

Abstract
Objective
The endoplasmic reticulum (ER) plays a critical role in ensuring proper folding of newly synthesized proteins. Aberrant ER stress is reported to play a causal role in cardiovascular diseases. However, the effects of ER stress on vascular smooth muscle contractility and blood pressure remain unknown. The aim of this study was to investigate whether aberrant ER stress causes abnormal vasoconstriction and consequent high blood pressure in mice.
Methods and Results
ER stress markers, vascular smooth muscle contractility, and blood pressure were monitored in mice. Incubation of isolated aortic rings with tunicamycin or MG132, 2 structurally unrelated ER stress inducers, significantly increased both phenylephrine-induced vasoconstriction and the phosphorylation of myosin light chain (Thr18/Ser19), both of which were abrogated by pretreatment with chemical chaperones or 5-Aminoimidazole-4-carboxamide ribonucleotide and metformin, 2 potent activators for the AMP-activated protein kinase. Consistently, administration of tauroursodeoxycholic acid or 4-phenyl butyric acid, 2 structurally unrelated chemical chaperones, in AMP-activated protein kinase-α2 knockout mice lowered blood pressure and abolished abnormal vasoconstrictor response of AMP-activated protein kinase-α2 knockout mice to phenylephrine. Consistently, tunicamycin (0.01 μ/g per day) infusion markedly increased both systolic and diastolic blood pressure, both of which were ablated by coadministration of 4-phenyl butyric acid. Furthermore, 4-phenyl butyric acid or tauroursodeoxycholic acid, which suppressed angiotensin II infusion–induced ER stress markers in vivo, markedly lowered blood pressure in angiotensin II–infused mice in vivo.
Conclusion
We conclude that ER stress increases vascular smooth muscle contractility resulting in high blood pressure, and AMP-activated protein kinase activation mitigates high blood pressure through the suppression of ER stress in vivo.
Keywords: AMPK, ER stress, hypertension, vascular smooth muscle
The endoplasmic reticulum (ER) is a membranous intracellular network where a wide range of proteins are synthesized, processed, and prepared for trafficking. Under stress conditions, proteins become misfolded and accumulated within the ER to provoke the unfolded protein response.1–4 After detecting such accumulation, the ER activates transcriptional and translational pathways leading to the increased expression of ER-resident chaperones, inhibition of protein translation, and acceleration of protein degradation.5 However, prolonged ER stress is considered a pathogenic mechanism in human diseases, including aging,6–11 obesity,12–14 atherosclerosis,15–18 and diabetes mellitus.19–21 However, a causal link between ER stress and hypertension remains to be established.
The AMP-activated protein kinase (AMPK) is an evolutionarily conserved serine/threonine protein kinase, which is a heterotrimeric complex composed of α, β, and γ subunits. The α subunit of AMPK contains the catalytic domain and has 2 isoforms, α1 and α2, which are phosphorylated at threonine 172 on enzyme activation. AMPK is ubiquitously expressed in tissues. It is activated by multiple stress signals including nutrient deprivation, hypoxia, and oxidants. On its activation, AMPK exerts multiple effects other than its conventional effects on energy metabolism.
Recent studies from us and others have established that AMPK is essential in maintaining vascular homeostasis. In vascular smooth muscle cells (VSMCs), AMPK activation is reported to suppress VSMC proliferation,22 migration,23 and hypertrophy.24 AMPK is also reported to exert a direct vasorelaxing effect in isolated aortic rings.25 Indeed, AMPKα2 knockout mice (AMPKα2−/−) are hypertensive exhibiting increased responses to vasoconstrictors.26 In endothelial cells, AMPKα2 deficiency causes aberrant ER stress resulting in endothelial dysfunction and atherosclerosis in vivo.17,18 ER stress is reported to play causative roles in cardiovascular diseases, including diabetes mellitus, atherosclerosis, and heart failure. AMPK is reported to be essential in maintaining ER homeostasis,27 and AMPK inhibition causes aberrant ER stress in endothelial cells, hepatocytes,18,28 and other cell types. 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) is reported to cause both endothelium-dependent and endothelium-independent relaxation.29,30 Whether ER stress contributes to the initiation and progression of hypertension is unknown. In this study, we found that aberrant ER stress increases phenylephrine (PE)-induced vascular contractility and hypertension, both of which were suppressed by AMPK activation. Conversely, we found that AMPKα2 deletion caused aberrant ER stress, abnormal vascular contractility, and high blood pressure in vivo.
Materials and Methods
The ER stress marker, vascular smooth muscle contractility, and blood pressure were monitored in cultured vascular cells and wildtype (WT) or mice deficient in AMPKα1 or AMPKα2. A full description of materials, animals, and methods used, including cell culture, blood pressure measurement, osmotic pump implantation, organ chamber, and Western blots, can be found in the online-only Data Supplement.
Results
Infusion of Tunicamycin Induces ER Stress in Mouse Aortas In Vivo
Tunicamycin is reported to trigger ER stress in cultured cells. Whether Tunicamycin caused ER stress in mice was unknown. We first determined whether Tunicamycin caused ER stress in vivo. To this end, C56BL/6 wild-type (WT) mice were infused with Tunicamycin (10 pg/g per day) for 7 days. Mouse aortas were isolated for immunohistochemical staining of p-elf2α, spiced XBP-1 (XBP-1s), and KDEL, 3 well-characterized ER stress markers. The staining with p-elf2α, XBP-1s, or KDEL was barely detectable in vehicle-infused WT aortas (Figure 1A). However, Tunicamycin infusion markedly increased the levels of p-elf2α, XBP-1s, and KDEL (Figure 1A), indicating that Tunicamycin is a potent ER stress inducer in vascular walls.
Figure 1.
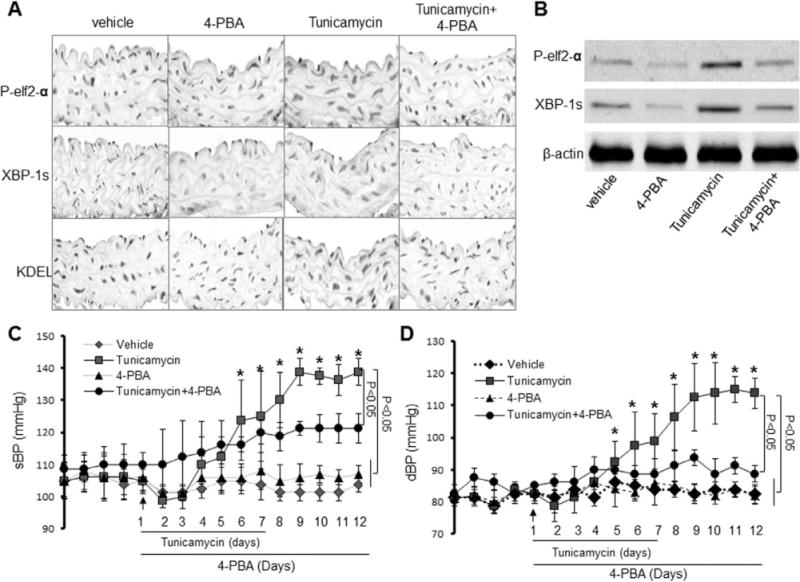
Acute infusion of Tunicamycin causes a 4-Phenyl butyric acid (4-PBA)-inhibitable high blood pressure in vivo. Mice were infused with Tunicamycin with or without 4-PBA. Blood pressures and heart rates were monitored by telemetry, as described in Materials and Methods. A, Effects of 4-PBA on Tunicamycin-induced endoplasmic reticulum (ER) stress in aortas in vivo. The image is a representative of 5 images from ≥5 mice. B, 4-PBA reduces ER stress markers in Tunicamycin-induced ER stress in aortas in vivo. The blot is a representative of 6 blots from 6 individual experiments. C, Effects of 4-PBA on Tunicamycin-induced elevation of systolic blood pressure (sBP); n=12 to 16 in each group. *P<0.05. D, Effects of 4-PBA on Tunicamycin-induced changes of diastolic blood pressure (dBP). n=12 to 16 in each group, *P<0.05. The data from C and D were analyzed using the 2-way ANOVA for repeated measures followed by the Bonferroni post hoc test.
4-Phenyl butyric acid (4-PBA) is a potent ER chaperone. Infusion of 4-PBA did not alter the levels of p-elf2α, spiced XBP-1, and KDEL (Figure 1A) in WT mice. However, 4-PBA markedly reduced Tunicamycin-increased ER stress markers (p-elf2α, XBP-1s, and KDEL) in the aortas (Figure 1A).
We further validated the markers of ER stress in Tunicamycin-infused mouse aortas in Western blots. In line with increased immunohistochemical stainings shown in Figure 1A, Tunicamycin infusion markedly increased the levels of p-elf2α and XBP-1s (Figure 1B). Consistently, 4-PBA administration ablated Tunicamycin-enhanced levels of p-elf2α and XBP-1s (Figure 1B).
Tunicamycin Infusion Increases Systolic and Diastolic Blood Pressures In Vivo
We next evaluated whether Tunicamycin infusion altered systolic (sBP) and diastolic blood pressure (dBP) in mice in vivo. Blood pressure was continuously monitored in vehicle-or Tunicamycin-infused mice for 16 days by using telemetry transmitters. Tunicamycin infusion lowered sBP at days 5 to 7 (Figure 1C). After this initial reduction, Tunicamycin caused a gradual elevation of sBP from day 7 to 9 (Figure 1C). After day 10, sBPs in Tunicamycin-infused mice were significantly higher than those in vehicle-infused mice (Figure 1C). sBP in Tunicamycin-treated mice reached maximal at day 13 and remained elevated until the end of the experiments (day 16; Figure 1C). Tunicamycin infusion did not alter heart rate (data not shown). Thus, Tunicamycin-induced systolic hypertension appeared to be independent of heart rates (data not shown).
We also monitored the effects of Tunicamycin on dBP. Similar to sBP, after an initial drop in pressure between days 5 and 7, dBP in Tunicamycin-infused mice gradually increased at day 8 and reached maximal at day 13 (Figure 1C). The dBP remained significantly elevated at day 16 (Figure 1C).
4-PBA Abolishes Tunicamycin-Enhanced sBP and dBP in Mice
To establish the causal link of ER stress and hypertension, we treated Tunicamycin-infused mice with ER chaperone 4-PBA. As shown in Figure 1C and 1D, 4-PBA alone did not affect either sBP or dBP in vehicle-treated WT mice. 4-PBA alone did not alter heart rate in mice (data not shown). Interestingly, 4-PBA administration partially but significantly reduced Tunicamycin-induced sBP increase (Figure 1C). In contrast to a partial reduction of sBP, 4-PBA completely normalized dBP in Tunicamycin-infused mice (Figure 1D), suggesting that ER stress might preferentially cause the elevation of dBP in vivo.
Exposure of Isolated Aortic Rings to Tunicamycin or MG132 Induces ER Stress Ex Vivo
Next, we assayed whether exposure of isolated aortas to Tunicamycin or MG132 caused ER stress in isolated aortas. To this end, isolated aortas were exposed to Tunicamycin or MG132 with or without the addition of 4-PBA or AICAR. As expected, exposure of aortic rings to either Tunicamycin or MG132 markedly increased the levels of p-elf2α, GRP78, and XBP-1s (Figure 2A and 2B). In addition, 4-PBA attenuated the levels of p-elf2α, GRP78, and XBP-1s enhanced by Tunicamycin or MG132 (Figure 2A and 2B). Furthermore, preincubation of aortic rings with AICAR, an AMPK activator, abolished ER stress markers enhanced by Tunicamycin or MG132 (Figure 2A and 2B). These results suggest that AICAR and 4-PBA can effectively lower Tunicamycin- and MG132- induced ER stress in vessels.
Figure 2.
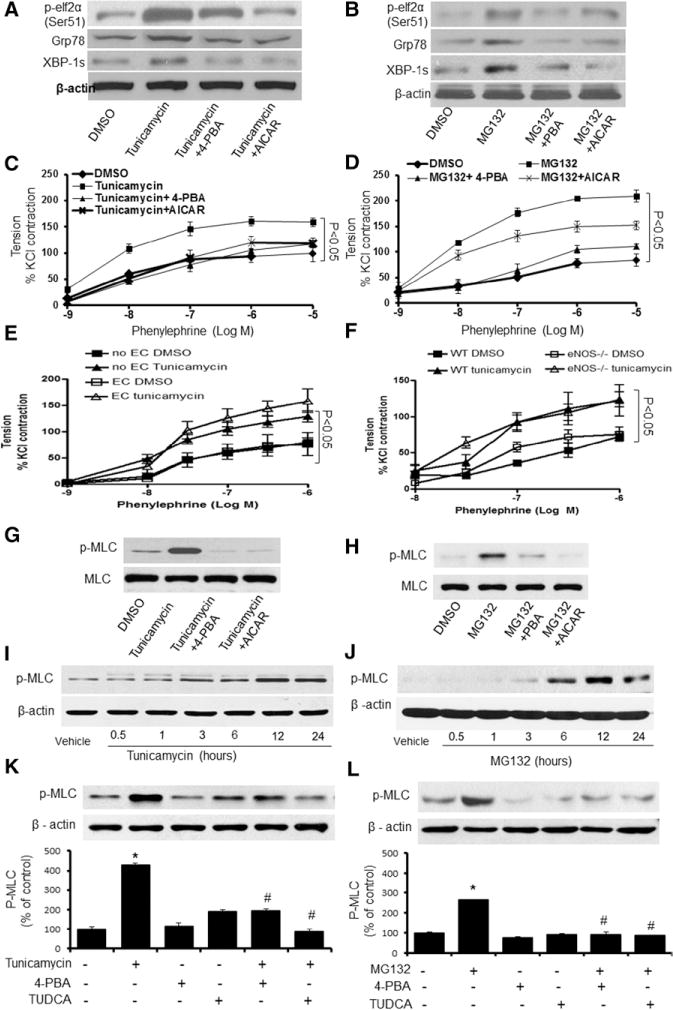
Endoplasmic reticulum (ER) stress enhances vasoconstriction and the phosphorylation of myosin light chain (MLC) in isolated vessels. A, Effects of 4-phenyl butyric acid (4-PBA) and 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) on Tunicamycin-induced ER stress in isolated mouse aortas; n=24 measurements from 6 mice per group. B, Effects of 4-PBA and AICAR on MG132-induced ER stress in isolated mouse aortas; n=24 measurements from 6 mice per group. C, Effects of 4-PBA and AICAR on Tunicamycin-induced vasoconstriction. Results are mean±SEM (n=24 from 6 mice per group). D, Effects of 4-PBA and AICAR on MG132-induced vasoconstriction; vessel contraction in isolated aortas from WT mice treated with 4-PBA and AICAR after MG132 incubation for 2 hours; n=24 measurements from 6 mice per group. E, Effects of Tunicamycin on phenylephrine (PE)-induced vasoconstriction in endothelium-denuded aortic rings. Contraction to phenylephrine in aortic segments of 8- to 10-week–old wild-type (WT) mice scraped endothelial cells and treatment with Tunicamycin. Results are mean±SEM (n=6 from 6 mice per group). *P<0.05 vs Tunicamycin. F, Effects of endothelial nitric oxide synthase (eNOS) deficiency on Tunicamycin-induced vasoconstriction in isolated aortas. Aortic segments of 10-week—old WT mice or eNOS−/− mice were exposed to PE after being treated with Tunicamycin; n=24 measurements from 6 mice per group. *P<0.05 vs Tunicamycin. G and H, Effects of AICAR on Tunicamycin- or MG132-induced phosphorylation of MLC in isolated mouse aortas. I and J, Immunoblots showing the levels of MLC phosphorylation in human smooth muscle cells (HSMCs) treated with Tunicamycin or MG132 for indicated hours. The blot is a representative of 6 blots from 6 mice; n=6. K and L, Immunoblots showing the levels of phosphorylated (p-)MLC in HSMCs treated with 4-PBA or tauroursodeoxycholic acid (TUDCA) for 3 hours after incubation with Tunicamycin (G) or MG132 (H) for 2 hours; n=5. *P<0.05 Tunicamycin- or MG132-treated vs vehicle-treated; #P<0.05 Tunicamycin- or MG132-treated vs 4-PBA plus Tunicamycin or 4-PBA plus MG132. The data from A to F were analyzed using the 2-way ANOVA for repeated measures followed by the Bonferroni post hoc test. Statistical analysis for I to L was performed using a 2-tailed Student t test between 2 groups.
ER Stress Inducers Accentuate PE-Induced Vasoconstriction Ex Vivo
dBP is mainly determined by the contractility of peripheral resistant arteries. Increased vasocontractility was found to be similar in the aortas and peripheral resistant arteries from AMPKα2−/− mice.26 Although mouse aortas are not resistant arteries, we used isolated mouse aortas to assay the effects of ER stress inducers on vasocontractility. As depicted in Figure 2C and 2D, PE caused vasoconstriction in a dose-dependent manner. Incubation of aortic rings with either Tunicamycin or MG132 markedly accentuated PE-induced vasoconstriction (Figure 2C and 2D). In addition, coincubation with 4-PBA, an ER stress inhibitor, markedly lowered vasoconstriction enhanced by either 4-PBA or MG132 (Figure 2C and 2D), implying that ER stress potentiates PE-dependent vasoconstriction.
AICAR Attenuates ER Stress-Accentuated Vasoconstriction
Because AICAR reduced Tunicamycin- and MG132-induced ER stress (Figure 2A and 2B), we hypothesized that AICAR might suppress Tunicamycin-enhanced vasoconstriction in response to PE. As shown in Figure 2C, AICAR abrogated the Tunicamycin-enhanced PE response in isolated aortas. Similarly, AICAR abolished MG132-potentiated PE-induced vasoconstriction (Figure 2D).
ER Stress-Enhanced PE-Induced Vasoconstriction Is Endothelium Independent
We had reported previously that AMPK activation suppressed ER stress in endothelial cells.17,18 It was important to determine whether ER stress in the endothelium contributed to ER stress–enhanced vasoconstriction in response to PE. To this end, endothelium-denuded aortas were incubated with Tunicamycin before its incubation with PE. As shown in Figure 2E, Tunicamycin-induced PE response was comparable between endothelium-intact aortas and the endothelium-denuded aortas.
One of the most important factors in maintaining vasotone is nitric oxide (NO). We next investigated the effects of Tunicamycin in the aortas isolated from endothelial NO synthase (eNOS)-deficient mice (eNOS−/−). PE caused a dose-dependent vasoconstriction in the aortas from WT and eNOS−/− mice (Figure 2F). Exposure of mouse aortas to Tunicamycin markedly increased PE-triggered vasoconstriction in WT and eNOS−/− mice (Figure 2F), suggesting that ER stress in the endothelium was not involved in Tunicamycin-enhanced PE response.
ER Stress-Dependent Phosphorylation of Myosin Light Chain in Isolated Aortas
The phosphorylation of myosin light chain (MLC) facilitates the stimulation of myosin ATPase activity by α-actin, leading to cross-bridge cycling and contraction. Thus, it was important to evaluate the effects of ER stress on MLC phosphorylation in isolated aortas. As expected, exposure of isolated aortic rings to Tunicamycin or MG132 markedly increased the levels of phosphorylated (p-)MLC (Figure 2G and 2H). Importantly, inhibition of ER stress with 4-PBA abolished the effects of Tunicamycin and MG132 on p-MLC (Figure 2G and 2H), implying that ER stress mediated the increase of p-MLC. Similar to 4-PBA, AICAR attenuated Tunicamycin-or MG132-mediated MLC phosphorylation (Figure 2G and 2H), suggesting that the effects of AICAR on p-MLC are likely via its inhibition on ER stress.
ER Stress Triggers MLC Phosphorylation in Cultured VSMCs
Because vasocontractility is mainly dictated by VSMCs in vasculature, we next determined whether Tunicamycin or MG132 altered p-MLC in cultured VSMCs. As depicted in Figure 2I, Tunicamycin-enhanced MLC phosphorylation occurred at 3 hours and reached maximal at 12 hours (Figure 2I). Similarly, MG132 (500 nmol/L) markedly increased the levels of p-MLC at 2 hours and reached a peak at 12 hours (Figure 2J). Importantly, pretreatment of tauroursodeoxycholic acid (TUDCA) or 4-PBA for 2 hours attenuated the effects of Tunicamycin or MG132 on MLC phosphorylation in VSMCs (Figure 2L and 2K), suggesting an ER stress-dependent MLC phosphorylation in VSMCs.
AMPK Activation Suppresses ER Stress–Enhanced MLC Phosphorylation in VSMCs
We next determined whether AMPK activators, AICAR and metformin, altered ER stress–induced MLC phosphorylation. As depicted in Figure 3A and 3B, treatment with AICAR or metformin markedly reduced Tunicamycin-induced ER stress in cultured VSMCs. Pretreatment with metformin or AICAR for 1 hour attenuated the levels of p-elf2α, Grp78, and XBP-1s (Figure 3A and 3B). The maximal effect of metformin was observed at 2 hours. The maximal effect of AICAR was seen at 4 hours after the treatment (data not shown). Concomitantly, AICAR and metformin also markedly suppressed Tunicamycin- or MG132-enhanced MLC phosphorylation in VSMCs (Figure 3C and 3D).
Figure 3.
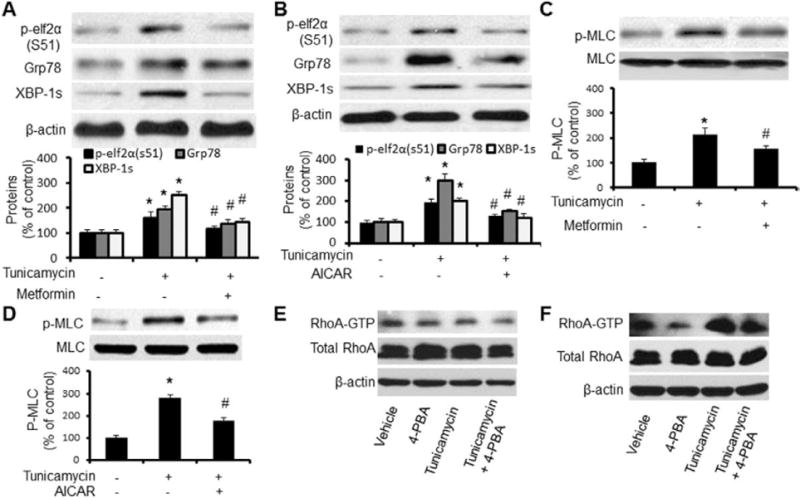
AMP-activated protein kinase (AMPK) activation suppresses endoplasmic reticulum (ER) stress-potentiated myosin light chain (MLC) phosphorylation in cultured vascular smooth muscle cells (VSMCs). A and B, Immunoblots showing the levels of ER stress relative protein P-elf2α(s51), Grp78, and spliced XBP-1 in human smooth muscle cells (HSMCs) treated with 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR; A) and metformin (B) after Tunicamycin incubation for 6 hours; n=5. *P<0.05, Tunicamycin vs vehicle; #P<0.05, Tunicamycin vs Tunicamycin plus AICAR or metformin. C and D, Effects of AICAR and metformin on Tunicamycin-induced MLC phosphorylation in VSMCs. VSMCs were incubated with indicated concentrations of Tunicamycin for 6 hours; n=5. *P<0.05 Tunicamycin vs vehicle; #P<0.05 Tunicamycin vs Tunicamycin plus AICAR or metformin. E and F, Effects of Tunicamycin on RhoA kinase. Confluent VSMCs were treated with tunicamycin for 4 hours (E) or 6 hours (F) with or without 4-phenyl butyric acid (4-PBA). After the incubation, cells were collected and GTP-bound RhoA was pull-down/detected by total RhoA antibody. The blot is representative of 5 blots from 5 independent experiments. Statistical analysis for A to C was performed using a 2-tailed Student t test between 2 groups.
p-MLC is regulated through myosin light-chain kinase and myosin light-chain phosphatase. Thus, it was interesting to determine whether ER stress affected MLC phosphatase. Because RhoA and Rho kinase, components of the Ca2+-independent pathway for maintaining muscle contraction, inhibited MLC phosphatase activity, we further determined whether RhoA kinase was required for ER stress-enhanced MLC phosphorylation in VSMCs. Cultured VSMCs were stimulated with Tunicamycin with or without 4-PBA, and GTP-bound RhoA was pulled down and Western blotted with the antibody against RhoA antibody. Short incubation of Tunicamycin (<6 hours) did not alter RhoA-GTP contents (Figure 3E). Thus, increased p-MLC was before RhoA activation by Tunicamycin. Longer incubation of Tunicamycin (>6 hours) markedly increased the level of RhoA-GTP without altering total Rho A levels, which were partially attenuated by 4-PBA (Figure 3F). Taken together, RhoA activation unlikely contributed to early phosphorylation of p-MLC caused by ER stress.
AMPK Activation Suppresses ER Stress–Enhanced Intracellular Calcium but Increases Calcium in ER
Previous studies31,32 had demonstrated that elevation of intracellular Ca2+ is a common mechanism for aberrant ER stress and unfolded protein response activation. Importantly, p-MLC is regulated through MLC kinase via intracellular Ca2+ elevation, Therefore, we tested whether inhibition of AMPK increased ER stress by altering intracellular Ca2+ levels. As expected, Tunicamycin caused a rapid increase of cytosolic Ca2+ in parallel with a reduction of Ca2+ in ER (Figure 4A); pretreatment with AICAR or metformin significantly reduced Tunicamycin-induced rise of intracellular Ca2+, whereas they increased the levels of Ca2+ in ER (Figure 4A). Consistently, inhibition of AMPK with compound C markedly increased the levels of intracellular Ca2+ (Figure 4B).
Figure 4.
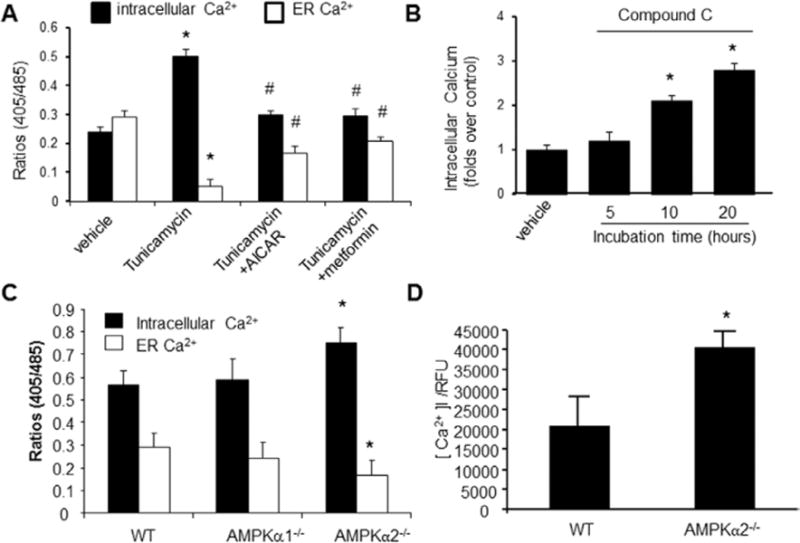
AMP-activated protein kinase (AMPK) activation on intracellular Ca2+ and endoplasmic reticulum (ER) Ca2+ in cultured vascular smooth muscle cells (VSMCs). After being loaded with Indo-1/AM dye, the cells were stimulated with ionomycin (10 μmol/L), which triggers Ca2+ release from the ER. A, 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) and metformin suppress the rise of intracellular [Ca2+]i, whereas they increase Ca2+ in the ER. *P<0.05 Tunicamycin vs vehicle; #P<0.05 AICAR or metformin plus Tunicamycin vs Tunicamycin alone; n=5. *P<0.05 Tunicamycin vs vehicle; #P<0.05 Tunicamycin vs Tunicamycin plus AICAR or metformin. B, Effects of compound C on ionomycin-induced Ca2+ release and intracellular Ca2+ store; *P<0.05 compound C vs vehicle; n=6, *P<0.05 compound C vs vehicle. C, Increased levels of intracellular Ca2+ and decreased Ca2+ in ER in VSMCs derived from AMPKα2−/−; AMPKα2−/− vs wild-type (WT); n=7; *P<0.05 WT vs AMPKα2−/−. D, Increased [Ca2+]i/relative fluorescence units (RFUs) in VSMCs from AMPKα2−/− mice. AMPKα2−/− vs WT. n=8. Statistical analysis was performed by using a 2-tailed Student t test between 2 groups.
To exclude the potential off-target effects of compound C, we assayed the intracellular Ca2+ concentration in VSMCs isolated from WT and AMPKα2−/− mice. As depicted in Figure 4C, intracellular Ca2+ levels in AMPKα2−/− VSMCs were significantly elevated when compared with VSMCs isolated from WT mice (Figure 4C). Consistently, ER Ca2+ levels were significantly lower in VSMCs from AMPKα2−/− mice, as compared with those obtained from WT mice (P<0.05; Figure 4C). Finally, free cytoplasmic calcium [Ca2+] I/relative fluorescence units were significantly elevated in VSMCs from AMPKα2−/− mice when compared with those from WT mice (Figure 4D). Taken together, these results suggest that reduction of AMPKα2 activity increased the intracellular Ca2+ levels but exhibited reduced levels of Ca2+ in the ER.
Aberrant ER Stress Contributes to Elevated Levels of MLC Phosphorylation in AMPKα2−/− Mice
It was interesting to determine whether aberrant ER stress altered the MLC phosphorylation in AMPKα2−/− mice in vivo. As depicted in Figure 5A, AMPKα2−/− mice exhibited higher levels of p-elf2α (Ser51), Grp78, and XBPs than those from WT mice, which were attenuated by the pretreatment of TUDCA or 4-PBA. Importantly, the levels of p-MLC were higher in AMPKα2−/− mice than those in WT mice (Figure 5B). Furthermore, inhibition of ER stress with TUDCA or 4-PBA normalized the levels of p-MLC in AMPKα2−/− mice, although it did not alter the levels of p-MLC in WT mice (Figure 5A and 5B). These results suggested that ER stress in AMPKα2−/− mice caused increased MLC phosphorylation.
Figure 5.

Inhibition of aberrant endoplasmic reticulum (ER) stress normalizes vasoconstriction and myosin light chain (MLC) phosphorylation in AMP-activated protein kinase (AMPK) α2−/− mice. Vessels were pretreated with tauroursodeoxycholic acid (TUDCA) or 4-phenyl butyric acid (4-PBA) for 2 hours. A, Immunoblots of P-elf2α (Ser51), Grp78, and spliced XBP-1 in aortas from mice treated with 4-PBA or TUDCA for 3 weeks. The blot is a representative of 3 blots from ≥3 mice. B, Immunoblots of phosphorylated (p-) MLC in aortas from mice treated with 4-PBA or TUDCA for 3 weeks. The blot is a representative of 3 blots from ≥3 mice. C, Vessel contraction in isolated aortas from wild-type (WT) and AMPKα2−/− mice pretreated with 4-PBA for 2 hours; n=6. D, Effects of TUDCA on vessel contraction in isolated vessels from WT and AMPKα2−/− mice; n=6. The data from C and D were analyzed by using the 2-way ANOVA for repeated measures followed by Bonferroni post hoc test.
Inhibition of ER Stress Normalizes PE-Induced Vasoconstriction in AMPKα2−/− Mice
We next determined whether aberrant ER stress was responsible for increased PE-induced vasoconstriction in AMPKα2−/− mice. As depicted in Figure 5C and 5D, PE caused greater vasoconstriction responses in AMPKα2−/− mice than those from WT mice. Incubation of 4-PBA or TUDCA alone did not alter the vasoconstriction response in WT aortas (data not shown). However, either 4-PBA or TUDCA markedly suppressed PE-induced vasoconstriction (Figure 5C and 5D), suggesting that heightened ER stress was likely responsible for increased vasoconstriction responses in AMPKα2−/− mice.
Inhibition of ER Stress Lowers High Blood Pressure in AMPKα2−/− Mice In Vivo
We had found previously that genetic deletion of AMPKα2 led to elevated blood pressure in C57BL6 mice26 and that this also caused an elevated level of ER stress and disturbed intracellular Ca2+ balance. We therefore reasoned that inhibition of ER stress might lower high blood pressure observed in AMPKα2−/− mice. To this end, WT and AMPKα2−/− mice were administered 4-PBA for 2 weeks. As expected, the levels of ER stress markers (p-elf2α, XBP-1s, and KDEL) were higher in AMPKα2−/− mice when compared with WT mice (Figure 6A). In addition, 4-PBA markedly decreased the levels of ER stress markers (p-elf2α, XBP-1s, and KDEL) in AMPKα2−/− mice in vivo (Figure 6A). As shown in Figure 6B and 6C, 4-PBA significantly lowered both sBP and dBP in AMPKα2−/− mice in vivo.
Figure 6.
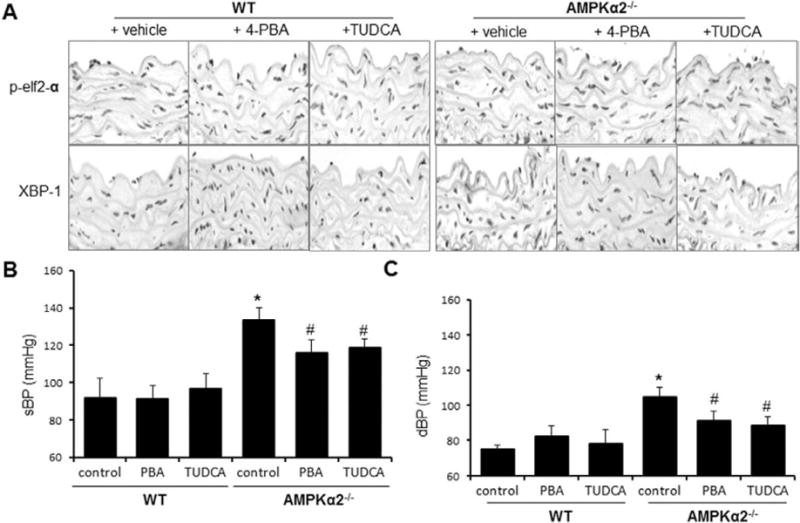
Endoplasmic reticulum (ER) stress chaperones normalize blood pressure in AMP-activated protein kinase (AMPK) α2−/− mice. Wild-type (WT) mice and AMPKα2−/− mice were orally administered with 4-phenyl butyric acid (4-PBA; 1 g/kg per day) or tauroursodeoxycholic acid (TUDCA; 0.5 g/kg per day) for 3 weeks, as described in Materials and Methods. A, 4-PBA and TUDCA suppress aberrant ER stress in AMPKα2−/− mice in vivo. ER stress markers were detected in immunohistochemical staining with the specific antibodies. The image is a representative of 6 images from 6 mice. B, Effects of 4-PBA and TUDCA on systolic blood pressure (sBP); n=12 in each group. *P<0.05 WT vs AMPKα2−/−. #P<0.05 vs AMPKα2−/− vs AMPKα2−/− mice treated with 4-PBA or TUDCA. C, Effects of 4-PBA and TUDCA on diastolic blood pressure (dBP); n=12 in each group. *P<0.05 WT vs AMPKα2−/−. #P<0.05 vs AMPKα2−/− vs AMPKα2–/– mice treated with 4-PBA or TUDCA. Statistical analysis for B and C was performed by using a 2-tailed Student t test between 2 groups.
Inhibition of ER Stress Lowers High Blood Pressure in Mice Infused With Angiotensin II In Vivo
The renin-angiotensin system is essential in maintaining blood pressure. Thus, it was important to determine whether ER stress contributes to angiotensin II (Ang II)-induced hypertension in vivo. As reported previously,33 Ang II infusion caused an increase in sBP and dBP in WT animals (Figure 7A and 7B). As expected, 4-PBA lowered both sBP and dBP at day 7 after Ang II infusion (Figure 7A and 7B). The effects of 4-PBA reached a maximal effect at day 11 for both parameters (Figure 7A and 7B). In contrast, administration of Ang II in AMPKα2−/− mice resulted in a more marked rise in sBP and dBP (Figure 7C and 7D). Similarly, 4-PBA cotreatment in AMPKα2−/− mice resulted in a partial normalization of both sBP and dBP starting at day 7. The effects of 4-PBA on both parameters reached maximal at day 11 (Figure 7C and 7D). The effects of 4-PBA in AMPKα2−/− mice were smaller than those in WT mice (Figure 7E and 7F). The pressures in AMPKα2−/− mice remained ≈ 5- to 10-mm|Hg higher than controls (4-PBA only; Figure 7C and 7D). These results might be explained by heightened ER stress levels in AMPKα2−/− mice.
Figure 7.
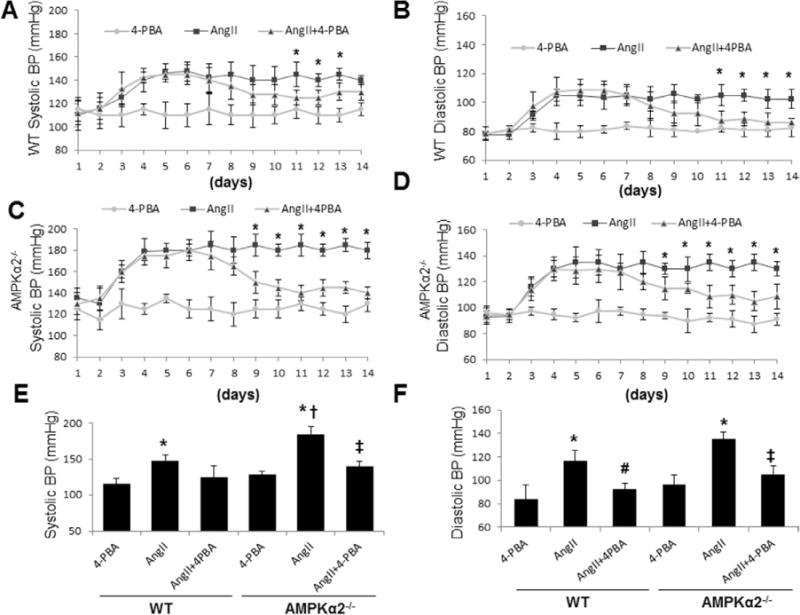
Suppression of endoplasmic reticulum (ER) stresses by 4-phenyl butyric acid (4-PBA) lowers angiotensin II (Ang II)–induced high blood pressure. The systolic blood pressure and diastolic blood pressure were monitored by telemetry system. Means±SEMs for each group (n=12) are presented for both AMP-activated protein kinase (AMPK) α2−/− and wild-type (WT) mice. *P<0.05 Ang II vs 4-PBA or 4-PBA plus Ang II. A and B, WT mice were treated with 4-PBA for 2 weeks after Ang II treatment for 5 days; n=10 in each group. *P<0.05 Ang II vs 4-PBA or 4-PBA plus Ang II. C and D, AMPKα2−/− mice were treated with 4-PBA for 2 weeks after Ang II treatment for 5 days; n=10 in each group. *P<0.05 Ang II vs 4-PBA or 4-PBA plus Ang II. E and F, Systolic and diastolic blood pressures in AMPKα2−/− and WT mice treated with or without 4-PBA. *P<0.05 Ang II vs 4-PBA, #P<0.05 Ang II vs 4-PBA plus Ang II, †P<0.05 Ang II in WT vs Ang II in AMPKα2−/−, and ‡P<0.05 Ang II vs Ang II plus 4-PBA. The data were analyzed by using the 2-way ANOVA for repeated measures followed by Bonferroni post hoc test.
Discussion
In this study, we have provided the first evidence that ER stress can significantly elevate blood pressure in vivo and that this effect is ameliorated by AMPK activation. ER stress inducer Tunicamycin increases VSMC phosphorylation of MLC, vasocontractility, and blood pressure. Importantly, chronic administration of ER stress chaperone lowers blood pressure in Ang II–infused mice and AMPKα2−/− mice, suggesting that AMPK inhibition–triggered ER stress in VSMCs might play a causative role in the development and progression of high blood pressure in vivo.
Blood pressure is maintained by both cardiac output and peripheral vascular resistance.32 ER stress might cause hypertension by increasing cardiac output and peripheral vascular resistance. Indeed, increased sBP and dBP were found in mice infused with Tunicamycin and in AMPKα2−/− mice in which aberrant ER stress was observed. Importantly, administration of ER chaperones significantly reduced both sBP and dBP in AMPKα2−/− mice and in mice infused with Ang II. The most provocative finding in this study is that ER stress causes abnormal contractility of vascular smooth muscle, resulting in increased blood pressure. This conclusion is supported by the following findings. First, induction of ER stress by Tunicamycin or MG132 markedly increased PE-mediated vasoconstriction, which was further confirmed by increased detection of p-MLC. These effects of Tunicamycin were ablated by ER chaperones such as 4-PBA. Second, acute infusion of Tunicamycin caused a marked elevation of both sBP and dBP. Coadministration of 4-PBA, which alone had no effect on blood pressure, abolished the effects of Tunicamycin, suggesting an ER stress-dependent elevation of blood pressure. Third, coinfusion of 4-PBA significantly lowered both sBP and dBP in mice infused with Ang II, suggesting that ER stress might, at least partially, contribute to high blood pressure caused by Ang II infusion, a well-characterized model of hypertension. Finally, AMPKα2−/− mice, which exhibited aberrant levels of ER stress, were hypertensive with increased contractility. Importantly, administration of 4-PBA and TUDCA markedly lowered sBP and dBP in AMPKα2−/− mice but not in WT mice, providing further support that ER stress is essential in maintaining normal blood pressure. Indeed, aberrant ER stress is also reported in hypertensive patients,32,33 or animals34 with metabolic syndrome and in hypertensive animals such as high salt intake-induced hypertensive rats35 and Ang II–infused hypertensive mice.36 Taken together, our results strongly suggest that 4-PBA via ER stress inhibition inhibited vascular contractility and high blood pressure. However, our results have not established a causative relation among ER stress, vascular contractility, and high blood pressure. Although we consider that this possibility is low, we cannot exclude the possibility that 4-PBA might have myriad effects on blood pressure that are independent of ER stress or vascular contractility. Further study on a causative effect of ER stress, vascular contractility, and blood pressure is warranted.
Recent studies have found that activation of AMPK is an important defensive response to ER stress in cardiomyocytes.37,38 Our own previous studies have also found that activation of AMPK exerts protective effects in endothelial cells by suppressing ER stress in the genesis of atherosclerosis.17,18 Here we have extended these observations in VSMCs and found that AMPK, at least in part via its suppression on ER stress in VSMCs, maintains normal vascular tones. AMPKα2 mice exhibited increased vasoconstriction to PE and exhibited high blood pressure, which were sensitive to ER chaperones. The molecular mechanisms by which AMPK inhibits vascular contractility and lowers blood pressure might be multiple. In this study, we found that ER stress promotes the phosphorylation of MLC, likely via the reduction of intracellular Ca2+. This finding is consistent with our previous study, which demonstrates that AMPK functions as a physiological suppressor of ER stress by maintaining sarco/ER Ca2+-ATPase activity and intracellular Ca2+ homeostasis in endothelial cells.18 In addition, AMPK has been shown to lower blood pressure by improving NO bioactivity and endothelial cell function.26 This has been mechanistically attributed to increasing NO release by enhancing the phosphorylation and activation of eNOS at Ser1177 and Ser633 and by blocking NO inactivation by reactive oxygen species.26 Although the contribution of the AMPK-eNOS-ER stress axis in maintaining normal blood pressure remains to be investigated, the present study provides new evidence in support of an ER stress-AMPK-MLC axis that partly explains the hypertensive phenotype of mice deficient in AMPKα2, thereby establishing a novel role of AMPK in the regulation of blood pressure.
Clinical data strongly support that AMPK might be a therapeutic target in treating hypertension. Metformin, one of mostly used antidiabetic drugs, is a potent activator of AMPK. AMPK-lowering effects are supported by blood-lowering effects of metformin.39–41 In women with polycystic ovarian syndrome, metformin reduced sBP, hyperinsulinemia, and insulin resistance and facilitated menstrual regulation and pregnancy.42 Metformin also improves blood pressure in hypertensive and obese women.43
In summary, our results have demonstrated that AMPK is essential in maintaining ER function in VSMCs, and aberrant ER stress might play a causative role in the development and progression of hypertension.
Supplementary Material
Acknowledgments
Sources of Funding
This study was supported by funding from the National Institutes of Health RO1 (HL074399, HL079584, HL080499, HL08920, HL096032, HL105157, and HL110488), the American Diabetes Association, and the Warren Endowed Chair of the University of Oklahoma Health Science Center (all to M. Zou). Part of this work was also supported by an international cooperation grant from the Chinese National Science Foundation (to Y. Zhu and M. Zou). M. Zou is a recipient of the National Established Investigator Award of the American Heart Association.
Footnotes
The online-only Data Supplement is with this article at available at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.112.300606/-/DC1.
Disclosures
None.
References
- 1.Chang WJ, Chehab M, Kink S, Toledo-Pereyra LH. Intracellular calcium signaling pathways during liver ischemia and reperfusion. J Invest Surg. 2010;23:228–238. doi: 10.3109/08941939.2010.496036. [DOI] [PubMed] [Google Scholar]
- 2.Trusina A, Tang C. The unfolded protein response and translation attenuation: a modelling approach. Diabetes Obes Metab. 2010;12(suppl 2):27–31. doi: 10.1111/j.1463-1326.2010.01274.x. [DOI] [PubMed] [Google Scholar]
- 3.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brewer JW, Hendershot LM, Sherr CJ, Diehl JA. Mammalian unfolded protein response inhibits cyclin D1 translation and cell-cycle progression. Proc Natl Acad Sci USA. 1999;96:8505–8510. doi: 10.1073/pnas.96.15.8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gregersen N, Bross P. Protein misfolding and cellular stress: an overview. Methods Mol Biol. 2010;648:3–23. doi: 10.1007/978-1-60761-756-3_1. [DOI] [PubMed] [Google Scholar]
- 6.Salminen A, Kauppinen A, Hyttinen JM, Toropainen E, Kaarniranta K. Endoplasmic reticulum stress in age-related macular degeneration: trigger for neovascularization. Mol Med. 2010;16:535–542. doi: 10.2119/molmed.2010.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alladi PA, Mahadevan A, Vijayalakshmi K, Muthane U, Shankar SK, Raju TR. Ageing enhances alpha-synuclein, ubiquitin and endoplasmic reticular stress protein expression in the nigral neurons of Asian Indians. Neurochem Int. 2010;57:530–539. doi: 10.1016/j.neuint.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 8.Salminen A, Kaarniranta K. ER stress and hormetic regulation of the aging process. Ageing Res Rev. 2010;9:211–217. doi: 10.1016/j.arr.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 9.Lee JH, Won SM, Suh J, Son SJ, Moon GJ, Park UJ, Gwag BJ. Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp Mol Med. 2010;42:386–394. doi: 10.3858/emm.2010.42.5.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–658. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- 11.Puzianowska-Kuznicka M, Kuznicki J. The ER and ageing II: calcium homeostasis. Ageing Res Rev. 2009;8:160–172. doi: 10.1016/j.arr.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 12.Boden G, Merali S. Measurement of the increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Meth Enzymol. 2011;489:67–82. doi: 10.1016/B978-0-12-385116-1.00004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKK[beta]/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135:61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG, Jr, Ozcan U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9:35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 15.Davies PF, Civelek M. Endoplasmic reticulum stress, redox, and a proinflammatory environment in athero-susceptible endothelium in vivo at sites of complex hemodynamic shear stress. Antioxid Redox Signal. 2011;15:1427–1432. doi: 10.1089/ars.2010.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT, Koschinsky ML, Harkewicz R, Witztum JL, Tsimikas S, Golenbock D, Moore KJ, Tabas I. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010;12:467–482. doi: 10.1016/j.cmet.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong Y, Zhang M, Liang B, Xie Z, Zhao Z, Asfa S, Choi HC, Zou MH. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation. 2010;121:792–803. doi: 10.1161/CIRCULATIONAHA.109.900928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong Y, Zhang M, Wang S, Liang B, Zhao Z, Liu C, Wu M, Choi HC, Lyons TJ, Zou MH. Activation of AMP-activated protein kinase inhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes. 2010;59:1386–1396. doi: 10.2337/db09-1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas SE, Dalton LE, Daly ML, Malzer E, Marciniak SJ. Diabetes as a disease of endoplasmic reticulum stress. Diabetes Metab Res Rev. 2010;26:611–621. doi: 10.1002/dmrr.1132. [DOI] [PubMed] [Google Scholar]
- 20.Wu J, Zhang R, Torreggiani M, Ting A, Xiong H, Striker GE, Vlassara H, Zheng F. Induction of diabetes in aged C57B6 mice results in severe nephropathy: an association with oxidative stress, endoplasmic reticulum stress, and inflammation. Am J Pathol. 2010;176:2163–2176. doi: 10.2353/ajpath.2010.090386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Görgün CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Igata M, Motoshima H, Tsuruzoe K, Kojima K, Matsumura T, Kondo T, Taguchi T, Nakamaru K, Yano M, Kukidome D, Matsumoto K, Toyonaga T, Asano T, Nishikawa T, Araki E. Adenosine monophosphate-activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circ Res. 2005;97:837–844. doi: 10.1161/01.RES.0000185823.73556.06. [DOI] [PubMed] [Google Scholar]
- 23.Liang KW, Yin SC, Ting CT, Lin SJ, Hsueh CM, Chen CY, Hsu SL. Berberine inhibits platelet-derived growth factor-induced growth and migration partly through an AMPK-dependent pathway in vascular smooth muscle cells. Eur J Pharmacol. 2008;590:343–354. doi: 10.1016/j.ejphar.2008.06.034. [DOI] [PubMed] [Google Scholar]
- 24.Zhang M, Dong Y, Xu J, Xie Z, Wu Y, Song P, Guzman M, Wu J, Zou MH. Thromboxane receptor activates the AMP-activated protein kinase in vascular smooth muscle cells via hydrogen peroxide. Circ Res. 2008;102:328–337. doi: 10.1161/CIRCRESAHA.107.163253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goirand F, Solar M, Athea Y, Viollet B, Mateo P, Fortin D, Leclerc J, Hoerter J, Ventura-Clapier R, Garnier A. Activation of AMP kinase alpha1 subunit induces aortic vasorelaxation in mice. J Physiol (Lond) 2007;581:1163–1171. doi: 10.1113/jphysiol.2007.132589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang S, Liang B, Viollet B, Zou MH. Inhibition of the AMP-activated protein kinase-α2 accentuates agonist-induced vascular smooth muscle contraction and high blood pressure in mice. Hypertension. 2011;57:1010–1017. doi: 10.1161/HYPERTENSIONAHA.110.168906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yeh CH, Chen TP, Wang YC, Lin YM, Fang SW. AMP-activated protein kinase activation during cardioplegia-induced hypoxia/reoxygenation injury attenuates cardiomyocytic apoptosis via reduction of endoplasmic reticulum stress. Mediators Inflamm. 2010;2010:130636. doi: 10.1155/2010/130636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Wu Z, Li D, Wang D, Wang X, Feng X, Xia M. Involvement of oxygen-regulated protein 150 in AMP-activated protein kinase-mediated alleviation of lipid-induced endoplasmic reticulum stress. J Biol Chem. 2011;286:11119–11131. doi: 10.1074/jbc.M110.203323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ford RJ, Rush JW. Endothelium-dependent vasorelaxation to the AMPK activator AICAR is enhanced in aorta from hypertensive rats and is NO and EDCF dependent. Am J Physiol Heart Circ Physiol. 2011;300:H64–H75. doi: 10.1152/ajpheart.00597.2010. [DOI] [PubMed] [Google Scholar]
- 30.Wang S, Zhang M, Liang B, Xu J, Xie Z, Liu C, Viollet B, Yan D, Zou MH. AMPKalpha2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: role of 26S proteasomes. Circ Res. 2010;106:1117–1128. doi: 10.1161/CIRCRESAHA.109.212530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beevers G, Lip GY, O’Brien E. ABC of hypertension: the pathophysiology of hypertension. BMJ. 2001;322:912–916. doi: 10.1136/bmj.322.7291.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cornier MA, Dabelea D, Hernandez TL, Lindstrom RC, Steig AJ, Stob NR, Van Pelt RE, Wang H, Eckel RH. The metabolic syndrome. Endocr Rev. 2008;29:777–822. doi: 10.1210/er.2008-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu J, Wang S, Wu Y, Song P, Zou MH. Tyrosine nitration of PA700 activates the 26S proteasome to induce endothelial dysfunction in mice with angiotensin II-induced hypertension. Hypertension. 2009;54:625–632. doi: 10.1161/HYPERTENSIONAHA.109.133736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galán M, Kassan M, Choi SK, Partyka M, Trebak M, Henrion D, Matrougui K. A novel role for epidermal growth factor receptor tyrosine kinase and its downstream endoplasmic reticulum stress in cardiac damage and microvascular dysfunction in type 1 diabetes mellitus. Hypertension. 2012;60:71–80. doi: 10.1161/HYPERTENSIONAHA.112.192500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Isodono K, Takahashi T, Imoto H, Nakanishi N, Ogata T, Asada S, Adachi A, Ueyama T, Oh H, Matsubara H. PARM-1 is an endoplasmic reticulum molecule involved in endoplasmic reticulum stress-induced apoptosis in rat cardiac myocytes. PLoS ONE. 2010;5:e9746. doi: 10.1371/journal.pone.0009746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kassan M, Galán M, Partyka M, Saifudeen Z, Henrion D, Trebak M, Matrougui K. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler Thromb Vasc Biol. 2012;32:1652–1661. doi: 10.1161/ATVBAHA.112.249318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M, Kawase I, Hirota H. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol. 2005;25:9554–9575. doi: 10.1128/MCB.25.21.9554-9575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang J, Holman GD. Insulin and contraction stimulate exocytosis, but increased AMP-activated protein kinase activity resulting from oxidative metabolism stress slows endocytosis of GLUT4 in cardiomyocytes. J Biol Chem. 2005;280:4070–4078. doi: 10.1074/jbc.M410213200. [DOI] [PubMed] [Google Scholar]
- 39.Landin K, Tengborn L, Smith U. Treating insulin resistance in hypertension with metformin reduces both blood pressure and metabolic risk factors. J Intern Med. 1991;229:181–187. doi: 10.1111/j.1365-2796.1991.tb00328.x. [DOI] [PubMed] [Google Scholar]
- 40.Giugliano D, De Rosa N, Di Maro G, Marfella R, Acampora R, Buoninconti R, D’Onofrio F. Metformin improves glucose, lipid metabolism, and reduces blood pressure in hypertensive, obese women. Diabetes Care. 1993;16:1387–1390. doi: 10.2337/diacare.16.10.1387. [DOI] [PubMed] [Google Scholar]
- 41.Giugliano D, Quatraro A, Consoli G, Minei A, Ceriello A, De Rosa N, D’Onofrio F. Metformin for obese, insulin-treated diabetic patients: improvement in glycaemic control and reduction of metabolic risk factors. Eur J Clin Pharmacol. 1993;44:107–112. doi: 10.1007/BF00315466. [DOI] [PubMed] [Google Scholar]
- 42.Glueck CJ, Wang P, Fontaine R, Tracy T, Sieve-Smith L. Metformin-induced resumption of normal menses in 39 of 43 (91%) previously amenorrheic women with the polycystic ovary syndrome. Metab Clin Exp. 1999;48:511–519. doi: 10.1016/s0026-0495(99)90113-0. [DOI] [PubMed] [Google Scholar]
- 43.Velazquez EM, Mendoza S, Hamer T, Sosa F, Glueck CJ. Metformin therapy in polycystic ovary syndrome reduces hyperinsulinemia, insulin resistance, hyperandrogenemia, and systolic blood pressure, while facilitating normal menses and pregnancy. Metab Clin Exp. 1994;43:647–654. doi: 10.1016/0026-0495(94)90209-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.