

Abstract
Deregulation of the cell cycle and genome instability are common features of cancer cells and various mechanisms exist to preserve the integrity of the genome and guard against cancer. The cullin 4-RING ubiquitin ligase (CRL4) with the substrate receptor Cdt2 (CRL4Cdt2) promotes cell cycle progression and prevents genome instability through ubiquitylation and degradation of Cdt1, p21, and Set8 during S phase of the cell cycle and following DNA damage. Two recently published studies report the ubiquitin-dependent degradation of Cdt2 via the cullin 1-RING ubiquitin ligase (CRL1) in association with the substrate specificity factor and tumor suppressor FBXO11 (CRL1FBXO11). The newly identified pathway restrains the activity of CRL4Cdt2 on p21 and Set8 and regulates cellular response to TGF-β, exit from the cell cycle and cellular migration. Here, we show that the CRL1FBXO11 also promotes the degradation of Cdt2 during an unperturbed cell cycle to promote efficient progression through S and G2/M phases of the cell cycle. We discuss how this new method of regulating the abundance of Cdt2 participates in various cellular activities.
Keywords: FBXO11, Cdt2, Set8, cullin, ubiquitylation
The rapid and specific polyubiquitylation and degradation of key cell cycle-regulatory proteins via the ubiquitin-proteasome system (UPS) drives the ordered and irreversible transition of cells through the various stages of the cell cycle and is critical in regulating cellular proliferation. The Cullin RING E3 ubiquitin ligase 1 complexes (also known as SCF complexes; SKP1-CUL1-F-box protein complexes) represent some of the best-characterized E3 ubiquitin ligases today.1,2 Cullin 1 (CUL1) recruits, through interaction of its C-terminal end with a small RING finger protein Rbx1/2, the E2 ubiquitin-conjugating enzyme (E2) charged with an ubiquitin ready for transfer to a substrate. The N-terminal domain of CUL1 binds the substrate adaptor Skp1, which recruits over 60 F-box containing substrate specificity factors or substrate receptors (F-box proteins) to constitute a large family of distinct SCF (or CRL1) E3 ubiquitin ligases. Several F-box proteins, such as Skp2, β-TrCP, and FBXW7, exhibit altered expression, or their genes are mutated or deleted in a number of human diseases, including cancer.1,3,4 CRL1Skp2 is best known for promoting the degradation of the cyclin-dependent kinase inhibitors p21, p27, and p57 and is essential for cell cycle progression. CRL1β-TrCP degrades cell cycle regulators CDC25A and Wee1 as well as claspin, while CRL1FBXW7 degrades several positive regulators of cell cycle progression: c-myc, cyclin E, Notch, and c-jun.5 Additional CRL1 ligases, such as the CRL1cyclin F, CRL1FBXO4, CRL1FBXL2, and CRL1FBXO11 are less characterized, and few substrates for these E3 ligases have been identified.6-12
CRL4Cdt2: Master Regulator of Cell Cycle Progression and Genome Stability
Similar to the CRL1 family of E3 ubiquitin ligases, the cullin 4 (CUL4) RING ligases (CRL4) are emerging as important regulators of various physiologic activities, such as cellular responses to DNA damage and the regulation of the cell cycle and genomic stability.13 Like CUL1, CUL4A, or CUL4B interacts through Rbx1/2 with an E2, while the N-terminal domain binds the DNA damage binding protein 1 (DDB1, functionally homologous to the Skp1 adaptor protein in the CRL1 complexes). The DDB1 adaptor associates with WD-repeat containing substrate specificity factors (DCAFs, DDB1, and Cullin 4-associated factors), assembling about 90 distinct CRL4 E3 ubiquitin ligases,14-17 many of which are involved in the regulation of various chromatin-related DNA metabolic activities such as DNA transcription and DNA repair.13,18 For example, the Xerodema pigmintosum group E gene product DDB2 (DNA damage-binding protein 2) is a DCAF protein participates in nucleotide excision repair (NER),19 primarily through its ability to assemble with CRL4 (CRL4DDB2) to ubiquitylate the NER component XPC and histone H2A at sites of DNA damage.20-24
Another DCAF, Cdt2 (Cdc10-dependent transcript 2, also known as DTL/RAMP) is a central regulator of cell cycle progression and genomic stability.25 CRL4Cdt2 promotes the degradation of the replication-licensing factor Cdt1 (Cdc10 transcript 1), the Cdk2 inhibitor p21, and the epigenetic modifier and histone H4 lysine 20 (H4K20) monomethyl transferase Set8/Pr-Set7 during S-phase of the cell cycle and following DNA damage (Fig. 1).14,25-36 The ability of CRL4Cdt2 to target these substrates for degradation and to promote the monoubiquitylation of PCNA37 is critical for cell cycle progression, for preventing aberrant DNA re-replication, and for PCNA-dependent translesion DNA synthesis (TLS) (Fig. 1).25 CRL4Cdt2 recognizes many of its substrates when they interact with chromatin-bound PCNA through a conserved and specialized PCNA-interacting peptide (PIP box), a condition only established during S-phase of the cell cycle and following DNA damage.25,38 Overexpression of Cdt2 is sufficient to destabilize at least two of its substrates: p21 and Set8.29,39,40 However, very little information about the regulation of CRL4Cdt2 or its assembly or disassembly is known. Two recent studies identified a mechanism for regulating the level of Cdt2 through ubiquitylation and degradation to impact various cellular activities.39,40
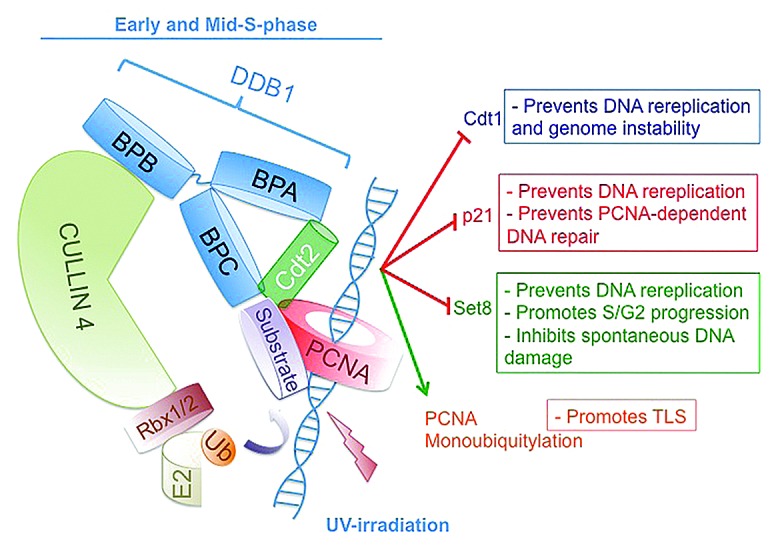
Figure 1. Schematic illustration of cullin 4 (CRL4)-based E3 ubiquitin ligase with the substrate receptor Cdt2 (CRL4Cdt2) and its various substrates and physiological functions. The scaffold cullin 4 (CUL4A or CUL4B) proteins (light green) in complex with the small RING finger protein Rbx1/2 form the catalytic core of CRL4. Rbx1/2 proteins recruit the ubiquitin-conjugating enzyme E2 to mediate the covalent attachment of ubiquitin (orange) to the substrate (light purple). DDB1 (damage-specific DNA binding protein 1) (light blue) is an adaptor protein that functions to bridge the cullin proteins with the substrate receptor Cdt2 (green). Cdt2 recognizes its substrates only when they are bound to the trimeric PCNA ring (red) encircling DNA (blue helix). For DDB1, one β-propeller domain (BPB) interacts with CUL4, whereas the two other β-propeller domains (BPA and BPC) make contacts with Cdt2.
CRL4A and CRL1FBXO11 Promote the Polyubiquitylation and Degradation of Cdt2
An si-RNA screen for E3 ubiquitin ligases that regulate Cdt2 abundance in proliferating cells identified CUL4A and CUL1 as independent regulators of Cdt2 abundance and stability (Fig. 2).39 CUL4A, but not its paralog CUL4B, promotes the autoubiquitylation of Cdt2 both in vivo and in vitro (Fig. 2, left panel). Thus, similar to other substrate receptors of CUL441,42 or CUL1,43 Cdt2 undergoes autoubiquitylation and degradation. The autoubiquitylation of Cdt2 may recycle the CUL4A complex for its reassembly with other DCAFs or terminate the Cdt2 activity following the polyubiquitylation of its substrates, but this necessitates further investigation.
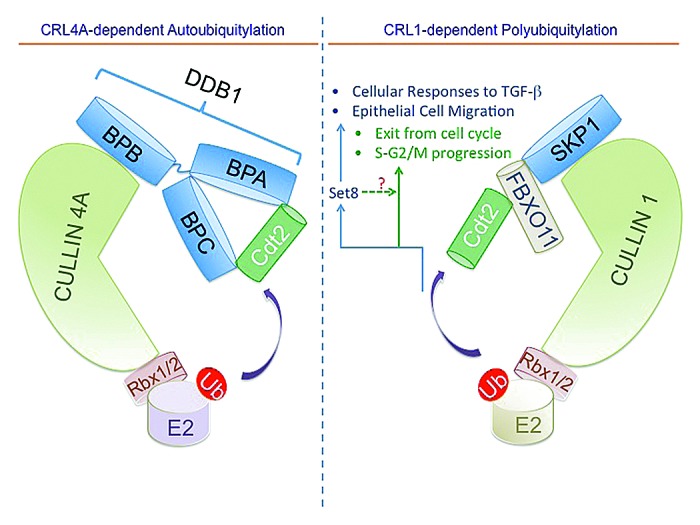
Figure 2. Schematic illustration of CRL4A and CRL1FBXO11-dependent ubiquitylation of Cdt2 and the regulation of various cellular activities. The scaffold CUL4A and CUL1 proteins (light green) in complex with the small RING finger protein (Rbx1/2) form the catalytic core of CRL4 and CRL1 (also known as SCF), respectively. CRL4A promotes the autoubiquitylation and degradation of the CUL4 substrate receptor Cdt2 (green).39 The adaptor protein SKP1 (light blue) bridges CUL1 with the substrate receptor FBXO11 (beige), recognizing Cdt2 and promoting its ubiquitylation.39,40 FBXO11-mediated ubiquitylation and degradation of Cdt2 is important for regulating cellular responses to TGF-β and cellular migration, primarily through its ability to upregulate Set8. It additionally regulates cell cycle progression (S and G2) and promote exit from the cell cycle.
The regulation of Cdt2 abundance and stability by CUL1 was more surprising, and suggested cross-talk between CRL1 and CRL4 ligases. An si-RNA screen of F-box proteins identified FBXO11, a tumor suppressor protein frequently mutated or deleted in a subset of diffuse large B cell lymphoma (DLBCL),12 as a major regulator of Cdt2 stability (Fig. 2, right panel).39 A similar conclusion was reached independently by the Pagano group while searching for potential substrates of FBXO11 by affinity purification and mass spectrometry of FBXO11-interacting proteins.40
How Does FBXO11 Recognize Cdt2?
Overexpression of FBXO11 decreased Cdt2. Deletion mutagenesis of Cdt2 in this assay identified a small peptide (aa 456–464 in human Cdt2) necessary for FBXO11-mediated degradation.39 The same region was identified by the other study, based on coimmunoprecipitation of Cdt2 mutants with FBXO11,40 suggesting that this peptide is an FBXO11-specific degron, though this has not been tested formally by adding the degron to a heterologous substrate. Two highly conserved residues within this peptide, Ser-462 and Thr-464, were essential for interaction with FBXO11 and for FBXO11-mediated degradation of Cdt2 in vivo. Our study additionally identified Asp-457 as an essential residue for FBXO11-mediated degradation of Cdt2 in vivo, although its substitution to alanine did not impact binding to or polyubiquitylation by FBXO11 in vitro. The Cdt2-specific putative degron is conserved from worm to man, suggesting that FBXO11-mediated Cdt2 degradation may also be conserved.
Because most F-box proteins recognize their substrates through binding to phosphodegrons,1 we hypothesized that Cdt2 may be similarly phosphorylated before recognition by FBXO11. However, our mass spectrometry of immune-purified Cdt2 form proliferating 293T cells did not detect phosphorylation of Cdt2 within this peptide, although the phosphorylation of several other residues was readily detectable. Furthermore, pharmacologic inhibitors of various kinases that may phosphorylate Cdt2 on several of the identified sites did not inhibit FBXO11-mediated destabilization of Cdt2 (data not shown). The Pagano group found that immobilized and unphosphorylated synthetic peptides containing the FBXO11 binding sequence (aa 457–470 in human Cdt2) efficiently bound FBXO11. Interestingly, phosphorylation of Thr-464 on Cdt2 inhibited FBXO11 binding in vitro and in vivo. CDK activity was required for Thr-464 phosphorylation in vivo, and both cyclin A-Cdk2 and cyclin B-Cdk1 kinases, but not cyclin E-Cdk2, phosphorylated this residue in vitro. Thus, unlike most degrons recognized by CRL1, the Cdt2-degron may not require prior phosphorylation, although this cannot be formally ruled out. In addition, phosphorylation of Cdt2 within its degron inhibits, rather than stimulates, FBXO11 binding. It is currently unknown whether the phosphorylation of other substrates of FBXO11 also inhibits binding/recognition by this E3 ligase.
FBXO11 Promotes the Degradation of Cdt2 during S and G2/M Phases of the Cell Cycle and Limits the Degradation of the Cdt2-Dependent Substrates p21 and Set8 to Promote Cell Cycle Progression
CRL4Cdt2 regulates the steady-state levels of various cell cycle-regulated proteins, such as Cdt1, p21, and Set8.25 Because cyclin A and cyclin B-dependent kinase activity is high in S and G2 phases of the cell cycle, respectively, their ability to phosphorylate Cdt2 and inhibit its degradation via CRL1FBXO11 may ensure high levels of Cdt2 during S and G2. We find, however, that the total Cdt2 protein decreases as cells progress through S-phase to reach 50% of the starting levels by 10 h post-release from G1/S block, as cells approach G2/M (Fig. 3A–E). FBXO11, on the other hand, increased to about 4-fold at 4 h after release from G1/S, but subsequently declined to reach basal levels as cells progress through G2 to enter G1 of the next cell cycle (Fig. 3A, B, and E). In cells depleted of FBXO11, Cdt2 level was about 3-fold higher at G1/S and progressively increased thereafter (Fig. 3C and D). Thus, contrary to our expectation, FBXO11 seems to promote the degradation of Cdt2 in S and G2, when Cdt2 is most active, most likely due to an increase in FBXO11.

Figure 3. FBXO11 promotes the degradation of Cdt2 during mid/late S and G2 phases of the cell cycle and is required for efficient progression through S/G2. (A and B) Expression of Cdt2 and FBXO11 proteins during S/G2 phases of the cell cycle. (A) Western blot of endogenous Cdt2 and FBXO11 in cell lysates from U2OS cells synchronized by double thymidine block (DTB) as described in the “Experimental Procedures” and released for the indicated hour. Asterisk indicates a cross-reactive band in the FBXO11 immunoblot. β-actin is shown for loading control. (B) Quantitation of Cdt2 (normalized to β-actin) and FBXO11 (isoforms 4, 1) proteins (normalized to the cross-reactive band from the anti-FBXO11 blot (marked by asterisk)) from (A), and expressed relative to the 0 h time point (G1/S). (C and D) Similar to (A and B), except cells were either mock depleted (si-GL2), or depleted of FBXO11 by si-RNA (si-FBXO11). (E) Inactivation of FBXO11 delays progression of cells through S/G2 phases of the cell cycle. Propidium iodide staining and FACS analysis of control U2OS cell or U2OS cells depleted of FBXO11 (si-FBXO11) and synchronized at the G1/S transition by double thymidine block (DTB) (0 h) and released from the block for the indicated time points. The results demonstrate that FBXO11 is required for efficient progression through the S and G2 phases of the cell, although its depletion does not affect the cell cycle distribution in asynchronous culture or cell cycle proliferation.39 (F) The stable overexpression of wt-Cdt2, Cdt2D457A, or Cdt2S462A in U2OS cells synchronized at the G1/S transition by double thymidine (0 h) and released from the block for 10 h. The histogram shows the percentage of cells in various phases of the cell cycle (G1, S, and G2/M) as determined by propidium iodide and FACS analysis. (G) Depletion of FBXO11 or CUL4A results in increased Cdt2 mostly in the soluble fraction. Western blot of Cdt2 in whole cell extracts or in cytoplasmic (S100), soluble nuclear (Nuclear), or chromatin fractions (Chromatin) of U2OS cells. Asterisk indicates a cross-reactive band in the FBXO11 immunoblot. Western blot with anti-Orc2 or p42/p44 antibody ensures proper fractionation. Low and high exposures of the anti-Cdt2 blot are shown for clarity.
FBXO11 depletion delayed the progression of cells through S and G2 phases of the cell cycle in cells released from G1/S (Fig. 3E), but this delay did not impact cell proliferation, probably due to a compensatory shortening of some other phase of the cell cycle. We tested whether the delayed progression through S phase could be attributed to stabilization of Cdt2. Either wt-Cdt2 or Cdt2 mutant proteins insensitive to FBXO11, namely Cdt2D457A and Cdt2S462A, were overexpressed in U2OS cells. Ten hours after release from a G1/S block, 30% of control cells were still in S-phase, 20% in G2 and 40% in the G1 phase of the next cell cycle (Fig. 3F). Interestingly, we found that more than 40% of cells overexpressing wt-Cdt2, Cdt2D457A, or Cdt2S462A remained in S-phase, with fewer cells in G2 or in G1 of the next cell cycle (Fig. 3F). Thus the delay in progression through S and G2 induced by depletion of FBXO11 is at least partly dependent on increase of Cdt2.
Importance of FBXO11-Mediated Restraint of CRL4Cdt2 Activity in Proliferating Cells
If FBXO11 downregulates Cdt2 when Cdt2 is most active, it appears that an important function of FBXO11 is to restrain the activity of Cdt2. Indeed, there was significant shortening of the half-lives of p21 and Cdt1 in cells with inactivated FBXO11 following UV irradiation.39 Set8 half-life following UV irradiation was already too short (<10 min), and inactivation of FBXO11 did not reduce this further. In actively proliferating un-irradiated cells, knockdown of FBXO11 destabilized p21 and Set8, but not Cdt1, presumably because Cdt1 degradation during a normal S-phase is predominantly executed by CRL1Skp2.44 Although Set8 half-life was altered, there was not a significant decrease in the level of Set8 in these cells.
Because overexpression of wt-Cdt2 or of FBXO11-resistant Cdt2 delayed progression through S phase (Fig. 3E), the question arises as to what is the substrate of Cdt2 whose degradation leads to this delay. p21 is a cell cycle inhibitor, and so its degradation is not expected to delay cell cycle progression. We do not know all substrates of Cdt2 yet, but although we do not see a reduction of total Set8 in cells depleted of FBXO11 under normal growth condition in culture, we speculate that loss of a small pool of Set8 at a critical site in the cell may be responsible for the delay in S phase progression. Previous studies demonstrated that the depletion of Set8 by si-RNA in mammalian cells, or deletion of the Set8 gene, resulted in spontaneous DNA damage and induced an S/G2 cell cycle arrest.45-47 We too observed spontaneous DNA damage in U2OS and A549 cells depleted of Set8 by si-RNA (data not shown). Therefore the delayed progression through S and G2/M in FBXO11-depleted cells may be due to DNA damage from a reduction of a critical pool of Set8 due to the stabilized Cdt2.
FBXO11 is a Chromatin-Bound Protein, and its Inactivation Increases Both Chromatin and Soluble Cdt2 Protein
Cdt2 promotes the degradation of its substrates when these substrates are bound to PCNA on chromatin.25 Cdt2 is localized primarily in the chromatin fraction, although detectable levels also exist in the soluble (cytoplasmic [S100] and soluble nuclear) fraction (Fig. 3G). On the other hand, FBXO11 is localized almost exclusively to chromatin. Depletion of FBXO11 slightly increased Cdt2 in the chromatin but significantly in the soluble pool (Fig. 3G). Depletion of CUL4A, had similar results, and the co-depletion of CUL4A and FBXO11 together substantially increased the soluble and chromatin-bound Cdt2. Because FBXO11 is exclusively chromatin-associated (Fig. 3G), and because inactivation of FBXO11 increases functionally active Cdt239 (which is believed to act on its substrates on chromatin), we conclude that FBXO11 degrades chromatin-bound Cdt2, but when FBXO11 is inactivated, the excess Cdt2 rapidly re-localizes to the soluble fraction. Alternatively, because CRL4Cdt2 binds to chromatin through PCNA that is loaded on DNA, the stabilized Cdt2 saturates the DNA-associated PCNA, leaving the newly synthesized Cdt2 to accumulate in the soluble pool.
FBXO11-Mediated Degradation of Cdt2, Allows the Accumulation of Set8 in TGFβ-Treated Cells and Limits the Phospho-Smad2 Signal
Consistent with a role of FBXO11 in restraining the activity of Cdt2, we found that FBXO11-mediated degradation of Cdt2 to be essential for restraining the cellular response to TGFβ.39 Depletion of FBXO11 by siRNA or the expression of FBXO11-insensitive mutants of Cdt2 resulted in sustained phospho-Smad2 signaling and increased N-cadherin expression. Normally after 10–12 h of TGFβ treatment, there is an FBXO11-dependent reduction of Cdt2 that leads to an upregulation of the Cdt2 substrates p21 and Set8 and that is followed a few hours later by a decrease in phospho-Smad2 and a cessation of N-cadherin induction.39 We speculate that the upregulation of Set8 normally seen after TGFβ treatment (and dependent of FBXO11 degrading Cdt2) is important for restraining the response of a cell to TGFβ. Although it remains unclear how Set8 promotes Smad2 de-phosphorylation, our results provide a biochemical basis of the genetic link between FBXO11 and Smad2 signaling that may underlie the developmental defects observed in mice with FBXO11 deletion.48
The Set8 protein has also been implicated in regulating the invasion of breast cancer cells in vivo and in vitro and has been shown to interact with Twist and to promote Twist-dependent EMT through the regulation of N-cadherin and E-cadherin promoters.49 Although inactivation of FBXO11 increased Cdt2 and decreased Set8, we did not notice a delay in EMT induced by TGFβ. This may be because our studies were in A549 lung cancer cells and not in breast cancer cells, or because we did not titrate down the TGFβ levels to ascertain whether a delay in EMT becomes apparent at low levels of the cytokine.
FBXO11-Mediated Degradation of Cdt2 Promotes Exit from the Cell Cycle
TGF-β treatment or serum starvation also results in FBXO11-mediated reduction of Cdt2 to promote exit from the cell cycle. Depletion of FBXO11 increased Cdt2, decreased Set8, and delayed cell cycle exit.40 Deletion of FBXO11 in C. elegans (DRE-1) or in mice results in lethality.50,51 During development, epidermal stem cells (seam cels) in the worm undergo multiple rounds of asymmetric cell division before they exit the cell cycle early in L4 and fuse into long syncytium, forming a continuous ridged cuticular structure known as adult alae. Partial loss of function mutations in dre-1 cause precocious fusion of seam cells, altered temporal patterning of gonadal outgrowths, and gaps in the adult alae.50 The latter phenotype results from failure to exit the cell cycle and differentiate properly.40,52 Importantly, the knockdown of cdt-2 (the worm ortholog of Cdt2) by si-RNA reduced the number of dre-1 (dh99) mutant animals with alae gaps from 56–13%, demonstrating that the alae gaps were due to the accumulation of Cdt2 following the genetic inactivation of DRE-1. These results confirm that FBXO11-mediated degradation of Cdt2 pathway is conserved from worm to man, and that in worms, too, this pathway is important for exit from the cell cycle.40
Cells exiting the cell cycle, either in response to TGF-β treatment, serum withdrawal or differentiation signals appear to require FBXO11-mediated degradation of Cdt2. Again, we do not know which substrate of Cdt2 needs to be stabilized during cell cycle exit, but Set8 is a plausible candidate. This can be formally proven if ectopic expression of Set8 prevents the delay of exit from the cell cycle observed in cells with inactivated FBXO11-Cdt2 pathway or in dre-1 mutant fly. An alternative, but not mutually exclusive possibility, is that the decrease in Cdt2 increases p21, leading to cell cycle exit. Again, a role of p21 can be tested experimentally as described above for Set8. Finally, it remains possible that other unknown substrates of Cdt2 need to be stabilized before cells can exit from the cell cycle.
FBXO11 Promotes Set8-Dependent Cellular Migration
FBXO11-mediated degradation of Cdt2 is critical for the migration of epithelial cells.39 Epithelial cells depleted of FBXO11 or expressing FBXO11-insensitive mutants of Cdt2 fail to migrate efficiently, and this defect can be rescued by overexpressing catalytically active, but not catalytically inactive, Set8 protein.39 We speculate that low levels of Set8 lead to migration defects that cause the developmental defects seen in mice with homozygous mutations of the FBXO11 gene, namely cleft palate and eyes open at birth.51 Furthermore, because Set8 promotes the invasion of breast cancer cells,49 it will be interesting to test whether FBXO11 may have similar pro-invasive phenotype in breast cancer through downregulating Cdt2 and upregulating Set8.
Does Inhibition of Cdt2 Contribute to the Tumor Suppressor Functions of FBXO11?
Recently, several loss-of-function mutations and deletions were found in the FBXO11 gene in a large subset of diffuse large B-cell lymphomas (DLBCL).12 The loss of FBXO11 led to the disease due to failure to polyubiquitylate and degrade the B cell-specific oncoprotein and transcriptional regulator BCL6, commonly overexpressed in DLBCL. Because many of the mutations/deletions in FBXO11 found in this disease are monoallelic, FBXO11 may function as a haplo-insufficient tumor suppressor protein.12 Other loss-of-function mutations in FBXO11 are also seen in head and neck, ovarian, colon, and lung cancers,53-57 but the mechanism by which FBXO11 contributes to the development of these cancers remains to be identified. On the other hand, Cdt2 may function as a tumor oncogene, because it exhibits high expression in multiple human cancers, including liver, breast, gastric, and colon cancers, and its elevated expression correlates positively with advanced disease state, metastasis, and poor patient survival.58-61 In addition, the Cdt2 gene was amplified in a subset of Ewing sarcomas.62 However, it is unclear how Cdt2 may exhibit its oncogenic activity, although its ability to assemble CRL4Cdt2 E3 ubiquitin ligase and promote the polyubiquitylation and degradation of p21 as well as its ability to monoubiquitylate PCNA and regulate TLS in proliferating cells may play an important role.25,37 The identification of the FBXO11-Cdt2 degradation axis raises an interesting hypothesis that FBXO11 may suppress tumor formation through the destabilization of Cdt2 (and concurrent stabilization of some Cdt2 substrates, such as p21). Investigating the level and activity of CRL4Cdt2 and its downstream targets in tumors with deletion/mutation in the FBXO11 will shed more light on this possibility.
Experimental Procedures
Cell culture, antibodies, and reagents
U2OS cells were obtained from ATCC and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics. Antibodies against ORC2 and actin were purchased from Santa Cruz Biotechnologies. Anti-FBXO11 (Bethyl Laboratories) and anti-p42/44 (Cell Signaling). Other reagents and methods were previously described.39
Chromatin Fractionation and protein-protein interactions
The cell fractionation method was adapted from ref. 63. 3.0 × 106 cells were resuspended in 80 ul buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 0.1% Triton X-100, 1 mM DTT, 1X protease inhibitor cocktail, 1 mM PMSF, 10 mM NaF, and 1mM Na3VO4) and incubated on ice for 5 min and centrifuged at 1300 × g for 5 min to yield S1 and P1. S1 fractions were centrifuged at 13000 rpm for 15 min to yield soluble fraction (cytoplasmic, S2). The pellet was washed with 500 μl of buffer A and resuspended in 80 μl buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, 1× protease inhibitor cocktail, 1 mM PMSF, 10 mM NaF, and 1 mM Na3VO4). Samples were incubated on ice 30 min. and centrifuged at 1700 × g for 5 min to yield supernatant (soluble nuclear, S3) and chromatin fraction (P3). Chromatin fraction was suspended in 1× sample buffer and sonicated for 10 sec at 10%.
Cell cycle synchronization and FACS analysis
U2OS cells were synchronized by double thymidine block (DTB) and released from the block as described previously.30 For flow cytometry analysis (FACS), cells were washed with phosphate buffer saline (PBS) and fixed in 70% ethanol. Fixed cells were resuspended in 1 ml of PBS with 60 μg/ml propidium iodide and 50 μg/ml RNase A, rotated at 4°C for 30 min and analyzed by FACScalibur flow cytometer (Becton Dickinson).
Acknowledgment
This work was supported by NIH grants, R00CA140774 to TA and R01CA60499 to AD.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25314
References
- 1.Lipkowitz S, Weissman AM. RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat Rev Cancer. 2011;11:629–43. doi: 10.1038/nrc3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 3.Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8:438–49. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skaar JR, D’Angiolella V, Pagan JK, Pagano M. SnapShot: F Box Proteins II. Cell. 2009;137:1358–, e1. doi: 10.1016/j.cell.2009.05.040. [DOI] [PubMed] [Google Scholar]
- 5.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 6.D’Angiolella V, Donato V, Vijayakumar S, Saraf A, Florens L, Washburn MP, et al. SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature. 2010;466:138–42. doi: 10.1038/nature09140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D’Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y, et al. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell. 2012;149:1023–34. doi: 10.1016/j.cell.2012.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin DI, Barbash O, Kumar KG, Weber JD, Harper JW, Klein-Szanto AJ, et al. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Mol Cell. 2006;24:355–66. doi: 10.1016/j.molcel.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen BB, Glasser JR, Coon TA, Zou C, Miller HL, Fenton M, et al. F-box protein FBXL2 targets cyclin D2 for ubiquitination and degradation to inhibit leukemic cell proliferation. Blood. 2012;119:3132–41. doi: 10.1182/blood-2011-06-358911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen BB, Glasser JR, Coon TA, Mallampalli RK. F-box protein FBXL2 exerts human lung tumor suppressor-like activity by ubiquitin-mediated degradation of cyclin D3 resulting in cell cycle arrest. Oncogene. 2012;31:2566–79. doi: 10.1038/onc.2011.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watanabe T, von der Kammer H, Wang X, Shintani Y, Horiguchi T. Neuronal expression of F-box and leucine-rich-repeat protein 2 decreases over Braak stages in the brains of Alzheimer’s disease patients. Neurodegener Dis. 2013;11:1–12. doi: 10.1159/000336016. [DOI] [PubMed] [Google Scholar]
- 12.Duan S, Cermak L, Pagan JK, Rossi M, Martinengo C, di Celle PF, et al. FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature. 2012;481:90–3. doi: 10.1038/nature10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson S, Xiong Y. CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem Sci. 2009;34:562–70. doi: 10.1016/j.tibs.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell. 2006;23:709–21. doi: 10.1016/j.molcel.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 15.Angers S, Thorpe CJ, Biechele TL, Goldenberg SJ, Zheng N, MacCoss MJ, et al. The KLHL12-Cullin-3 ubiquitin ligase negatively regulates the Wnt-beta-catenin pathway by targeting Dishevelled for degradation. Nat Cell Biol. 2006;8:348–57. doi: 10.1038/ncb1381. [DOI] [PubMed] [Google Scholar]
- 16.He YJ, McCall CM, Hu J, Zeng Y, Xiong Y. DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev. 2006;20:2949–54. doi: 10.1101/gad.1483206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Higa LA, Wu M, Ye T, Kobayashi R, Sun H, Zhang H. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat Cell Biol. 2006;8:1277–83. doi: 10.1038/ncb1490. [DOI] [PubMed] [Google Scholar]
- 18.Lee J, Zhou P. Pathogenic Role of the CRL4 Ubiquitin Ligase in Human Disease. Front Oncol. 2012;2:21. doi: 10.3389/fonc.2012.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang J, Chu G. Xeroderma pigmentosum complementation group E and UV-damaged DNA-binding protein. DNA Repair (Amst) 2002;1:601–16. doi: 10.1016/S1568-7864(02)00052-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Wang QE, Zhu Q, El-Mahdy MA, Wani G, Praetorius-Ibba M, et al. DNA damage binding protein component DDB1 participates in nucleotide excision repair through DDB2 DNA-binding and cullin 4A ubiquitin ligase activity. Cancer Res. 2006;66:8590–7. doi: 10.1158/0008-5472.CAN-06-1115. [DOI] [PubMed] [Google Scholar]
- 21.Shiyanov P, Nag A, Raychaudhuri P. Cullin 4A associates with the UV-damaged DNA-binding protein DDB. J Biol Chem. 1999;274:35309–12. doi: 10.1074/jbc.274.50.35309. [DOI] [PubMed] [Google Scholar]
- 22.Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, et al. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113:357–67. doi: 10.1016/S0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- 23.Kapetanaki MG, Guerrero-Santoro J, Bisi DC, Hsieh CL, Rapić-Otrin V, Levine AS. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc Natl Acad Sci USA. 2006;103:2588–93. doi: 10.1073/pnas.0511160103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, et al. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell. 2005;121:387–400. doi: 10.1016/j.cell.2005.02.035. [DOI] [PubMed] [Google Scholar]
- 25.Abbas T, Dutta A. CRL4Cdt2: master coordinator of cell cycle progression and genome stability. Cell Cycle. 2011;10:241–9. doi: 10.4161/cc.10.2.14530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Higa LA, Banks D, Wu M, Kobayashi R, Sun H, Zhang H. L2DTL/CDT2 interacts with the CUL4/DDB1 complex and PCNA and regulates CDT1 proteolysis in response to DNA damage. Cell Cycle. 2006;5:1675–80. doi: 10.4161/cc.5.15.3149. [DOI] [PubMed] [Google Scholar]
- 27.Senga T, Sivaprasad U, Zhu W, Park JH, Arias EE, Walter JC, et al. PCNA is a cofactor for Cdt1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. J Biol Chem. 2006;281:6246–52. doi: 10.1074/jbc.M512705200. [DOI] [PubMed] [Google Scholar]
- 28.Sansam CL, Shepard JL, Lai K, Ianari A, Danielian PS, Amsterdam A, et al. DTL/CDT2 is essential for both CDT1 regulation and the early G2/M checkpoint. Genes Dev. 2006;20:3117–29. doi: 10.1101/gad.1482106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, Dutta A. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 2008;22:2496–506. doi: 10.1101/gad.1676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol Cell. 2010;40:9–21. doi: 10.1016/j.molcel.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim Y, Starostina NG, Kipreos ET. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 2008;22:2507–19. doi: 10.1101/gad.1703708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishitani H, Shiomi Y, Iida H, Michishita M, Takami T, Tsurimoto T. CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J Biol Chem. 2008;283:29045–52. doi: 10.1074/jbc.M806045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Centore RC, Havens CG, Manning AL, Li JM, Flynn RL, Tse A, et al. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol Cell. 2010;40:22–33. doi: 10.1016/j.molcel.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oda H, Hübner MR, Beck DB, Vermeulen M, Hurwitz J, Spector DL, et al. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol Cell. 2010;40:364–76. doi: 10.1016/j.molcel.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jørgensen S, Eskildsen M, Fugger K, Hansen L, Larsen MS, Kousholt AN, et al. SET8 is degraded via PCNA-coupled CRL4(CDT2) ubiquitylation in S phase and after UV irradiation. J Cell Biol. 2011;192:43–54. doi: 10.1083/jcb.201009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, et al. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat Cell Biol. 2010;12:1086–93. doi: 10.1038/ncb2113. [DOI] [PubMed] [Google Scholar]
- 37.Terai K, Abbas T, Jazaeri AA, Dutta A. CRL4(Cdt2) E3 ubiquitin ligase monoubiquitinates PCNA to promote translesion DNA synthesis. Mol Cell. 2010;37:143–9. doi: 10.1016/j.molcel.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Havens CG, Walter JC. Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 2011;25:1568–82. doi: 10.1101/gad.2068611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abbas T, Mueller AC, Shibata E, Keaton M, Rossi M, Dutta A. CRL1-FBXO11 promotes Cdt2 ubiquitylation and degradation and regulates Pr-Set7/Set8-mediated cellular migration. Mol Cell. 2013;49:1147–58. doi: 10.1016/j.molcel.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rossi M, Duan S, Jeong YT, Horn M, Saraf A, Florens L, et al. Regulation of the CRL4(Cdt2) ubiquitin ligase and cell cycle exit by the SCF(Fbxo11) ubiquitin ligase. Mol Cell. 2013;49:1159–66. doi: 10.1016/j.molcel.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen X, Zhang Y, Douglas L, Zhou P. UV-damaged DNA-binding proteins are targets of CUL-4A-mediated ubiquitination and degradation. J Biol Chem. 2001;276:48175–82. doi: 10.1074/jbc.M106808200. [DOI] [PubMed] [Google Scholar]
- 42.Nag A, Bondar T, Shiv S, Raychaudhuri P. The xeroderma pigmentosum group E gene product DDB2 is a specific target of cullin 4A in mammalian cells. Mol Cell Biol. 2001;21:6738–47. doi: 10.1128/MCB.21.20.6738-6747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deshaies RJ. SCF and Cullin/Ring H2-based ubiquitin ligases. Annu Rev Cell Dev Biol. 1999;15:435–67. doi: 10.1146/annurev.cellbio.15.1.435. [DOI] [PubMed] [Google Scholar]
- 44.Nishitani H, Sugimoto N, Roukos V, Nakanishi Y, Saijo M, Obuse C, et al. Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J. 2006;25:1126–36. doi: 10.1038/sj.emboj.7601002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jørgensen S, Elvers I, Trelle MB, Menzel T, Eskildsen M, Jensen ON, et al. The histone methyltransferase SET8 is required for S-phase progression. J Cell Biol. 2007;179:1337–45. doi: 10.1083/jcb.200706150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oda H, Okamoto I, Murphy N, Chu J, Price SM, Shen MM, et al. Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Mol Cell Biol. 2009;29:2278–95. doi: 10.1128/MCB.01768-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Houston SI, McManus KJ, Adams MM, Sims JK, Carpenter PB, Hendzel MJ, et al. Catalytic function of the PR-Set7 histone H4 lysine 20 monomethyltransferase is essential for mitotic entry and genomic stability. J Biol Chem. 2008;283:19478–88. doi: 10.1074/jbc.M710579200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tateossian H, Hardisty-Hughes RE, Morse S, Romero MR, Hilton H, Dean C, et al. Regulation of TGF-beta signalling by Fbxo11, the gene mutated in the Jeff otitis media mouse mutant. Pathogenetics. 2009;2:5. doi: 10.1186/1755-8417-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang F, Sun L, Li Q, Han X, Lei L, Zhang H, et al. SET8 promotes epithelial-mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J. 2012;31:110–23. doi: 10.1038/emboj.2011.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fielenbach N, Guardavaccaro D, Neubert K, Chan T, Li D, Feng Q, et al. DRE-1: an evolutionarily conserved F box protein that regulates C. elegans developmental age. Dev Cell. 2007;12:443–55. doi: 10.1016/j.devcel.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 51.Hardisty-Hughes RE, Tateossian H, Morse SA, Romero MR, Middleton A, Tymowska-Lalanne Z, et al. A mutation in the F-box gene, Fbxo11, causes otitis media in the Jeff mouse. Hum Mol Genet. 2006;15:3273–9. doi: 10.1093/hmg/ddl403. [DOI] [PubMed] [Google Scholar]
- 52.Slack FJ, Basson M, Liu Z, Ambros V, Horvitz HR, Ruvkun G. The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol Cell. 2000;5:659–69. doi: 10.1016/S1097-2765(00)80245-2. [DOI] [PubMed] [Google Scholar]
- 53.Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–73. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 54.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–9. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 57.Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci USA. 2012;109:3879–84. doi: 10.1073/pnas.1121343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pan HW, Chou HY, Liu SH, Peng SY, Liu CL, Hsu HC. Role of L2DTL, cell cycle-regulated nuclear and centrosome protein, in aggressive hepatocellular carcinoma. Cell Cycle. 2006;5:2676–87. doi: 10.4161/cc.5.22.3500. [DOI] [PubMed] [Google Scholar]
- 59.Ueki T, Nishidate T, Park JH, Lin ML, Shimo A, Hirata K, et al. Involvement of elevated expression of multiple cell cycle regulator, DTL/RAMP (denticleless/RA-regulated nuclear matrix associated protein), in the growth of breast cancer cells. Oncogene. 2008;27:5672–83. doi: 10.1038/onc.2008.186. [DOI] [PubMed] [Google Scholar]
- 60.Li J, Ng EK, Ng YP, Wong CY, Yu J, Jin H, et al. Identification of retinoic acid-regulated nuclear matrix-associated protein as a novel regulator of gastric cancer. Br J Cancer. 2009;101:691–8. doi: 10.1038/sj.bjc.6605202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baraniskin A, Birkenkamp-Demtroder K, Maghnouj A, Zöllner H, Munding J, Klein-Scory S, et al. MiR-30a-5p suppresses tumor growth in colon carcinoma by targeting DTL. Carcinogenesis. 2012;33:732–9. doi: 10.1093/carcin/bgs020. [DOI] [PubMed] [Google Scholar]
- 62.Mackintosh C, Ordóñez JL, García-Domínguez DJ, Sevillano V, Llombart-Bosch A, Szuhai K, et al. 1q gain and CDT2 overexpression underlie an aggressive and highly proliferative form of Ewing sarcoma. Oncogene. 2012;31:1287–98. doi: 10.1038/onc.2011.317. [DOI] [PubMed] [Google Scholar]
- 63.Méndez J, Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol. 2000;20:8602–12. doi: 10.1128/MCB.20.22.8602-8612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]