

Abstract
Described is the construction of a large array of releasable microstructures (micropallets) along with screening and isolation protocols for sorting rare, approximately 1 in 10,000, cancer stem cells (CSCs) from a heterogeneous cell population. A 10.1 × 7.1 cm array of micropallets (50 × 50 × 75 μm structures and 25 μm micropallet gap) was fabricated on a large glass substrate, providing an array of approximately 1.3 million releasable microstructures. Image analysis algorithms were developed to permit array screening for identification of fluorescently labeled cells in less than 15 minutes using an epifluorescent wide-field microscope with a computer controlled translational stage. Device operation was tested by culturing HeLa cells transfected with green fluorescent protein (GFP) admixed with wild-type HeLa cells at ratios of 1:104 to 1:106 on the array followed by screening to identify flourescent cells. Micropallets containing cells of interest were then selectively released by a focused laser pulse and collected on a numbered poly(dimethylsiloxane (PDMS) substrate with high viability. A direct comparison of this technology with fluorescence-activated cell sorting (FACS) demonstrated that micropallet arrays offered enhanced post sorting purity (100%), yield (100%) and viability (94 – 100%) for rare cell isolation. As a demonstration of the technology’s value, pancreatic tumor cells from Panc-1 cell lines and patient-derived xenografts were screened for the presence of CD24, CD44 and CD326; surface markers of pancreatic CSCs. Following cell isolation and culture, 63 ± 23% of the isolated Panc-1 cells and 35% of sorted human xenograft cells formed tumor spheroids retaining high expression levels of CD24, CD44 and CD326. The ability to isolate rare cells from relatively small sample sizes will facilitate our understanding of cell biology and the development of new therapeutic strategies.
INTRODUCTION
Cancer accounts for approximately 25% of deaths in the United States with most mortality due to metastases.1,2 Growth of tumors at sites distant from the primary location arises from intravasation of tumor cells followed by extravasation and growth in new locations. Only a small percentage of tumor cells circulating in the blood stream are competent to engraft and form new tumors.3,4,5 These successful cells are thought to possess stem cell-like attributes which enable the cells to divide, reproducing additional cancer stem cells (CSCs). Additionally, CSCs can differentiate into the proliferating cells comprising the tumor. CSCs have been identified in many tumor systems including: breast cancer,6 prostate cancer,7 the hematopoietic system8 and the central nervous system.9 Pancreatic ductal adenocarcinoma is an important tumor clinically because death rates from individuals diagnosed with pancreatic cancer remain high. A rare subset of cells with stem-like properties in pancreatic cancer is characterized by expression of CD44, CD24 and CD326 surface markers.10 CD44+/CD24+/CD326+ cells have recently been reported to exhibit greater invasive and proliferative properties than other cell populations and are competent to form tumors in mouse xenograft models.10
Difficulties in monitoring and characterizing these CSCs occur due to their low abundance in the heterogeneous tumor cell population. The majority of research directed at analyzing and sorting these stem-like cells employs fluorescence-activated cell sorting (FACS).10,11 For successful FACS sorting of CSCs, the adherent tumor cells are stripped from their growth surfaces, labeled with surface-marker specific antibodies and then isolated by FACS. While FACS possesses high throughput (>10,000 cells/s), these systems are not effective at isolating very rare target cells (frequencies below 0.01%).12 Isolation of rare cells by FACS is often preceded by an enrichment step, such as magnetic-activated cell sorting (MACS), prior to FACS for the best outcome.13,14,15 However, MACS becomes complicated when selection of multiple surface markers is necessary, such is the case for the CD44+/CD24+/CD326+ pancreatic CSCs. Several microfluidic technologies have recently been developed to achieve isolation and analysis of rare cells. These systems utilize a wide variety of sorting strategies including: immunocapture,16 magnetism,17 size,18 and dielectrophoresis.19,20 These strategies often offer low yields especially when sorting cells that normally grow adherent to a surface. The methods require removal of adherent cells from their growth surface which is accompanied by a change in the cell morphology, removal cellular surface markers and markedly altered cell physiology. Sample preparation, cell manipulation and loss of cell-surface contacts all lead to low recovery and viability when adherent cell types are separated by these flow-based sorting strategies.21
Microscopy-based cell imaging devices eliminate challenges associated with examining adherent cells in suspension by allowing analysis of cells while still attached to their growth surfaces. Additionally, these methods permit evaluation of subcellular components, temporal responses and cell-cell interactions. Several imaging cytometry systems have shown success at enumerating rare cells.22,23 Unfortunately, few devices have successfully incorporated cell sorting capabilities with high-throughput microscopy-based detection. The Allbritton group has previously demonstrated the utility of arrays of releasable elements microfabricated on glass substrates termed ‘micropallets’ for sorting single adherent cells and small colonies while the cells remain attached to the micropallet suface.24 This technology has shown success in sorting single cells present at a rarity down to 1% in a mixed cell population with low reagent requirements and with a high post sorting yield and viability.25 However, the minute quantity of many tumorigenic cancer cells intermixed with a very large, heterogeneous cellular population makes standard micropallet arrays (comprising 10,000 – 50,000 elements) ineffective platforms for isolating these cells. In the present work, the potential for using micropallet arrays to sort rare cell types i.e. 1 cell per 104 – 106 non-target cells, is examined. For these purposes, a large array of approximately 1.3 million micropallets was developed along with a high-throughput array screening procedure. A high-resolution wide-field microscope and automated image processing were utilized to identify low abundance target cells on the array. Isolation of viable rare cells by the micropallet arrays was achieved and results were directly compared to FACS sorting. This system was then employed to sort and culture viable CSCs from both the Panc-1 cell line and pancreatic tumor cells acquired from patient-derived xenograft mice models.
Experimental Section
Fabrication of micropallet arrays and PDMS chambers
Large arrays of micropallets composed of 0.25% magnetic 1002F photoresist were fabricated on 150 mm diam. B270 glass slides (Valley Design Corp., Santa Cruz, CA) as described in the Supporting Information.26 Large arrays were composed of a 1350 × 950 array of micropallets with dimensions of 50 × 50 × 75 μm (L × W × H) and a 25 μm gap between micropallets. This generated an array with a total size of 101.2 × 71.3 mm consisting of 1,280,448 micropallets with 513 marker or numbered micropallets. Following micropallet fabrication, a plastic cassette was glued around the micropallet array with poly(dimethylsiloxane (PDMS). The array was coated with hydrophobic perfluoroalkylsilane layer ((heptadecafluoro-1,1,2,2-tetrahydrodecyl) trichlorosilane) by chemical vapor deposition as described previously.27 The arrays were sterilized by rinsing with 95% ethanol and dried in a tissue culture hood. Excess ethanol was removed with 5 phosphate buffered saline (PBS) rinses. Top surfaces of the micropallets on the array were then coated with fibronectin by incubation with 5 mL of 25 μg/mL fibronectin in PBS for one hour at room temperature. For attachment of primary cells, micropallets were pre-incubated with 1% matrigel in PBS for at least one hour at 37°C. Following surface coating, the array was rinsed ×5 with 1X PBS.
Cell culture
HeLa (a human ovarian carcinoma cell line), GFP-HeLa (HeLa cells stably transfected with a fusion protein of histone-H1 and green fluorescent protein [GFP]) and Panc-1 (an epitheliod carcinoma cell line from human pancreas) cells were cultured on a 75 cm2 cell culture flask in DMEM supplemented with FBS (10%), L-glutamine (584 mg L−1), penicillin (100 units mL−1) and streptomycin (100 μg ml−1) in a 37°C incubator with a 5% CO2 atmosphere. Cells were detached from the substrate with 0.25% trypsin in media followed by centrifugation at 800 rcf for 2 min. The supernatant was subsequently removed and replaced by 10 mL media. At this point the cell suspension was passed through a 40 μm sterile cell strainer and cells counted with a hemocytometer.
To culture cells on the micropallet arrays, the 1X PBS was replaced with cell culture media and suspensions of HeLa cells intermixed with various quantities of GFP-HeLa cells were added to the array at a cell:micropallet ratio yielding <1 cell per micropallet (10 mL of 40,000 cells/mL). Cells were allowed to settle and adhere to the array for 8 h unless otherwise stated in the text. Arrays were then rinsed with PBS and imaged for the presence of target cells. Following cell identification, micropallets carrying cells of interest were released from the array with a laser pulse and magnetically collected, as described in Supporting Information. Waste collected from the arrays following cell imaging was collected in a petri dish and manually imaged to count any non-adherent cells. The number of cells counted on the array divided by the total number of cells loaded (enumerated cells plus cells counted in waste) was calculated to determine capture efficiencies.
Microscopy
A computer-controlled (ProScan III motorized stage system, Prior Scientific Inc., Rockland, MA) XY translational stage (H138A/C ProScan upright microscope stage, Prior Scientific Inc., Rockland, MA) was mounted on an Olympus MVX10 MacroView microscope (Olympus, Center Valley, PA) with a Hamamatsu ORCA-Flash4.0 CMOS camera (Hamamatsu, Bridgewater, NJ) for imaging. Focus adjustments were controlled by a motorized focus drive (ProScan™ III motorized focus control, Prior Scientific Inc., Rockland, MA). Automated array screening was achieved through a custom MATLAB program (Figure S1). The boundaries and focal plane of the array were identified, then the array was screened with a 1X objective and 2X zoom which generated 166 individual 6.85 × 6.85 mm images consisting of 8,100 micropallets each. Micropallet arrays were imaged in a raster scan pattern with brightfield microscopy and fluorescence microscopy using blue (Ex. 350±25nm, Dichroic 400, Em. 460±25nm), green (Ex. 470±20, Dichroic 495, Em. 525±25), red (Ex. 545±15, Dichroic 570 Em. 620±30) and/or far red (Ex. 620±60, Dichroic 660, Em. 810±75) fluorescent filter sets; fluorescence illumination was achieved via a Lumen 200 arc lamp (Prior Scientific Inc., Rockland, MA). Following image acquisition, images were processed and analyzed for the presence of target cells by a custom MATLAB program as described in Supporting Information (Figure S6).
Fluorescence-activated cell sorting (FACS)
GFP-HeLa cells were mixed with HeLa cells at ratios of 1:104, 1:105 and 1:106 in complete media. For the 1:104 and 1:105 ratios, a total of 1,000,000 cells were used while 4 × 106 cells were employed for the 1:106 mixture. The cell mixtures were then split into two aliquots, one to be separated by FACS and the other cultured on the micropallet arrays with the goal of isolating the GFP-HeLa cells. For FACS, cells were separated based on forward and side scatter and GFP fluorescence using a singlet-cell gate and 100 μm tip (MoFlo, Beckman-Coulter, Brea, CA). Single cells were deposited into wells of a 96 well glass bottom plate preloaded with 100 μL of conditioned media (Auto Clone, Beckman-Coulter, Brea). Wells with cells were identified by microscopy and cultured in conditioned media for 7 days. After that time, the cells were again examined and colony formation was determined. In addition to the aliquot of cells to be sorted, the FACS system also utilized an additional 10,000 cells (50:50 HeLa/GFP-HeLa) to set the sort parameters.
Mouse Pancreatic Tumor Models
All animal studies were approved by University of North Carolina at Chapel Hill Animal Care and Use Committee and comply with National Institute of Health guidelines. Cells were obtained from patient-derived xenograft mouse models of pancreatic adenocarcinoma.28 Tumors were harvested and washed with PBS. The sample was then disaggregated by mincing with a sterile razor followed by incubation for 20 min at 37°C in 100 μL of combined collagenase D + Dispase II solution (40 mg/mL each) in RPMI media with 10% FBS and 1X Penicillin/Streptomicin (P/S). Following incubation, an additional 1 mL media was added and the cells were centrifuged at 1800 rcf for 2 min in a conical tube. The supernate was then removed and cells resuspended in 10 mL of DMEM containing 10% FBS, 1X P/S, 1 ng/mL insulin growth factor (IGF) and 1 ng/mL epidermal growth factor (EGF) followed by passage through a 40 μm sterile cell strainer (BD Falcon, Franklin Lakes, NJ).
Cell Staining
Following attachment of Panc-1 or xenograft cells to the micropallet tops, the arrays were washed 3× with PBS. An antibody cocktail (consisting of 10 μg/mL anti-CD24 [phycoerythrin (PE)], 10 μg/mL anti-CD44 [fluorescein isothiocyanate] and 10 μg/mL anti-CD326 [AlexaFluor 647]) in cell staining buffer was incubated on the arrays at 4°C for 20 min. Micropallet arrays were then rinsed twice with PBS and covered with a sterile glass slide for imaging.
RESULTS AND DISCUSSION
Design of large-scale micropallet arrays
To identify cells occurring at frequencies as low as 1 in 106, arrays accommodating large numbers of cells were fabricated. Arrays (10.1 × 7.1 cm) with 1.3 million micropallets (50 × 50 × 75 μm (L × W × H), 25 μm interpallet gap) were fabricated on a glass substrate (Figure 1B).24 Every 50th and 51st element was replaced with a single 125 × 125 μm square micropallet imprinted with numbers to assist in identifying micropallet location on the array. The micropallets were composed of 1002F photoresist with 0.25% γFe2O3 nanoparticles wt./wt. to enable efficient collection of released elements within a magnetic field.26 These large-scale arrays contained 130 times the number of micropallets as a standard size micropallet arrays (10,000 micropallets). This substantially reduced the fabrication costs, time and reagents necessary for assaying large numbers of cells.
Figure 1.

Image acquisition and analysis. (A) Schematic of the process flow for image acquisition, data analysis and cell identification. Imaging criteria for scanning the micropallet array is first entered into a ScanArray GUI that controls imaging by brightfield and fluorescence microscopy. Images are then processed with a combination of background subtraction, thresholding, size-based filtering and negative control filtering to identify target cells. Micropallets carrying specified cells are then selectively detached from the array and magnetically collected. (B) Photograph of large micropallet array. A U.S. quarter is shown next to the array for size comparison. (C–D) Micrographs of HeLa cells admixed with a low abundance of GFP-HeLa cells on micropallets. Brightfield and fluorescence images show identification of a single GFP-HeLa cell. Insets show the GFP-HeLa at higher magnification. The ‘star’ highlights array regions with water infiltration around the micropallets.
Image acquisition
Screening of large-scale arrays for rare events requires an efficient means of imaging the array and identifying cells of interest. Numerous factors should be addressed when developing a system for high-throughput imaging including: screening duration, cellular fluorescence intensity and pixel number per cell. A widefield fluorescence microscope was employed to generate high resolution images at low magnification. To automate image acquisition, the camera, microscope stage and focus adjustments were controlled by a customized MATLAB program. The efficiency of system automation was assessed by screening a 10.1 cm × 7.1 cm micropallet array as described in the Supporting Information (Image acquisition). Multiple combinations of microscope objectives and magnifications were examined to evaluate pixel resolution and scan speed. An array screening time of less than 5 min and a resolution of ≤5 μm/pixel were picked as metrics to ensure rapid acquisition of high quality images. A 1X objective with 2X magnification (166 images/array, 8,100 micropallets/image) yielded the most acceptable compromise between resolution (3.34 μm/pixel), image acquisition time (3 min 51 s) and detection sensitivity (100%) (Table S1). This configuration allowed four sequential images of the array in to be acquired in less than 15 min.
Imaging workflow
A defined series of steps were integral in automating micropallet array screening, image processing and cell identification (Figure 1A). Initially, the system parameters were user defined in a graphical user interface (GUI) that included selection of the number of wavelengths, exposure times, size exclusion limits and threshold method (Figure S1). The boundaries (X–Y axis) of the micropallet array were set by imaging and software-based marking of the array corners under brightfield microscopy. Although the glass slide and microscope stage have excellent flatness, micron size particles on either surface often skewed the surface plane of the array by several microns over the array length. Simultaneously, while marking the array boundary, the microscope was focused on the micropallet tops. Principal component analysis (PCA) was then utilized to define a planar fit of the array surface. This provided the Z position for each micropallet so that the image plane always coincided with the micropallet surface during array scanning. The micropallet array was then sequentially screened using both brightfield and fluorescence microscopy. Brightfield images were acquired to aid in cell identification and to report micropallet addresses using the numbered micropallets (Figure 1C). Fluorescence images were attained at the appropriate fluorescence wavelength for the fluorophore (EGFP in this case) within the target cells and at irrelevant wavelengths (blue and red emission). The irrelevant wavelengths were used to aid in identification of debris that was either highly scattering or fluorescent (Figure S3). In general the debris generated a light signal that reached the camera at all three excitation/emission wavelengths. Thus bright objects at the irrelevant wavelengths (blue and red) were likely artifacts that could be ignored despite producing a measurable signal at the same excitation/emission wavelengths as EGFP. The system parameters for imaging a 10.1 × 7.1 cm array were optimized by screening an array containing 100,000 GFP-HeLa cells intermixed with HeLa cells (Figure 1C-D). GFP-HeLa cells were utilized as target cells due to their high, stable fluorescence intensity. The array was imaged at three different excitation/emission wavelengths to create blue, green and red fluorescence images of the array. Imaging parameters were optimized for their effectiveness in correctly identifying GFP-HeLa cells while minimizing false positive reports as described below.
Image processing
Prior to cell identification, image acquisition and pre-processing of raw images was necessary to reduce background noise in the green image. Reduction in the background signals resulting from small air bubbles and autofluorescence of the micropallets were predicted to improve discrimination between target cells and artifacts during image analysis. Image acquisition and processing were optimized by controlling the fluorescence exposure intensity and by applying noise filtering and background subtraction on the images as described in detail in Supporting Information (Image processing). Exposure times were evaluated for their ability to generate the greatest S/N ratio in the shortest duration; an optimal exposure duration for the green image set was determined to be 182 ± 7 (see Supporting Information – Image processing) (Figure S2).
Increases in S/N were further attained by performing noise filtering and background subtraction on the green images.29 Following image capture, five different methods were tested to increase the S/N for the green images; adaptive Wiener filtering, top hat filtering, modified top hat filtering and a combination of these strategies (Table S2).30 The modified top hat filter without adaptive Wiener filtering provided the greatest increase in the S/N (77 ± 7) with respect to the original image (S/N 40 ± 3) (Figure S4D). In essence, the modified top hat filter generated the best background approximation that could be subtracted from the raw image. This image processing strategy was employed prior to data analysis in all subsequent experiments.
Image analysis
To quickly identify cells on the array, background subtracted images were thresholded to remove signals with low intensity and further processed to eliminate debris. Thresholding algorithms were screened for their effectiveness at achieving high sensitivity while minimizing false positive reports, as described in Supporting Information (Imaging analysis – Image thresholding). Four 11.7 mm2 micropallet array images were acquired possessing a total 159 ± 47 user-defined GFP-HeLa cells each. An ideal threshold was determined to be the largest value that yielded 0% false negatives. False positives were tolerated since the goal was to identify 100% of the target cells, while the true positives could be rapidly identified by manual screening of the objects exceeding the threshold value. Manual setting of the threshold yielded a false positives number of 5 ± 2 per image for the four images (Figure S5).
While manually setting a fluorescence threshold cutoff value allowed accurate identification of target cells, this strategy suffered from the same limitations as flow cytometry threshold selection methods: a high concentration of control cells were necessary to efficiently calibrate detection. This approach can be challenging when the target cells are extremely rare and positive controls are not available. For this reason we investigated automated thresholding techniques (Table S3). Six different thresholding algorithms (K-Means clustering, Histogram Trough, Kapur Entropy, Mean, Yen Entropy and Li Entropy) were examined for their ability to identify thresholds which provided 100% sensitivity and minimized false positives as described in Supporting Information (Image Analysis – Automated thresholding).31 The algorithms selected required minimal image processing times and were easily implemented in MATLAB. The ‘Kapur Entropy’ thresholding algorithm was the only algorithm to accurately select 100% of the true positives for the green image while maintaining a low numbers of false positive objects (13 ± 4). This entropic thresholding method iteratively selects a threshold value that maximizes the sum of the entropies above and below the threshold.32 The false positives identified in the green image were due to objects that intensely scattered light.
In addition to removal of objects below threshold cutoffs, intensely fluorescent or scattering debris was eliminated from the green image by identifying bright objects in the red and blue images. The red and blue images were processed in a manner similar to the green image to identify bright objects (Figure 1A). The coordinates of bright entities identified in the blue and red images were used to exclude these objects from consideration in the green image decreasing the number of false positives from the ‘Kapur Entropy’ thresholded image by 57 ± 10% (7 ± 4 false positives remaining).
Another common strategy for removing unwanted signals during imaging cytometry is to apply sized-based filtering to the thresholded image. When a size filter (5 μm < x < 40 μm diameter) was applied to the ‘Kapur Entropy’ thresholded images to remove objects outside of the size range of a GFP-HeLa cell, 40 ± 14% (8 ± 2 false positives remaining) of the false positive results were eliminated, while all true positives were detected. The minimal pixel count per cell, as determined above, must be >1 to permit removal of small artifacts. To further minimize false positive counts, both the scatter and size filters were applied in succession. This strategy reduced the false positive number by 75 ± 8% (4 ± 2 false positives remaining) while still identifying all true positives. Following imaging analysis, target cell coordinates were reported by correlating their absolute position on the XY stage relative to the numbered micropallets imprinted on the array. This allowed the user to quickly screen through the objects of interest at a higher magnification and confirm true positives.
Isolation of rare cells by micropallet technology vs FACS
A direct comparison of micropallet sorting with FACS for sorting 103 or 106 cells when the ratio of target fluorescent cells to background cells was 1:99 has been previously reported.25 The FACS system was unable to isolate cells when loaded with 103 cells but could efficiently isolate target cells from samples composed of 106 cells. Here a comparison was made between the micropallet technology and FACS for isolating very rare target cells. Standard mixtures of GFP-HeLa cells and HeLa cells at ratios of 1:104, 1:105 and 1:106 were generated and split into two aliquots for analysis by each technology. Half the samples (500,000 cells for the ratios of 1:104 and 1:105, and 2,000,000 cells for the ratio of 1:106) were sorted using the micropallet arrays, and in parallel experiments the other half of the sample was sorted by FACS.
Micropallet arrays were imaged and GFP-HeLa cells identified using the automated image analysis software described above (Fig. 2). The identified objects were then manually examined to determine whether the cells expressed GFP. GFP-HeLa cells were flagged in the samples with efficiencies of 100% (48 cells), 100% (5 cells) and 100% (2 cells) for the target to background cell ratios of 1:104, 1:105 and 1:106, respectively (Figure 2G). The optimized image processing and analysis gave on average 468 ± 80 false positive signals over a total of 6 arrays analyzed (7.8 million micropallets). Along with the fluorescence-based target identification, the array was imaged with brightfield microscopy to ascertain the coordinates of the target cells. Numbered micropallets were distributed throughout the array so that the micropallets with GFP-HeLa cell could be readily assigned addresses (Fig. 2A–B). The GFP-HeLa cells identified above were each detached from the array and magnetically collected into numbered PDMS multiwell plates with 100% collection efficiency (Fig. 2C–D). After 7 days of culture, 45, 5 and 2 of the single GFP-HeLa cells formed small colonies from the collected micropallets (for the target cell ratios of 1:104, 1:105 and 1:106, respectively) (Fig. 2E–G). No non-fluorescent HeLa cells were present on the collection plate, indicating that there were no false-negative identifications and a sorting purity of 100%.
Figure 2.
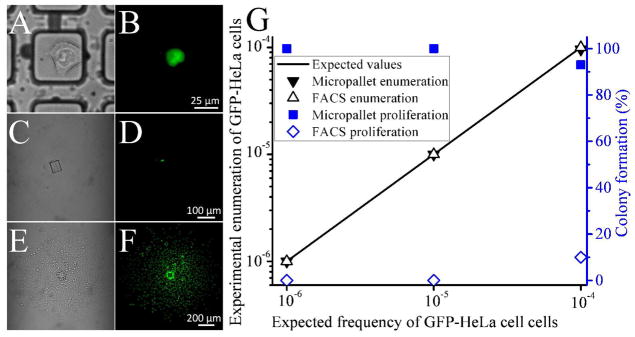
Isolation of low-abundance cells. (A–B) Brightfield and fluorescence images of a GFP-HeLa cell present in an excess of wild-type HeLa cells at a ratio of 1:106. The same cell is shown following micropallet release and collection (C-D) and culture for 7 days (E-F). (G) Efficiencies of low-abundance GFP-HeLa cell sorting by micropallet arrays and FACS. GFP-HeLa cells admixed into a population of HeLa cells at frequencies of 10−6 – 10−4 were detected using micropallet technology (solid black triangles) and FACS (open black triangles). Following sorting into a multiwell plate by micropallet techology (solid blue squares) or FACS (open blue diamonds) the proliferation was recorded as the percentage of cells that formed small colonies after 7 days incubation.
The FACS system was very accurate at identifying the GFP-HeLa cells; however, calibration with a concentrated cell suspension was required prior to cell sorting to set gating thresholds. Target cells were sorted into individual chambers of a 96 well plate and cultured for 7 days (Fig. 2G). When imaged for the presence of colony formation, 5 colonies were observed for the 1:104 mixture (50 cells sorted), no cell colonies were present for the 1:105 mixture (5 cells sorted) and no colonies formed from the 1:106 mixture (2 cells sorted) (Fig. 2G). High cell loss and low collection viability are common drawbacks of FACS which result in low sorting yields for very rare cells. These weaknesses are the basis for the general recommendation that FACS systems not be used for isolating single cells from mixtures at frequencies less than 0.01%.13 Instead, rare cell isolation by FACS typically requires repeated rounds of enrichment applying substantial stresses to cells. Conversely, cells sorted by micropallet arrays formed colonies with excellent efficiency (94 – 100%). The ability to adjust the imaging analysis parameters during or following image acquisition and prior to sorting decisions provides high sorting efficiencies of unknown rare specimens which are not feasible with single time-frame sorting techniques such as FACS and microfluidics. These results suggested that large arrays of micropallets combined with sensitive image acquisition and analysis can efficiently enumerate and isolate low abundance cells.
Isolation of Panc-1 cancer stem cells
The above results suggest that micropallet technology might be used to isolate low abundance cells such as CSCs from a heterogeneous cell population. Reduction to practice was implemented by sorting cells expressing surface markers indicative of being CSCs from the Panc-1 cell line. A single cell suspension of 400,000 Panc-1 cells was cultured on a micropallet array for 12 h (n = 3). Cells on the arrays were stained with antibodies defining CSCs: phycoerythrin (PE)-labeled anti-CD24, fluorescein-labeled anti-CD44 and Alexa Fluor 647-labeled anti-CD326.10 The arrays were imaged and CSCs identified with the automated software as those cells positive for PE, fluorescein and Alexa Fluor 647 but not fluorescent in the blue wavelengths (Fig. 3A). In these analyses, 0.003 ± 0.002% (approx. 1 in 30,000) of the cells were found to be CSCs. Positive cells (n = 43) were then released from the arrays and collected into a multiwell plate coated with matrigel. Following 7 days of culture, collected micropallets were examined for the presence of small CSC colonies. 32 of the isolated cells formed colonies (78 ± 10%). Of these 32 colonies, 24 colonies (63 ± 23%) exhibited a spheroid phenotype with high surface expression of CD24, CD44 and CD326 indicating that they were CSCs (Fig. 3B). The remaining colonies lost expression of at least one of the CSC surface markers and typically exhibited an adherent phenotype with cells spreading across the substrate. These data indicate that low-abundance cells with CSC characteristics can be isolated and cultured maintaining the CSC-marker antigens during culture.
Figure 3.
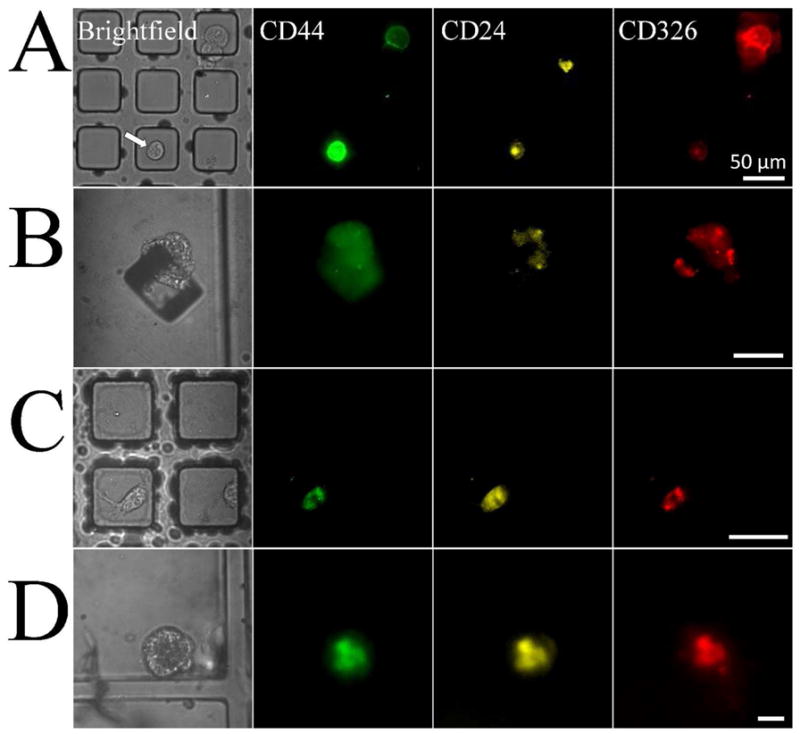
Isolation of pancreatic cancer stem cells. Brightfield and corresponding fluorescence images for fluorescein-labeled anti-CD44, PE-labeled anti-CD24 and Alexa Fluor 647-labeled anti-CD326. (A) Images of a CD44+/CD24+/CD326+ Panc-1 cell on a micropallet and (B) same cell after isolation and 7 days of culture. (C) Images of a CD44+/CD24+/CD326+ human pancreatic tumor xenograft cell on a micropallet and (D) resulting cell colony following isolation and 7 days of culture. Scale bars are 50 μm.
Isolation of pancreatic cancer stem cells from patient-derived xenografts
Clinical tumors possess considerably greater cellular heterogeneity than that of cultured cell lines. We therefore evaluated the ability of the micropallet technology to identify and isolate CSCs from patient-derived xenografts of pancreatic adenocarcinoma. Patient-derived xenografts retain the genetic and phenotypic properties of the primary human tumor making this xenograft system an excellent model system for human cancer.33 Tumors propagated in nude mice were enzymatically disaggregated and filtered to create a single cell suspension. Micropallets were coated with matrigel and 400,000 tumor cells were cultured for 2 days on the array. The array was then incubated with PE-labeled anti-CD24, fluorescein-labeled anti-CD44 and Alexa Fluor 647-labeled anti-CD326. The array was scanned using the automated software to identify cells that were CD24+/CD44+/CD326+. Fifty cells (0.0125% of the cells cultured) exhibited a CSC phenotype (Fig. 3C). Twenty micropallets with CSCs were then released and collected into a PDMS multiwell plate coated with matrigel. The wells were examined for the presence of small colonies following 7 days of culture. Of the collected cells, 12 (60%) formed small colonies of which 7 (35%) had a mammospheroid phenotype and retained a high surface expression of CD24, CD44 and CD326 (Fig. 3D). These results correlate well with previous studies demonstrating enhanced long term in vitro culture of stem-like cells by mammosphere cell culture.34 The procedures outlined above for isolating pancreatic CSCs could easily be adapted to sort CSCs from other tissues and by different surface markers. High efficiency isolation and culture of CSC colonies from individual primary tumor cells enabled by the micropallet technology could improve current research evaluating the biology and drug susceptibility of CSCs.
CONCLUSIONS
An array in excess of 1 million micropallets was developed for capturing and isolating rare cells from a heterogeneous mixture. Image processing and analysis were automated to rapidly identify cells on the array. Important factors in the successful identification of rare cells include the camera exposure time, image background subtraction, threshold selection and reduction of false positives by size-based filtering and negative control images at irrelevant wavelengths. Efficient sorting of adherent cells expressed at abundances down to one in a million was demonstrated with this technology. The ability to isolate rare cells without the need for multiple rounds of sorting will allow new biological applications since low-abundance cells are readily obtained. Though automated screening of the array was performed in <15 min, the precise micropallet positions were manually recorded and the arrays were transferred to an inverted microscope for user-controlled release of select micropallets. Incorporation of the laser-based released system and more advanced analysis software onto the same platform system would enable fully automated array screening and microstructure release. The strength of the micropallets arrays is the potential to maintain segregation between slow and fast growing cells due the very large size of the culture surface. This should permit the separation of slow growing or non-proliferating cells intermixed with fast growers, a feat not easily accomplished with other methods in which the rapidly growing cells overgrow other cell types, such as cells overexpressing a tumor suppressor or other growth inhibiting protein. The technology might also be adapted to isolate viable circulating tumor cells from whole blood without requiring lysis of the erythrocytes and lymphocytes. The strategies developed in this report to quickly scan and analyze a large area will be of utility in identifying cells on other large-scale culture platforms.
Supplementary Material
Acknowledgments
Pavak Shah, Jonathan Clark and Jadwiga Smyla are thanked for providing technical support. Barry Udis of the UNC-CH Flow Cytometry Core Facility is acknowledged for his assistance with FACS. We thank Chapel Hill Analytical and Nanofabrication Laboratory (CHANL) for providing access to the facility’s instrumentation. This research was supported by the NIH (EB012549, CA139599, and CA140424).
Footnotes
Further details and data are provided. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Siegel R, Naishadham D, Jemal A. CA-Cancer J Clin. 2012;62:10. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Chambers A, Groom A, MacDonald I. Nat Rev Cancer. 2002;2:563. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 3.Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, Groom AC. Am J Pathol. 1998;153:865. doi: 10.1016/S0002-9440(10)65628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weiss L. Adv Cancer Res. 1990;54:159. doi: 10.1016/s0065-230x(08)60811-8. [DOI] [PubMed] [Google Scholar]
- 5.Reya T, Morrison SJ, Clarke MF, Weissman IL. Nature. 2001;414:105. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 6.Al-Hajj M, Wicha M, Benito-Hernandez A, Morrison S, Clarke M. Proc Natl Acad Sci USA. 2003;100:3983. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Cancer Res. 2005;65:10946. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 8.Bonnet D, Dick JE. Nat Med. 1997;3:730. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 9.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Nature. 2004;432:396. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 10.Li C, Heidt D, Dalerba P, Burant C, Zhang L, Adsay V, Wicha M, Clarke M, Simeone D. Cancer Res. 2007;67:1030. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 11.Rasheed Z, Wang Q, Matsui W. J Vis Exp. 2010;43:e2169. doi: 10.3791/2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scriba TJ, Purbhoo M, Day CL, Robinson N, Fidler S, Fox J, Weber JN, Klenerman P, Sewell AK, Phillips RE. J Immunol. 2005;175:6334. doi: 10.4049/jimmunol.175.10.6334. [DOI] [PubMed] [Google Scholar]
- 13.Ibrahim S, van den Engh G, Kumar A, Galaev I, Mattiasson B. Cell Separation: Fundamentals, Analytical and Preparative Methods. Springer; Berlin: 2007. [Google Scholar]
- 14.Ibrahim S, van den Engh G. Curr Opin Biotech. 2003;14:5. doi: 10.1016/s0958-1669(02)00009-5. [DOI] [PubMed] [Google Scholar]
- 15.Cristofanilli M, Budd G, Ellis M, Stopeck A, Matera J, Miller M, Reuben J, Doyle G, Allard W, Terstappen L, Hayes D. N Engl J Med. 2004;351:781. doi: 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]
- 16.Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M. Nature. 2007;450:1235. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang JH, Krause S, Tobin H, Mammoto A, Kanapathipillai M, Ingber DE. Lab Chip. 2012;12:2175. doi: 10.1039/c2lc40072c. [DOI] [PubMed] [Google Scholar]
- 18.Vona G, Sabile A, Louha M, Sitruk V, Romana S, Schutze K, Capron F, Franco D, Pazzagli M, Vekemans M, Lacour B, Brechot C, Paterlini-Brechot P. Am J Pathol. 2000;156:57. doi: 10.1016/S0002-9440(10)64706-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gascoyne P, Noshari J, Anderson T, Becker F. Electrophoresis. 2009;30:1388. doi: 10.1002/elps.200800373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu X, Bessette P, Qian J, Meinhart C, Daugherty P, Soh H. Proc Nat Acad Sci USA. 2005;102:15757. doi: 10.1073/pnas.0507719102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seidl J, Knuechel R, Kunz-Schughart LA. Cytometry. 1999;36:102. doi: 10.1002/(sici)1097-0320(19990601)36:2<102::aid-cyto3>3.3.co;2-4. [DOI] [PubMed] [Google Scholar]
- 22.Krivacic R, Ladanyi A, Curry D, Hsieh H, Kuhn P, Bergsrud D, Kepros J, Barbera T, Ho M, Chen L, Lerner R, Bruce R. Proc Nat Acad Sci USA. 2004;101:10501. doi: 10.1073/pnas.0404036101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsieh H, Marrinucci D, Bethel K, Curry D, Humphrey M, Krivacic R, Kroener J, Kroener L, Ladanyi A, Lazarus N, Kuhn P, Bruce R, Nieva J. Biosens Bioelectron. 2006;21:1893. doi: 10.1016/j.bios.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Young G, Bachman M, Sims C, Li G, Allbritton N. Anal Chem. 2007;79:2359. doi: 10.1021/ac062180m. [DOI] [PubMed] [Google Scholar]
- 25.Xu W, Sims C, Allbritton N. Anal Chem. 2010;82:3161. doi: 10.1021/ac100434v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gach P, Sims C, Allbritton N. Biomaterials. 2010;31:8810. doi: 10.1016/j.biomaterials.2010.07.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Sims C, Marc P, Bachman M, Li G, Allbritton N. Langmuir. 2006;22:8257. doi: 10.1021/la061602k. [DOI] [PubMed] [Google Scholar]
- 28.Bang D, Wilson W, Ryan M, Yeh J, Baldwin A. Cancer Discov. 2013 doi: 10.1158/2159-8290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soille P. Morphological Image Analysis: Principles and Applications. Springer; New York: 1999. [Google Scholar]
- 30.Russ JC. The Image Processing Handbook. 6. CRC Press; 2011. [Google Scholar]
- 31.Sezgin M, Sankur B. J Electron Imaging. 2004;13:146. [Google Scholar]
- 32.Kapur J, Sahoo P, Wong A. Comput Gr Image Process. 1985;29:273. [Google Scholar]
- 33.Perez-Torras S, Vidal-Pla A, Miquel R, Almendro V, Fernandez-Cruz L, Navarro S, Maurel J, Carbo N, Gascon P, Mazo A. Cell Oncol. 2011;34:511. doi: 10.1007/s13402-011-0049-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA, Daidone MG. Cancer Research. 2005;65:5506. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.