047746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tabata Y, Ikada Y. Vascularization effect of basic fibroblast growth factor released from gelatin hydrogels with different biodegradabilities. Biomaterials. 1999;20(22):2169–75. doi: 10.1016/s0142-9612(99)00121-0. [DOI] [PubMed] [Google Scholar]
- 26.Yang TS, Cui Y, Wu CM, Lo JM, Chiang CS, Shu WY, et al. Determining the zero-force binding energetics of an intercalated DNA complex by a single-molecule approach. Chem Phys Chem. 2009;10(16):2791–4. doi: 10.1002/cphc.200900435. [DOI] [PubMed] [Google Scholar]
- 27.Wang MC, Uhlenbeck GE. On the theory of the Brownian motion II. Rev Mod Phys. 1945;17(2–3):323–42. [Google Scholar]
- 28.Chung A, Gao Q, Kao WJ. Macrophage matrix metalloproteinase-2/-9 gene and protein expression following adhesion to ECM-derived multifunctional matrices via integrin complexation. Biomaterials. 2007;28(2):285–98. doi: 10.1016/j.biomaterials.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 29.Cramer NB, Bowman CN. Kinetics of thiol–ene and thiol–acrylate photopolymerizations with real-time fourier transform infrared. J Polym Sci. Part A Polym Chem. 2001;39(19):3311–9. [Google Scholar]
- 30.Salinas CN, Anseth KS. Mixed mode thiol–acrylate photopolymerizations for the synthesis of PEG-peptide hydrogels. Macromolecules. 2008;41(16):6019–26. [Google Scholar]
- 31.Rydholm AE, Reddy SK, Anseth KS, Bowman CN. Controlling network structure in degradable thiol–acrylate biomaterials to tune mass loss behavior. Biomacromolecules. 2006;7(10):2827–36. doi: 10.1021/bm0603793. [DOI] [PubMed] [Google Scholar]
- 32.Calvet D, Wong JY, Giasson S. Rheological monitoring of polyacrylamide gelation: importance of cross-link density and temperature. Macromolecules. 2004;37(20):7762–71. [Google Scholar]
- 33.Mauck RL, Seyhan SL, Ateshian GA, Hung CT. Influence of seeding density and dynamic deformational loading on the developing structure/function relationships of chondrocyte-seeded agarose hydrogels. Ann Biomed Eng. 2002;30(8):1046–56. doi: 10.1114/1.1512676. [DOI] [PubMed] [Google Scholar]
- 34.Padmavathi NC, Chatterji PR. Structural characteristics and swelling behavior of poly(ethylene glycol) diacrylate hydrogels. Macromolecules. 1996;29(6):1976–9. [Google Scholar]
- 35.Witte RP, Blake AJ, Palmer C, Kao WJ. Analysis of poly(ethylene glycol)-diacrylate macromer polymerization within a multicomponent semi-interpenetrating polymer network system. J Biomed Mater Res Part A. 2004;71A(3):508–18. doi: 10.1002/jbm.a.30179. [DOI] [PubMed] [Google Scholar]
- 36.Peyton SR, Raub CB, Keschrumrus VP, Putnam AJ. The use of poly(ethylene glycol) hydrogels to investigate the impact of ECM chemistry and mechanics on smooth muscle cells. Biomaterials. 2006;27(28):4881–93. doi: 10.1016/j.biomaterials.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 37.Ghosh K, Pan Z, Guan E, Ge S, Liu Y, Nakamura T, et al. Cell adaptation to a physiologically relevant ECM mimic with different viscoelastic properties. Biomaterials. 2007;28(4):671–9. doi: 10.1016/j.biomaterials.2006.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewus KE, Nauman EA. In vitro characterization of a bone marrow stem cell-seeded collagen gel composite for soft tissue grafts: effects of fiber number and serum concentration. Tissue Eng. 2005;11(7–8):1015–22. doi: 10.1089/ten.2005.11.1015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.