

Abstract
Identification of measurable nontransient responses to low-dose radiation in human primary cell cultures remains a problem. To this end, circulating endothelial colony-forming (progenitor) cells (ECFCs) were examined as an experimental model. ECFCs were isolated from three cord blood donors. Cells were positive for endothelial cell markers and remained highly proliferative after long-term cryopreservation. A single dose of X-ray radiation (0.06–0.38 Gy) inhibited ECFC culture growth. This effect was evident at 48 hours and persisted up to 72 hr postirradiation. Such protracted cytostatic response of ECFCs to low-dose radiation suggests that ECFC primary cultures can be used to study low-dose radiation effects.
Keywords: Cancer biomarkers, Cancer prevention, Carcinogenesis
INTRODUCTION
Ionizing radiation is an established human carcinogen (1). The widespread use of advanced radiological imaging, such as X-ray computed tomography scans (CT-scans), raises a concern about the potential danger of exposure to low-dose ionizing radiation (LDIR) (2, 3). Humans are likely to have variable responses to LDIR, and therefore, medical exposure to LDIR may produce different effects depending on individual predisposition (1, 4–6). For example, a familial history of cancer has been identified as a modifying factor of cancer risk associated with diagnostic radiation response (7, 8). However, quantitative measures of an individual response to LDIR have not been developed (9). Studying variability of response to LDIR in human populations cannot be done by direct experimentation on humans. Traditionally, peripheral blood lymphocytes have been used to study individual differences in response to radiation (4, 9). These studies confirmed that individuals differ in their response to LDIR. However, the utility of these findings is hampered by the transient nature of the measured response indicators, such as DNA-damage response. Unfortunately, the search for other response indicators is limited by low-proliferation potential of the primary lymphocyte cultures. To overcome these limitations, we have proposed using primary cultures of circulating endothelial colony-forming (progenitor) cells (ECFCs) to study the effects of LDIR. These cells are known to produce long-term primary cultures (10). Human umbilical cord blood plasma can replace fetal bovine serum for in vitro expansion of functional human endothelial colony-forming cells. In this study, we isolated ECFCs from cord blood, characterized them, and demonstrated that a single exposure to LDIR induces an inhibition of ECFC growth.
MATERIALS AND METHODS
Reagents
Endothelial basal medium-2 (EBM-2) and endothelial cells growth supplement, SingleQuotsTM, were obtained from Lonza (Walkersville, MD). Tissue culture plates, Rat Tail Collagen I and Matrigel®, were obtained from BD Biosciences (San Jose, CA). Fetal bovine serum (FBS), 0.05% trypsin/EDTA, Hank’s balanced salt solution without calcium and magnesium (HBSS), and DiI complex of acetylated low-density lipoprotein (from human plasma, DiI AcLDL) were obtained from Life Technologies (Carlsbad, CA). Cell Counting Kit-8 (CCK-8), in vitro Toxicology Kit (TOX-7), paraformaldehyde, Histopaque® -1077, Ulex lectin-FITC, Hoechst 33258, and 0.4% Trypan Blue solution were obtained from Sigma-Aldrich (St. Louis, MO). MultiTox-Fluor Multiplex Cytotoxicity Assay was obtained from Promega (Madison, WI). E-Plates 96 were obtained from Roche Applied Sciences (Indianapolis, IN). CD34-PE, CD105 PE, CD45-FITC, CD14-FITC, CD31-FITC, and all other lineage-specific antibody were obtained from e-Bioscience (San Diego, CA).
Isolation and characterization of ECFCs
Anonymous cord blood specimens were obtained from the Carolinas Cord Blood Bank at Duke University Medical Center, Durham, NC. The protocol was approved by the Duke Institutional Review Board. ECFCs were purified from cord blood using a modified method from Lin et al. (11). A mononuclear cell fraction was prepared from approximately 100 mL of cord blood using Histopaque®-1077, washed twice with HBSS, and plated on a collagen-coated 10-cm dish in EBM-2 medium supplemented with SingleQuots™ kit. Cells were grown in a humidified 5% CO2 atmosphere at 37°C. The medium was exchanged every other day for 14 days, after which the emerged colonies were located under a microscope and collected by trypsinization. Cells were counted and further passaged without the collagen precoating of culture dishes. For long-term preservation, cells were frozen in 10% DMSO in a controlled rate freezing apparatus and stored at −156°C. For all experiments, frozen cells were thawed and then cultured for 1 week prior to further use. Cell population doubling time (Td) was calculated using the following formula (9): Td = Ln2/K′; K′ presents growth rate calculated as Ln(N2/N1)/(t2 − t1), where N1 corresponds to cell index at t1 and N2 corresponds to cell index at t2.
Irradiation
Cells were plated in 96-well plates at 1,000, 2,000, or 10,000 cells per well, as indicated in the figure text. Cells were left to attach for 4–4.5 hr and then irradiated in X-RAD 320 Biological Irradiator using a copper/aluminum filter (0.1 mm Cu + 2.5 mm Al) at a distance of 48 cm. All samples were irradiated at a constant electric potential of 120 kV and the following variable electric currents: for 0.06 Gy - 4 mA (6.9 ± 0.1 cGy/min), for 0.15–0.2 Gy - 10 mA (18.28 ± 0.3 cGy/min), and for 0.38–25 mA (34.66 ± 0.9 cGy/min).
Microscopy
Phase contrast and fluorescent images were obtained using a Zeiss Inverted Tissue Culture Fluorescence Microscope (Carl Zeiss Microscopy GmbH, Germany) equipped with Zeiss FL filter sets 02, 09, and 15. Phase contrast microscopy was used to obtain live cell culture images. For fluorescent microscopy, cells were grown on collagen-coated microscopy coverslips for 48 hours, fixed with 1% paraformaldehyde for 30 min at room temperature, washed with PBS, and blocked with 2% FBS in DMEM. Then, cells were stained for 30 min at room temperature with FITC-conjugated CD31 antibody diluted in 2% FBS/DMEM and counterstained with 1 μg/mL Hoechst 33258 for 3 min. Coverslips were washed twice with PBS, mounted with Fluoromount-G (SouthernBiotech, Birmingham, AL) and studied under the fluorescent microscope using appropriate filter sets. Images were acquired with a CCD camera. The images were processed in gray scale in Adobe Photoshop 7.0.
FACScan analysis
FACScan analysis was performed at the Flow Cytometry Core Facility of Duke Cancer Institute using FACSCalibur Flow Cytometer (BD Biosciences, San Jose, CA).
Cell culture impedance measurement
Cells were seeded in quadruplicates in 96-well plates equipped with gold electrodes allowing continuous measurement of cell culture impedance (E-plates 96). Cell growth was monitored using RT-CES Analyzer (Acea Biosciences, Inc., San Diego, CA) for indicated periods. Data were acquired using xCELLigence software (Roche Applied Science, Indianapolis, IN).
Lactate dehydrogenase (LDH) activity assay
The LDH activity was assayed using a TOX-7 kit (Sigma-Aldrich, St. Louis, MO) according to the manufacturer’s instructions. Briefly, cells were seeded at 10,000 per well, incubated for 4 hr, irradiated, and further grown for up to 72 hr without media change in a humidified CO2 incubator. An LDH substrate was added directly to the media and the reaction proceeded for 2 hr at 37°C. The LDH activity in the media was analyzed by absorbance measurement at 450 nm using 690 nm as a reference wavelength on a SpectraMax multimode plate reader (Molecular Devices, Sunnyvale, CA).
Dead/live cells assay
The ratio of dead/live cells was determined using the MultiTox-Fluor Multiplex Cytotoxicity Assay (Promega, Madison, WI). This assay allows for the simultaneous measurement of two different intra- and extracellular proteolytic activities. Cell-associated protease activity is proportional to the number of live cells, whereas the activity of a protease in the media correlates to the number of cells with damaged membranes (dead cells). Because both activities are measured simultaneously, it allows a ratiometric analysis of cell culture viability/cytotoxicity and the ratio is independent of the absolute number of cells. Briefly, cells were seeded in 96-well plates at 1,000 cells per well. Four hours later, the cultures were irradiated and incubated for another 3 days, as described above. After 72 hr of culture, MultiTox-Fluor assay substrates were added directly to the media. The reaction proceeded for 2 hr, after which the fluorescence was measured using SpectraMax plate reader. 400ex/505em and 485ex/520em filters were used to detect the products of live and dead cell-associated proteases, respectively.
WST-8 assay
Live cell content was determined using Cell Counting Kit-8 (CCK-8) containing 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-8). WST-8 was converted by live cells into a formazan, which dissolved directly into the culture medium, thus allowing simple quantification of viable cells in the culture. Cells were seeded, treated, and incubated as described above. Fresh media containing the WST-8 substrate was added to the cells after a 72-hr growth period. Cells were incubated for 2 hr in humidified CO2 incubator after which the absorbance was measured immediately at 450 nm using SpectraMax plate reader.
Statistics
A two-sided t test was used to assess the difference between control and irradiated cells. All graphs were built using GraphPad Prism 5.0 software.
RESULTS
Endothelial colony-forming cells exhibit robust growth in vitro
The ECFCs were isolated from mononuclear cells fractions of three cord blood samples designated as CB002, CB005, and CB006. The colony-forming cells could be identified under a microscope as early as 5 days after the plating of mononuclear cells on collagen-coated dishes. Well-developed colonies appeared between Days 9 and 12. Cells within the colonies exhibited a “cobblestone” morphology, typical for endothelial cells, and were positive for the endothelial cell marker CD31 [Figures 1(A) and (B)]. Based on an FACScan analysis, the isolated cells were highly positive for CD34 and negative for CD133/1 and CD133/2 (data not shown). ECFCs were also positive for other markers associated with endothelial cells, i.e., CD105 and CD73, and negative for markers characteristic to other blood cell lineages, i.e., CD2, CD 4, CD11B, CD14, CD15, CD19, CD 45, CD56, and CD90. Additional indicators of endothelial phenotype included the formation of a capillary-like network in Matrigel, absorption of Ulex lectin, and uptake of acetylated LDL (data not shown). Individual donor samples produced a variable number of colonies with CB002, CB005, and CB006 giving rise to 30, 45, and 3 colonies, respectively, which corresponded to 1.9 × 106, 1.1 × 106, and 6 × 104 of cells. The colonies from each donor were pooled, passaged two or three times, and frozen in aliquots until further use. After thawing, cell viability was 92–98% based on the trypan blue exclusion method. Cryopreservation of up to 12 months did not affect ECFC proliferation capacity and cells demonstrated a robust growth in standard cell culture conditions [Figure 1(C)]. Based on our data, 1 million of ECFCs can yield as many as 10 billion cells after 1 month of expansion in optimized cell culture conditions.
Figure 1.
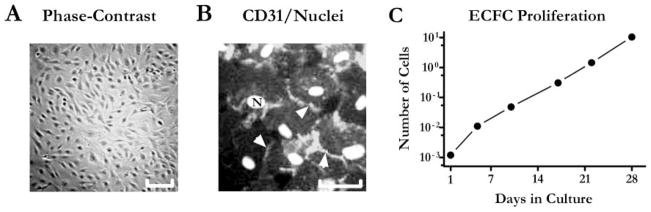
ECFCs demonstrate robust proliferative capacity. A, Phase contrast image of ECFCs colony after 14 days in culture. 5× objective. Scale bar: 50 μm. B, Fluorescence image of ECFCs stained with CD31-FITC (endothelial cells marker) indicated by arrowheads and Hoechst 33258 (nuclear dye) marked as N. The photographs were taken with green and blue filters, respectively, and then superimposed and processed as a gray scale images in Adobe Photoshop. 20× objective. Scale bar: 10 μm. C, ECFC growth chart: cells were passaged every 4–6 days for 4 weeks. Data were extrapolated assuming seeding one million cells on day one and further growing all of the cells. Mean values are plotted on the graph (n = 2). X-axis – days, Y-axis – calculated number of cells (in billions, log10 scale).
Low-dose ionizing radiation produces a cytostatic effect in actively growing ECFC cultures
A real-time cell-culture impedance measurement was used to monitor the effect of LDIR on ECFCs growth and proliferation. In actively growing cell cultures, the change of impedance correlates with the change in cell number (12). CB002, CB005, and CB006 ECFCs were seeded in quadruplicates in 96-well E-plates at 2,000 cells per well and then either subjected to a single radiation dose of 0.2 Gy (18.28 ±0.3 Gy/min) or left untreated. The cells continued to grow for up to 72 hr without media change. Within the next 48 hr, all three nonirradiated ECFC cultures demonstrated close to a linear growth curve with an average cell population doubling time of 19.5 ± 0.1 hr at 24 hr [Figures 2(Ctrl in A–C)]. Irradiated ECFC cultures initially grew at a rate comparable with the control cells with a mean population doubling time of 17.3 ± 3.5 hr at 24 hr [Figures 2(0.2 Gy in A–C)]. However, at 48 hr, irradiated ECFCs grew considerably slower than the control cells; mean population doubling times were 38.7 ± 3.5 hr in the irradiated cultures and 25.1 ± 2.0 hr in the controls. By the 72-hr time point, the irradiated cultures from all three donors had significantly lower cells indexes compared with the untreated cultures (p values for t tests < .05), indicating lower cell numbers.
Figure 2.

LDIR inhibits ECFC growth. A, CB002. B, CB005. C, CB006. Cells were seeded at 2,000 cells per well in 96-well E-plates and were growing for 72 hr. Impedance (expressed as nondimensional “Cell Index”) was measured every 15 min. Arrow indicates the time of irradiation (0.2 Gy X-rays, ~ 4.5 hr after the seeding). Graphs were generated by plotting Cell Index mean values ± SEM vs. time (n = 4). Note that means form a continuous white line. D, MultiTox-Fluor Assay demonstrates that radiation does not increase dead/live cell ratio. CB002, CB005, and CB006 were seeded in black-walled clear plates, irradiated, and incubated as described elsewhere. At 72 hr, fluorescent protease substrates (Promega, Madison, WI) were added directly to the media, and the reaction was incubated for 2 hr. 400ex/505em and 485ex/520em filters were used to detect the products of live and dead cell-associated proteases, respectively. Mean values ± SEM are shown (n = 4).
To determine whether the observed growth inhibition was a result of radiation-induced cell death, a dead/live cell viability assay was employed [Figure 2(D)]. The assay revealed that at 72 hr, the ratio of dead/live cells did not increase in the irradiated cultures as compared with the nonirradiated cultures, suggesting that at 0.2 Gy, X-rays were not cytotoxic to ECFCs. LDH activity measurements produced similar results, i.e., the ratio of LDH activity in conditioned media to intracellular LDH activity did not increase after irradiation (data not shown). Together, these data indicate that a single dose of LDIR inhibited the growth of ECFC cultures without causing cell death.
Cell growth inhibition by different LDIR doses was analyzed using CB005 ECFCs. To ensure linearity of cell growth during a longer period, cells were plated at a lower initial density (1000 cells/well). ECFCs were exposed to either a single dose of 0.06, 0.15, or 0.38 Gy or left untreated (control) and continued to grow for up to 72 hr. The initial cell growth rate of the irradiated cultures was similar to the untreated ones as demonstrated by growth curve [Figure 3(A)] and doubling time graphs [Figure 3(B)]. The population doubling time was 15.2 ± 1.3 hr at the 24-hr time point in both control and irradiated cells [Figure 3(B)]. However, during the next 24 hr the growth of the irradiated cells was considerably impaired. By the 50-hr time point, the population doubling time was 24.7 ± 2.6, 34.9 ± 1.7, 40.6 ± 4.3, and 49.2 ± 5.4 hr in cultures irradiated with 0, 0.06, 0.15, and 0.38 Gy, respectively [Figure 3(B)].
Figure 3.

Inhibition of ECFC growth by different doses of LDIR. A, CB005 growth curve: 0 – control, and after irradiation with 0.06, 0.015, and 0.38 Gy X-rays. CB005 cells were seeded at 1,000 cells per well in E-plates 96, irradiated, and grown for 72 hr. Impedance was measured every 15 min. Graphs were generated by plotting mean values ± SEM vs. time (n = 4). Note that means form a continuous white line. B, Calculated population doubling times. Twelve-hour window was used to calculate average population doubling times (Y-axis) at each indicated time point (X-axis). Mean values ± SEM are shown (n = 4). C, WST-8 assay. Relative viability of control and irradiated CB005 cultures was determined using WST-8 as a substrate. Cells were irradiated and grown as above. At 72 hr, fresh media containing WST-8 was added to cells. After 2 hr of incubation the amount of color product of formazan was determined at 450 nm. Mean values ± SEM are shown (n = 4), t test was used to calculate two-tailed p values. D, LDH activity in the conditioned media. Cells were plated, irradiated, and incubated as described in methods section. Conditioned media were collected at 72 hr. LDH substrate was added, and the reaction was incubated for 2 hr. The amount of product was measured at 450 nm using 690 nm as a reference wavelength. Mean values ± SEM are shown (n = 4), t test was used to calculate two-tailed p values.
WST-8 (cell counting) assay further confirmed these observations, demonstrating that the relative amounts of viable cells were lower in the irradiated cell cultures as compared with controls [Figure 3(C) (p values for t test < 0.05)]. In addition, LDH activity in the media was used as an indirect measure to assess ECFCs death in response to X-rays. The release of intracellular enzyme LDH into culture medium can serve as an indicator of cell membrane damage associated with cell death. In this experiment, cells were irradiated with low (0.15 and 0.38 Gy) and high (2 Gy) radiation doses. LDH activity in the media was determined at 72 hr [Figure 3(D) (68-hr postirradiation)]. There was no difference in LDH activity in media conditioned by irradiated versus control cells, suggesting that low and as high as 2 Gy doses are not cytotoxic to ECFCs (p values for t test > .05).
DISCUSSION
Our main observation is the protracted ECFC response to a single radiation at a dose as low as 0.06 Gy. ECFCs used in our experiments had typical endothelial cells morphology and surface markers and produced primary cultures with high proliferative potential (Figure 1). The EFCFC proliferative potential was not significantly affected by long-term cryopreservation [Figure 1(C)]. These unique qualities of ECFCs are important for the logistics of studies dealing with sample collection from human subjects. In epidemiological studies, biospecimen collection depends on availability of study subjects and cannot be easily synchronized. Ability to store ECFCs samples ensures feasibility of using ECFC-based assays in human studies.
Measurements of cell population growth in ECFC primary cultures demonstrated responsiveness of these cells to 0.2 Gy LDIR observed as a protracted deceleration of cell growth [Figures 2(A)–(C)]. The experiments with lower doses (0.06 and 0.15 Gy) confirmed responsiveness of ECFCs to LDIR at doses comparable with those produced by CTscans (2, 13). The observed deceleration of cell growth in response to a single LDIR dose persisted for days (Figures 2 and 3), as opposed transient character of the DNA damage response measured by gamma-H2AX foci (within minutes) (4, 6).
Although our results did not completely exclude radiation-induced cell death, there are several indications that the observed differences reflect an inhibition of cell growth [Figures 2(A)–(C) and 3(A)–(B)]. Three endpoint assays convincingly demonstrated the lack of cell damage after 3 days of cell culture: the release of live cell-associated protease activity [Figure 3(C)], intracellular dehydrogenase mediated conversion of WST-8 into formazan [Figure 3(C)], and change in LDH activity in conditioned media [Figure 3(D)]. In addition, microscopic examination did not reveal any sign of cell damage or apoptosis for up to 1 week after irradiation (data not shown).
Further investigation is needed to elucidate molecular mechanisms of the observed growth inhibition. Nonetheless, our data demonstrated that ECFCs present an excellent source of human primary cells for LDIR sensitivity testing. The observed LDIR effect presents a quantifiable (by cell index and/or doubling time) response. Most importantly, the ability to establish long-term primary cultures provides the basis for developing a variety of bioassays translatable to a high-throughput format.
Acknowledgments
The authors would like to thank Andrew Barker, Duke Cancer Institute, for his help in the experiments. We also thank the administrative assistant at the Cancer Prevention Detection and Control Research Program, and Ms. Katherine Zeph for her assistance in editing this paper.
Footnotes
DECLARATION OF INTEREST
Alexander Kinev is a founder and executive of Creative Scientist, Inc. He is also a majority shareholder of this company, which develops a radiation sensitivity test. No other conflicts of interest are reported. The authors alone are responsible for the content and writing of the paper.
This research was supported by an epidemiology grant from the National Brain Tumor Society and Duke Cancer Prevention, Detection and Control Research Program pilot study grants.
References
- 1.National Research Council Committee. Health Risks From Exposure to Low Levels of Ionizing Radiation: BEIR VII – Phase 2. 1. Washington, DC: National Academies Press; 2006. [PubMed] [Google Scholar]
- 2.Brenner DJ, Hall EJ. Computed tomography – an increasing source of radiation exposure. N Engl J Med. 2007;357(22):2277–2284. doi: 10.1056/NEJMra072149. [DOI] [PubMed] [Google Scholar]
- 3.Schauer DA, Linton OW. NCRP Report No. 160, Ionizing Radiation Exposure of the Population of the United States, medical exposure–are we doing less with more, and is there a role for health physicists? Health Phys. 2009;97(1):1–5. doi: 10.1097/01.HP.0000356672.44380.b7. [DOI] [PubMed] [Google Scholar]
- 4.Kato TA, Wilson PF, Nagasaw H, Peng Y, Weil MM, Little JB, Bedford JS. Variations in radiosensitivity among individuals: a potential impact on risk assessment? Health Phys. 2009;97(5):470–480. doi: 10.1097/HP.0b013e3181b08eee. [DOI] [PubMed] [Google Scholar]
- 5.Marcon F, Andreoli C, Rossi S, Verdina A, Galati R, Crebelli R. Assessment of individual sensitivity to ionizing radiation and DNA repair efficiency in a healthy population. Mutat Res. 2003;541(1–2):1–8. doi: 10.1016/s1383-5718(03)00171-2. [DOI] [PubMed] [Google Scholar]
- 6.Bishay K, Ory K, Olivier MF, Lebeau J, Levalois C, Chevillard S. DNA damage-related RNA expression to assess individual sensitivity to ionizing radiation. Carcinogenesis. 2001;22(8):1179–1183. doi: 10.1093/carcin/22.8.1179. [DOI] [PubMed] [Google Scholar]
- 7.Davis F, Il’yasova D, Rankin K, McCarthy B, Bigner DD. Medical diagnostic radiation exposures and risk of gliomas. Radiat Res. 2011;175(6):790–796. doi: 10.1667/RR2186.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ronckers CM, Doody MM, Lonstein JE, Stovall M, Land CE. Multiple diagnostic X-rays for spine deformities and risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2008;17(3):605–613. doi: 10.1158/1055-9965.EPI-07-2628. [DOI] [PubMed] [Google Scholar]
- 9.Maalouf M, Durante M, Foray N. Biological effects of space radiation on human cells: history, advances and outcomes. J Radiat Res. 2011;52(2):126–146. doi: 10.1269/jrr.10128. [DOI] [PubMed] [Google Scholar]
- 10.Huang L, Critser PJ, Grimes BR, Yoder MC. Human umbilical cord blood plasma can replace fetal bovine serum for in vitro expansion of functional human endothelial colony-forming cells. Cytotherapy. 2011;13(6):712–721. doi: 10.3109/14653249.2010.548380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin Y, Weisdorf DJ, Solovey A, Hebbel RP. Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest. 2000;105(1):71–77. doi: 10.1172/JCI8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Atienza JM, Yu N, Kirstein SL, Xi B, Wang X, Xu X, Abassi YA. Dynamic and label-free cell-based assays using the real-time cell electronic sensing system. Assay Drug Dev Technol. 2006;4(5):597–607. doi: 10.1089/adt.2006.4.597. [DOI] [PubMed] [Google Scholar]
- 13.Hricak H, Brenner DJ, Adelstein SJ, Frush DP, Hall EJ, Howell RW, McCollough CH, Mettler FA, Pearce MS, Suleiman OH, Thrall JH, Wagner LK. Managing radiation use in medical imaging: a multifaceted challenge. Radiology. 2011;258(3):889–905. doi: 10.1148/radiol.10101157. [DOI] [PubMed] [Google Scholar]