

Vascular endothelial growth factor-A (VEGF-A) plays a central role in formation of the blood vasculature. The growth factor acts on endothelial cells via a receptor tyrosine kinase, VEGF receptor 2 (VEGFR2) and a non-tyrosine kinase receptor neuropilin-1 (NRP1). Both of these are vital to VEGF-A signaling as a deletion or mutations in either receptor lead to a variety of vascular defects.1,2 Given the critical importance of the VEGF signaling system to vascular development and its exquisite sensitivity to the level of the ligand (even a 50% reduction in VEGF levels during development leads to embryonic lethality), it is not surprising that the system is tightly regulated at a number of levels.
One such recently appreciated regulatory mechanism is the spatial control of VEGFR2 signaling activities. Upon VEGF binding VEGFR2 undergoes endocytosis that is the beginning, rather than the ending, of a series of complicated events that control the downstream signaling pathways. Whereas some downstream pathways may reach full potential at the plasma membrane level, others approach their peak activity only after VEGFR2 enters a particular cellular compartment. VEGF-induced ERK activation is an example of a signaling pathway that mainly takes place in a specific endosomal compartment. While endosomal activation of ERK has been observed before, what is unusual about VEGFR2/ERK signaling is that the full activation of this pathway is regulated by the speed of trafficking of VEGFR2/NRP1 in the cytoplasm.3
Upon binding VEGF-A165, VEGFR2 and NRP1 undergo endocytosis as a complex. NRP1 possesses an N-terminal VEGF binding site that allows it to bind VEGF-A165 and a C-terminal PDZ binding domain. Once the VEGF-A165-VEGFR2-NRP1 complex enters the Rab5+ sorting endosomes, NRP1 via its PDZ binding site binds a PDZ domain protein synectin. Synectin, in turn, links the entire complex to myosin-VI, a reverse transport motor that allows rapid movement of the VEGFR2-containing endosomes away from the plasma membrane.4 This is a key step, as Rab5+ endosomes near the plasma membrane are also targeted by the protein tyrosine phosphatase PTP1b that can specifically dephosphorylate VEGFR2 at Tyr1175, the site involved in activation of the ERK signaling cascade. We have shown that deletion of the NRP1 cytoplasmic domain in arterial endothelial cells gives rise to delayed trafficking of VEGFR2, which causes prolonged exposure of VEGFR2 to PTP1b and a decrease in Tyr1175 phosphorylation (Fig. 1). This, in turn, leads to reduced ERK1/2 activation and impaired arteriogenesis.3 Therefore the dynamics of VEGFR2 trafficking influences its downstream signaling activities. The faster VEGFR2 escapes dephosphorylation by PTP1b, the higher downstream signaling activity it induces.
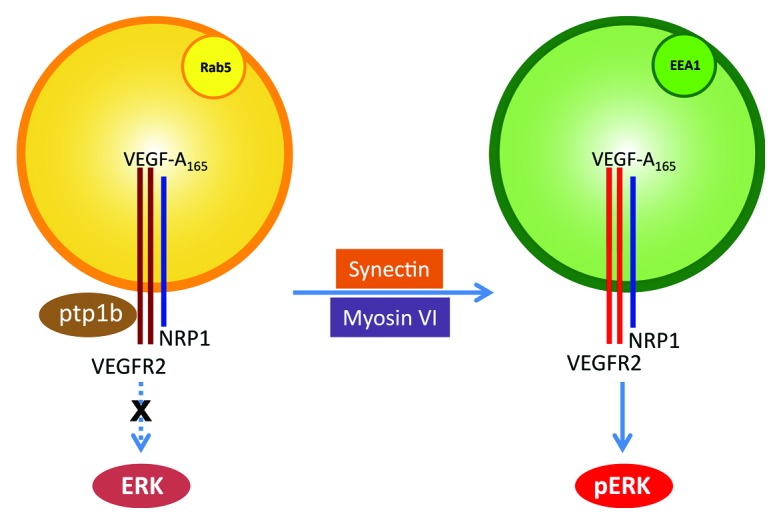
Figure 1. Schematic model of arteriogenic signaling induced by the co-trafficking of VEGFR2 and NRP1. VEGFR2 and NRP1 form a receptor complex upon VEGF-A165 stimulation. NRP1 facilitates VEGFR2 trafficking by binding synectin and linking the complex to myosin-VI. Delayed trafficking of VEGFR2 traps the receptor at Rab5+ endosomes where it is dephosphorylated by PTP1b. In EEA1+ endosomes, VEGFR2 escapes PTP1b dephosphorylation and induces ERK signaling, which is required for arteriogenesis.
What is particularly interesting is that a partial reduction in ERK activation leads to a decrease in arteriogenesis, while formation of the rest of the vasculature is not affected. This is in contrast to the full failure of ERK activation, where formation of the entire vasculature is impaired. One can conclude that arteriogenesis requires higher levels of ERK activation, but why that is the case is not clear. Part of the answer may lie in the importance of ERK signaling for lumen formation. A reduction in NRP1 levels leads to impaired tube formation by endothelial cells, which is rescued by the expression of a constitutively active ERK adenovirus.3 Similarly, impaired arterial morphogenesis in synectin−/−, myosin-VI−/−, and Nrp1cyto mice is characterized not only by reduced branching and arterial network size, but also by reduced lumen size. It is interesting to speculate that lumen size is the key parameter controlled by ERK activation, and that a certain lumen size is necessary for vascular branching.
VEGF-A165 and VEGF-A121 are the two major circulating forms of VEGF-A. VEGF-A165 induced higher ERK activation than VEGF-A121 in HUVECs.5 Since ERK1/2 activation is tightly related to VEGFR2 trafficking, it is reasonable to infer that VEGF-A165 induces more efficient VEGFR2 trafficking than VEGF-A121, probably because of the engagement of NRP1 in the process. The differential functions of the VEGF isoforms have been studied in transgenic mice that only express single VEGF isoforms. Transgenic mice that expressed only VEGF-A164 showed normal arteriolar development,6 whereas mice that expressed only VEGF-A120 exhibited severe defects in arterial development.6,7 These findings further support the link between full ERK activation and normal arteriogenesis.
It seems likely that PTP1b offers a buffering mechanism to prevent excessive VEGFR2 signaling near the cell membrane. A similar mechanism has been identified in EGFR signaling.8 Furthermore, PTP1b is tyrosine phosphorylated by activated EGFR resulting in 3-fold enhanced catalytic activity.8 Such an increase in PTP1b activity may be a general mechanism to downregulate receptor tyrosine kinases activities in certain endosomal compartments. Our observation that VEGFR2 and PTP1b interact in early endosomes does not rule out the interaction between these two proteins in other cellular compartments. Indeed, it is likely that at a later time point and in a different endosomal compartment, PTP1b, or some other PTP, completely shuts down VEGFR2 activity, preparing the receptor for recycling to the plasma membrane and another round of VEGF stimulation.
In conclusion, NRP1-driven VEGFR2 trafficking has emerged as a new and important regulatory mechanism controlling VEGF-induced ERK activation and the extent of arterial morphogenesis. It further provides insights into how VEGFR2 trafficking dynamics affects its signaling activity.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25536
References
- 1.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–6. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 2.Kawasaki T, Kitsukawa T, Bekku Y, Matsuda Y, Sanbo M, Yagi T, et al. A requirement for neuropilin-1 in embryonic vessel formation. Development. 1999;126:4895–902. doi: 10.1242/dev.126.21.4895. [DOI] [PubMed] [Google Scholar]
- 3.Lanahan A, Zhang X, Fantin A, Zhuang Z, Rivera-Molina F, Speichinger K, et al. The neuropilin 1 cytoplasmic domain is required for VEGF-A-dependent arteriogenesis. Dev Cell. 2013;25:156–68. doi: 10.1016/j.devcel.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lanahan AA, Hermans K, Claes F, Kerley-Hamilton JS, Zhuang ZW, Giordano FJ, et al. VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev Cell. 2010;18:713–24. doi: 10.1016/j.devcel.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Furumura M, Morita E. Distinct signaling pathways confer different vascular responses to VEGF 121 and VEGF 165. Growth Factors. 2008;26:125–31. doi: 10.1080/08977190802105909. [DOI] [PubMed] [Google Scholar]
- 6.Stalmans I, Ng YS, Rohan R, Fruttiger M, Bouché A, Yuce A, et al. Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J Clin Invest. 2002;109:327–36. doi: 10.1172/JCI14362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carmeliet P, Ng YS, Nuyens D, Theilmeier G, Brusselmans K, Cornelissen I, et al. Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Nat Med. 1999;5:495–502. doi: 10.1038/8379. [DOI] [PubMed] [Google Scholar]
- 8.Liu F, Chernoff J. Protein tyrosine phosphatase 1B interacts with and is tyrosine phosphorylated by the epidermal growth factor receptor. Biochem J. 1997;327:139–45. doi: 10.1042/bj3270139. [DOI] [PMC free article] [PubMed] [Google Scholar]