

Abstract
Glioblastoma is a disease with poor survival rates after diagnosis. Treatment of the disease involves debulking of the tumor, which is limited by the degree of invasiveness of the disease. Therefore, a treatment to halt the invasion of glioma is desirable for clinical implementation. There have been several candidate compounds targeting specific aspects of invasion, including cell adhesions, matrix degradation, and cytoskeletal rearrangement, but they have failed clinically for a variety of reasons. New targets against glioma invasion include upstream mediators of these classical targets in an effort to better inhibit invasion with more specificity for cancer. Included in these treatments is a new class of compounds inhibiting the generation of reactive oxygen species by targeting the NADPH oxidases. These compounds stand to inhibit multiple pathways, including nuclear factor kappa B and Akt. By conducting a screen of compounds thought to inhibit these pathways, a new compound to halt invasion was found that may have a beneficial effect against glioma, based on recent publications. Further, there are still limitations to the treatment of glioblastoma regardless of the discovery of new targets and compounds that should be addressed to better the therapies against this deadly cancer.
Keywords: glioma, drug screen, NADPH oxidase, cancer, invasion
Small Molecule Anti-Invasive Compounds for Glioblastoma Therapy
Brain tumors, in particular, glioblastoma (GBM), are a significant clinical problem due to their poor prognosis. The incidence of brain tumors is about 14 in every 100,000 people in the United States.1 However, the 5-y survival rate after brain tumor diagnosis is around 35%, as compared with an average 5-y survival rate in breast cancer of 89% and prostate of nearly 100%. Current treatment of glioblastoma (GBM) is difficult and results in high recurrence rates.2
Brain tumor invasion is an important factor in failure of therapy. The invasive nature of gliomas has long been documented. Yet only recently has a significant effort been put forth to try and observe or treat the invasive front of tumors.3 With MRI scans and advanced MRI techniques such as diffusion tensor imaging, it has been possible to determine the course of treatment based on degree of invasion.4 With more attention being paid to the morphology of the tumor pre-treatment, the development of anti-invasive treatments becomes more important in the clinic.
Current Treatment of Glioma
There have been changes in the clinical treatment of brain tumors, but there have not been any major increases in the survival rate and time of the patients.5-7 The first line of treatment includes surgical resection, followed by rounds of radiotherapy and chemotherapy with temozolomide. This treatment fails in the bulk of cases, largely due to the invasive nature of the tumor.8 Resection is limited by surgical experience and availability of advanced equipment for intraoperative monitoring of tumor removal. These limitations result in the inability to remove all tumor cells. Similarly, due to the invasion of cells, radiotherapy will miss many of the tumor cells in the brain in order to prevent damage to healthy tissue. Chemotherapy is delivered systemically and enters the tumor via the blood vessels and thus is limited by diffusion through the tumor.9,10 This means that often chemotherapeutics cannot reach invading cells and thus do not kill them. In cases of recurrence, anti-angiogenic therapeutics are applied to prolong life, though this treatment rarely offers a cure.
Induction of Invasion by Treatment
It has been suggested that each treatment approach may lead to invasion of tumor cells into healthy tissue, resulting in therapy failure and difficulty in future treatment. Though seen in animal models and humans, it is difficult to determine what is attributable to the treatment vs. progression of the tumor after longer survival in the patient. The most direct links of invasion with treatment are in radiotherapy11-13 and anti-angiogenic therapy.14 In some cases, resection has been shown to increase malignancy of tumors through a stem cell proliferation effect termed repopulation.15 Chemotherapy causes a large range of alterations in malignancy of cancer cells.6 However, the degree to which this process is accelerated as opposed to tumors that are untreated is unknown due to survival effects.
Radiation is closely linked to increased invasiveness in vitro and in vivo in brain, breast, and prostate cancer.16 This effect occurs in response to low radiation levels that activate both the tumor cells and the tumor stroma. Using human breast carcinoma cells, it was seen that irradiating the injection site before injecting tumor cells enhanced the invasion and metastasis of the cells upon implantation.17,18 Further, it has been shown that irradiation causes activation of fibroblasts in the tumor area, enhancing the invasive potential of prostate cancer cells.19 In glioma, in vitro studies have indicated increased invasion when low-dose radiation is applied in culture in both U87MG and C6 tumor cell lines.11,13,20,21 Irradiation of glioma cells leads to activation of Rho GTPases, which induce invasion of cancer cells.12 Inhibition of MMP-2 with siRNA leads to a reduction of this invasion effect, implicating multiple pathways in the radiation effect.22 Radiation therapy of glioma affects healthy tissue due to ill-defined borders. This can cause priming of the stroma and adjacent tissue, including activation of astrocytes and microglia, leading to enhancement of the invasive potential of the tumor.20
Anti-angiogenic treatment increases invasion in many tumor types, though in glioma, this effect is particularly pronounced. Used as a last line of treatment in glioblastoma, because it extends life by a few months, the invasiveness of the tumor into the brain is apparent postmortem. It is thought that the invasion is enhanced due to the removal of blood supply, yielding increased hypoxia in regions of high proliferation.23 This effect not only results in cell death and necrosis, but also causes switching on of pro-invasive factors.24 This change can also be seen after treatment with chemotherapeutics that affect blood vessels, though not as strong as with the anti-angiogenics.25,26
Chemotherapy of brain tumors leads to far-distant metastases in humans and animal models. This may be attributed to increases in invasion related to the blood supply decrease; however, it is often attributed to the clonal selection that can result from treatment.27,28 It is known that glioma stem cells, a subpopulation of proliferative and invasive tumor cells, show resistance to apoptosis induced by etoposide, camptothecin, cisplatin, temozolomide, doxorubicin, vincristine, and methotrexate at doses that are toxic to comparable non-stem cell populations.29 Further, these cells are commonly found at the invasive front of tumors.30 They also have been shown to interact with endothelial cells in the brain and non-stem glioma cells to promote an invasive phenotype via a Tie2/TEK mechanism.31 The ability of chemotherapy to actually induce invasion is thought to be largely through this selection mechanism of the most deadly of the cancer cells. It is apparent from the building clinical evidence that the invasion of glioma cells is closely connected with the success and future outcomes of treatments. Therefore, determination and development of therapeutics targeted against invasion of glioma cells has been and continues to be important for the advancement of patient outcomes.
Mechanisms of Invasion and Potential Targets
For cancer cells to invade, there are three primary mechanisms that must be manipulated: cell adhesion to the matrix and cells, matrix degradation, and cytoskeletal rearrangement. There have been many small-molecule inhibitors of these three aspects of cancer cell invasion that have shown success preclinically though fewer clinically (Fig. 1A). There still exists no clinically used anti-invasive agent for glioma invasion regardless of the potential of such an agent to make a significant impact.

Figure 1. Anti-invasive compounds. (A) More traditional routes to halt invasion of cancer cells include targeting the cytoskeleton (microtubules, actin, and associated myosin machinery), cell adhesions and their activators, and matrix degradation enzymes. (B) Newer treatment options for halting invasion include more diverse targets, including upstream inactivation routes including receptor tyrosine kinases and chemokine receptors, ion, and water channels, transcription factors such as HIF-1α, and inflammation driven progression through reactive oxygen species (ROS).
Targeting Matrix Degradation to Inhibit Invasion
In order to move through the matrix, cancer cells often need to manipulate the host environment. This can often be avoided by changing route which leads to formation of many of the typical glioma invasion structures. Regardless, cells need to degrade the extracellular matrix in the basement membrane and interstitial space in order to move, release growth factors bound to the matrix, and grow. Matrix metalloproteinases (MMPs), which are a class of zinc-dependent proteases that degrade a variety of membrane proteins, including collagen and gelatin, are the best-studied of degradation proteins in brain cancer.32 These proteins are upregulated in all cancers, and in brain tumors, MMP-2 and MMP-9, the two gelatinases, are commonly overexpressed.33-35 MMPs are regulated upstream by multiple signaling molecules including, Src kinase, PI3 kinase, protein kinase C, epidermal growth factor receptor, platelet-derived growth factor receptor, and other chemokine/growth factor receptors. Further, matrix metalloproteinases can be expressed by and stimulated by cell-cell interactions with normal glial cells such as astrocytes and microglia.36
Broad spectrum MMP inhibitors, such as marimastat,37 prinomastat,38 and MMI270B,39 have gone through extensive clinical trials in glioma and other invasive cancers. Marimastat affects all matrix metalloproteinases and, thus, in clinical trials, it showed many unwanted side effects without much efficacy, leading to termination.40 Prinomastat has more selectivity, inhibiting MMPs 2, 3, 9, 13, and 14 only, but still yields some off target effects and has not shown definitive efficacy when combined with chemotherapy.38 MMI270B is an orally available MMP inhibitor still under investigation clinically. This last compound is a hydroxyamate-based compound and has led to development of similarly structured molecules with better specificity and bioavailability including MMI-166, a gelatinase-specific inhibitor with good blood brain barrier crossing41 that has shown some promise in other cancers clinically and preclinically in glioma. Due to the failure of broad spectrum MMP inhibitors, this more specific approach has been popular; however, other matrix degradatory enzymes such as hyaluronidases (degrades hyaluronan) and cathepsin B (degrades laminin) are overexpressed by glioma42,43 to deal with the specific matrix heterogeneity of the brain.44 Further, glioma cells express and secrete a host of ECM constituents, including hyaluronan, collagen I and IV, laminin, versican, brevican, and chondroitin sulfate proteoglycans to rebuild the matrix for enhanced motility.45,46 Therefore, targeting of MMPs may not be the best approach for small molecule anti-invasive compounds in brain tumors.
Targeting Cell Adhesion to Inhibit Invasion
Glioma cells, like many cancer cells, have increased expression of cell adhesion molecules (CAMs) as compared with healthy brain tissue and this increase is correlated with malignancy of disease.47 In particular there is increased integrin αvβ3 expression and ICAM-1 expression, indicating both a need for increased cell-matrix and cell-cell interactions in glioma cells. As one of the earliest identified components of cell invasion in brain cancer and for their similar dysregulation in other cancers, small-molecule compounds targeting integrins are the best developed. The best example of these compounds is cilengitide, which inhibits both αvβ3, involved in vitronectin adhesion, and αvβ5, more associated with endothelial adhesion.48 This drug showed promise in inhibiting invasion of glioma cells and angiogenesis in vitro, though it has shown moderate effects in vivo in phase II clinical trials depending on scheduling.49
Cell adhesion molecules are regulated by, among other upstream molecules, focal adhesion kinase (FAK), which acts to promote assembly and disassembly of adhesion complexes.50,51 FAK can recruit cytoskeletal proteins and activate RhoGTPases integrating the adhesion process with cellular cytoskeleton assembly.52 It is known that FAK is overexpressed in almost every type of cancer, including brain, and the degree to which FAK is overexpressed correlates with degree of malignancy in breast, colon, and brain cancer.53 Several small-molecule inhibitors of FAK have been developed, including PF-573,228 and similar derivatives that act by blocking autophosphorylation of FAK in cancer cells54,55 and subsequent invasion. Further molecules are targeted for invasion inhibition by alteration of cellular adhesions, including CD44, the hyaluronan binding receptor,56 and junctional adhesion molecules, which both are upregulated in glioma.
Targeting Cytoskeletal Rearrangement to Inhibit Invasion
Adhesion disassembly and assembly is closely linked with the cytoskeleton. For the cell to protrude forward, there must be a large degree of actin polymerization and depolymerization, leading to movement of the cell body. Further, the use of myosin to contract the cell as it moves is vital to movement. Microtubules and intermediate filaments act to maintain the cell structure internally and hold the organelles in place.57 Formation of podosome structures and invadopodia, protrusions from the cell body that release high amounts of matrix degradation enzymes and form the leading structure for invasion, is dependent on cytoskeletal assembly and lipid bilayer rearrangement.58 Therefore, compounds targeting these pathways are desirable for their anti-invasive effect. However, specificity is an issue with cytoskeletal targeting agents as compounds that directly interact with these cell structures affect all cell types, and most specificity results from efficacy at a lower dose in cancer cells than in their healthy counterparts. Direct inhibitors of actin polymerization and depolymerization include cytochalasin, phalloidin, and jasplakinolide, and while good inhibitors of cancer cell invasion when using in vitro assays, in vivo use is limited by effects on healthy cells.59 Similarly, inhibition of microtubule assembly and disassembly, such as paclitaxel60 and vincristine61 at non-cytotoxic doses, prevents glioma invasion in vitro and tumor growth in vivo. Inhibitors of myosin II, involved in contraction of the cell for forward movement, are similarly inhibitory of invasion in the brain.62 These drugs, most notably blebbistatin, inhibit invasion of glioma cells in vitro though not a viable in vivo option for treatment.63 Due to the non-specificity of most direct inhibitors of the cytoskeleton and cell dynamics, there has been more interest in targeting upstream proteins involved in cytoskeletal signaling pathways such as the rho guanosine triphosphatases (Rho GTPases) and their regulatory elements64 as well as other signaling proteins and receptor tyrosine kinases.65 Inhibition of phosphatidylinositol-3- kinases, such as PIK3CA and PIK3R1, is known to inhibit glioblastoma proliferation and invasion in vitro.66 Often, growth factor receptors also affect all of these migratory pathways. Growth factors known to be involved in glioma invasion include many of the same pathways as in other cancer, such as epidermal growth factor (EGF/EGFR), platelet-derived growth factor (PDGF/PDGFR), Met tyrosine kinase, and transforming growth factor β.67 Inhibitors of these molecules exist and are at varying stages of development as discussed in reference 65. Further, these drugs have the added benefit of inhibiting multiple components of glioma invasion, including adhesion, cytoskeletal dynamics, and matrix degradation, which are more closely linked than alluded to here.
New Targets and Treatments That Effect Invasion of Glioma
As the knowledge of mechanisms that contribute to cancer cell invasion expand, so do the potential targets and agents that might be clinically viable. Most of these treatment strategies are in the experimental to pre-clinical (animal model) stages yet show distinct promise in contributing to efficacy of more aggressive antitumor treatments. Some of the strategies with compounds are summarized in Figure 1B.
Ion channels and water transporters
Recently, it has been shown that cancer cells upregulate both aquaporins68 and ion channels.69 Aquaporins are an active component of astrocyte function in healthy brain tissue and increase activity after injury or in presence of a tumor.70 More invasive glioma have increased expression of Aquaporin-1, -4, and -9, which correlates with grade of disease and are instrumental in formation of lamellipodia and invadopodia.71,72 Targeting aquaporin-4 with the small molecule AQN4, a prodrug, decreases glioma invasion and sensitizes the tumor to further treatments including radiotherapy and chemotherapy.73,74 Ion channels are upregulated in brain tumors as well, being involved in homeostasis and transport into the tumor cells.69 Use of chloride or potassium channel blockers such as tetraethylammonium (TEA) chloride, chlorotoxin, and tamoxifen, leads to inhibition of invasion of multiple models of glioma in vitro.75,76 There are no direct inhibitors used clinically for glioma yet, for either aquaporins or ion channels, though there are many compounds that have clinical approval for other indications, such as cerebral ischemia and seizure.
Chemotaxis and chemokine gradients
A hypothesis for the pattern of invasion of cancer cells in the brain is that they are following chemokine gradients inherent to the brain due to secretion by ependymal or endothelial cells.77 Chemokines known to trigger glioma invasion include CXCL12 (with receptor CXCR4 and CXCR7), PDGF (PDGF receptor), CXCL10 (with receptor CXCR3), and CXCL13 (with receptor CXCR5).78 The best-studied of these pathways is the CXCL12/CXCR4 axis with developed inhibitors such as AMD3100, a small-molecule antagonist of the CXCR4 that leads to decreased invasion and in vivo growth of glioma.79 This compound has been used in clinical trials for HIV but has not been tried in human glioblastoma clinically,80 though it shows promise as a novel small-molecule anti-invasive agent and adjuvant for chemotherapy.81 Of interest, many of these cytokines, are capable of inducing reactive oxygen species, which can lead to further cancer cell progression and invasion.
Hypoxia-induced invasion
Glioblastoma shows a distinct histological phenotype known as pseudopalisading necrosis, which results from areas of hypoxia in the tumor,82 leading to invasion of cancer cells away from a hypoxic center and the resulting necrosis of the central region. Hypoxia is also induced after anti-angiogenic therapy and is thought to partially lead to the invasive response.83 Cancer cells overexpress hypoxia-inducible factor-1 α (HIF-1α), which acts as a transcription factor for invasion machinery (as well as cell cycle, survival, chemoresistance, and angiogenesis genes). Blocking of HIF-1α or its downstream transcriptional activity yields inhibition of invasion of glioma cells and under hypoxic conditions.84 There are many small-molecule inhibitors of HIF-1α,85,86 though none in clinical trials yet. However, many clinically approved small molecules show hypoxia sensitivity enhancement in cancer cells, including cisplatin, doxorubicin,87 flavonoids such as green tea extract,88 and geldanamycin.89 Hypoxia obviously causes many other tumoral changes besides invasion; however, small molecules that specifically inhibit the invasion associated with hypoxia are strong candidates for adjuvant therapy. HIF-2α is also associated with poor prognosis, and may be the major mediator of chronic hypoxia, as opposed to HIF-1α, which may be a response to acute hypoxia.
Inflammation and reactive oxygen species (ROS)-driven invasion
Looking to nature is a commonality among anticancer research as noted above with many of the compounds already identified for glioma. Honokiol is a compound derived from the magnolia tree and is an ancient herbal remedy. It has been shown to inhibit growth and progression of multiple types of cancer including melanoma, prostate, and colon cancers.90-92 Through these studies it was also determined that honokiol works via modulation of NFkB activity though the direct target is still unknown. NFkB is constitutively activated in 75% of cancers.93,94 This is particularly dangerous, because NFkB directs both invasive and antiapoptotic activities of tumor cells, including metastasis of cancer cells including actin regulatory elements, cell adhesion proteins such as integrins and focal adhesion kinases, and secretion of matrix metalloproteinases.95 The upregulation of these factors contributes to the migration of cancer cells. In particular, these attributes are necessary for the invasion of glioblastoma into the surrounding brain. Hence, the known inhibitory effects of honokiol provided a rationale for testing honokiol derivatives for their potential anti-invasive activity.
Triphenylmethanes (TPM) are a class of compounds modeled after diphenyleneiodonium (DPI) and have NADPH oxidase (Nox) antagonistic activity.96 We had hypothesized that the ability of DPI to inhibit Nox activity was linked to its structure of a positive charge that could be delocalized among aromatic rings. TPM differ from DPI in that the central atom is a carbon instead of an iodine, as in DPI. They were explored in the early 1990s as chemopreventive agents though not pursued extensively.97 The Nox family of proteins is generally active in cancers, leading to enhanced presence of reactive oxygen species in the tumor cells and stroma. This leads to activation of a host of intracellular pathways, including Src kinase, p38/MAPK, and NFkB, which can lead to downstream cytoskeletal rearrangement as well as directly activating matrix metalloproteinases (MMPs).95,98,99 Compounds such as DPI have previously been seen to inhibit invadopodia formation, inflammatory response, and cancer cell migration in vitro.100 Therefore, this class of compounds was of interest for development of novel therapeutics against glioma invasion.
A screen of novel compounds from the triphenylmethane and honokiol classes of drugs (Fig. 2) resulted in identification of multiple anti-invasive agents against glioma. These are all novel compounds except honokiol, and all show antitumor activity to some extent. The compounds were synthesized through simple methods to yield derivatives of the original two compounds and thus are divided into Triphenylmethanes (TPMs) and honokiol derivatives (HDs). In this study we had two primary criteria for the selection of the lead compound to be used in further testing: (1) anti-invasive at non-cytotoxic doses in vitro as determined through Boyden chamber matrigel-based invasion assays; (2) no cytotoxicity toward primary rat astrocytes at doses ten times that used in invasion assays. In this way, we could select a compound that was both selective for glioma cells and had definite anti-invasive effects. Compounds were first screened at three different doses (0.1 µM, 1 µM, and 10 µM) to determine the cytotoxic dose over 72 h in RT2 glioma cells. The invasion screen was then run with highest dose shown to be non-cytotoxic to prevent confounding effects of cell death on invasion quantification (Fig. 3A). In parallel, the lowest cytotoxic dose was used to determine astrocyte cytotoxicity over 96 h (Fig. 3B). Though several compounds showed anti-invasive and antitumor effects with limited cytotoxicity toward healthy astrocytes, it was determined through this screen that the best anti-invasive candidate at non-cytotoxic doses to astrocytes or RT2 is imipramine blue, a novel triphenylmethane compound. This agent has recently demonstrated efficacy in inhibiting invasion of multiple primary and glioma cell lines in vitro as well as complete survival in rats bearing aggressively invasive RT2 glioma when combined with the potent chemotherapeutic doxorubicin.101 In this study, then, we outlined the move from in vitro compound, development of a drug delivery system, testing in vivo, and combination with a powerful antitumor treatment. Further, the mechanism of action was elucidated to show the involvement of both Src kinase and NADPH oxidases.

Figure 2. Compounds based on honokiol and triphenyliodide screened for anti-invasive activity. Included compounds in the screen for a novel anti-invasive compound were based on honokiol and diphenyliodide.

Figure 3. Results of compound screen. (A) Invasion assay conducted with compounds at concentrations listed through matrigel in tissue culture inserts, RT2 glioma. *P < 0.05 compared with control after 24 h (B) Viability of astrocytes as compared with initial measurements based on metabolic activity at 5 µM concentration after 96 h in culture.
These compounds work through inhibition of reactive oxygen species (ROS)-mediated activation of NFkB and associated pathways. ROS are capable of inducing NFkB activation by oxidatively inactivating IkB. In addition, ROS can activate Akt by oxidizing PTEN. Finally, ROS can inactivate wild-type p53. Thus, inhibition of NADPH oxidases results in decreased NFkB, decreased Akt, and activation of wild-type p53 in a single intervention (Fig. 4). Mutant p53 is associated with induction of pro-invasive phenotypes of cancer cells through use of a p63 molecular chaperone, while wild-type p53 can prevent this activity.102 These molecules have also been implicated in ICAM-1 expression, actin reassembly, and MMP production, thus inhibition of ROS-mediated pathways is beneficial to tumor invasion inhibition.
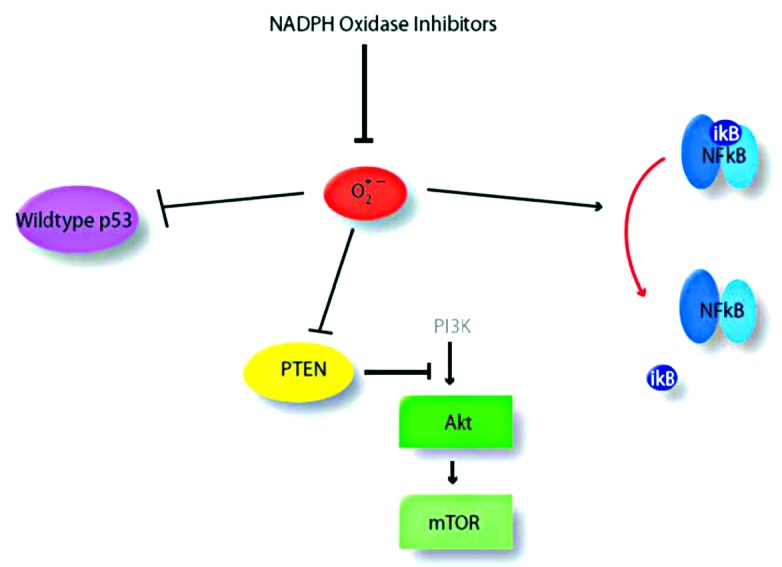
Figure 4. Mechanisms of reactive oxygen species-mediated activation of cancer cells. Reactive oxygen species (red) can activate several pathways of cancer progression and invasion including wild-type p53, and NFkB.
Challenges to Anti-Invasive Compound Development and Clinical Implementation
Small-molecule anti-invasive compounds face many of the same challenges as other new small-molecule compounds. Drug delivery issues with small molecules are significant and often stops the development of a compound at the in vitro level. Though compounds can accumulate via the compromised tumor vasculature, it is more desirable to have compounds that will also cross the blood brain barrier to reach invading cells. This can lead to development of hydrophobic compounds that result in poor circulation times and off-target accumulation. To avoid such issues, it is possible to locally deliver drugs in polymers (Carmustine in PLGA scaffolds) or directly inject drugs into the tumor via convection enhanced delivery (CED).103 An alternative is to encapsulate small-molecule drugs with poor half-life and accumulation in nanocarriers such as liposomes or micelles to passively accumulate at the tumor site. This method has shown repeated success with both chemotherapeutics104,105 and with some small-molecule anti-invasive agents (such as imipramine blue101 and curcumin106). Determination of the possible complications in delivery and design of a drug delivery approach with the development of the small-molecule inhibitor may circumvent failure from the in vitro to the in vivo stage and offer a viable approach to avoid side effects.
Further difficulty arises in the incorporation of anti-invasive drugs into therapy regimens and the assessment of effectiveness alone. Some compounds show increased chemosensitization with particular chemotherapeutics or radiosensitization but may not work for all therapy regimens or for the standard of care treatment of glioblastoma. These difficulties lead to an inability to determine how to actually incorporate these new types of treatments into the standard of care and if they, in fact, should be incorporated with traditional therapy. Additionally, the majority of anti-invasive compounds affect angiogenesis of the tumors.41,93,107-109 This can lead to the need for adjustments of future treatments based on accessibility of the tumor via the bloodstream depending on the efficiency of delivery.
Lastly, small-molecule inhibitors of invasion have difficult outcome measures as compared with antitumor counterparts. Many compounds show anti-invasive effects on glioma cells but do not necessarily have cytotoxicity effects alone. Therefore, determination of the validity of a new compound as compared with the standard of care can be difficult to assess. Further, a particular anti-invasive compound (such as the anti-inflammatory types of compounds) may work well with therapies that lead to inflammation-related invasion (such as radiation), whereas a compound inhibiting hypoxia-induced invasion may work better with an anti-angiogenic compound. Therefore, it seems that the best way to use these types of novel compounds can become personalized based on both the genotype of the tumor and the best course of antitumor treatment in order to enhance the overall survival and prevention of recurrence.
In conclusion, there are many different types of anti-invasive compounds available for clinical use. Though many traditional compounds for targeting invasion (i.e., adhesions, matrix degradation, and cytoskeletal dynamics) show poor performance in the clinic, newer agents that affect all of these pathways are showing great promise both alone and in conjunction with therapies. With such a variety of small molecules to target invasion of brain cancer cells with many beneficial side effects, such as breaking of chemoresistance and inhibition of angiogenesis, these compounds provide a positive outlook for the treatment of brain tumors. Further, with the rising popularity of the concept of personalized therapy for cancer treatment, the diversity of mechanisms involved in invasion and ability to inhibit them specifically offers an opportunity to affect therapy in a positive way. We should be able to advance the treatment of glioma to a point of increased survival and minimized recurrence unseen before if we can determine how best to assess and incorporate this new type of treatment.
Acknowledgments
The authors would like to acknowledge Franck Amblard for technical assistance and S Balakrishna Pai for discussion. Funding was provided to JLA and RVB by the Wallace Coulter Translational Research Fund and to RVB by the Nora L Redman Foundation, Georgia Cancer Coalition, and Ian’s Friends Foundation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25334
References
- 1.National Cancer Institute Brain Tumor Study in Adults: Fact. 2000.
- 2.Tran B, Rosenthal MA. Survival comparison between glioblastoma multiforme and other incurable cancers. J Clin Neurosci. 2010;17:417–21. doi: 10.1016/j.jocn.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Mikkelsen T. (1998) Brain tumor invasion: biological, clinical, and therapeutic considerations (Wiley-Liss, New York):464. [Google Scholar]
- 4.Price SJ, Jena R, Burnet NG, Hutchinson PJ, Dean AF, Peña A, et al. Improved delineation of glioma margins and regions of infiltration with the use of diffusion tensor imaging: an image-guided biopsy study. AJNR Am J Neuroradiol. 2006;27:1969–74. [PMC free article] [PubMed] [Google Scholar]
- 5.Chaichana KL, McGirt MJ, Laterra J, Olivi A, Quiñones-Hinojosa A. Recurrence and malignant degeneration after resection of adult hemispheric low-grade gliomas. J Neurosurg. 2010;112:10–7. doi: 10.3171/2008.10.JNS08608. [DOI] [PubMed] [Google Scholar]
- 6.Dinnes J, Cave C, Huang S, Milne R. A rapid and systematic review of the effectiveness of temozolomide for the treatment of recurrent malignant glioma. Br J Cancer. 2002;86:501–5. doi: 10.1038/sj.bjc.6600135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nabors LB, Fiveash J. Treatment of adults with recurrent malignant glioma. Expert Rev Neurother. 2005;5:509–14. doi: 10.1586/14737175.5.4.509. [DOI] [PubMed] [Google Scholar]
- 8.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups. National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 9.Jain RK. Transport of molecules, particles, and cells in solid tumors. Annu Rev Biomed Eng. 1999;1:241–63. doi: 10.1146/annurev.bioeng.1.1.241. [DOI] [PubMed] [Google Scholar]
- 10.Jain RK. Delivery of molecular and cellular medicine to solid tumors. Adv Drug Deliv Rev. 2001;46:149–68. doi: 10.1016/S0169-409X(00)00131-9. [DOI] [PubMed] [Google Scholar]
- 11.Gliemroth J, Feyerabend T, Gerlach C, Arnold H, Terzis AJ. Proliferation, migration, and invasion of human glioma cells exposed to fractionated radiotherapy in vitro. Neurosurg Rev. 2003;26:198–205. doi: 10.1007/s10143-003-0258-9. [DOI] [PubMed] [Google Scholar]
- 12.Zhai GG, Malhotra R, Delaney M, Latham D, Nestler U, Zhang M, et al. Radiation enhances the invasive potential of primary glioblastoma cells via activation of the Rho signaling pathway. J Neurooncol. 2006;76:227–37. doi: 10.1007/s11060-005-6499-4. [DOI] [PubMed] [Google Scholar]
- 13.Wild-Bode C, Weller M, Rimner A, Dichgans J, Wick W. Sublethal irradiation promotes migration and invasiveness of glioma cells: implications for radiotherapy of human glioblastoma. Cancer Res. 2001;61:2744–50. [PubMed] [Google Scholar]
- 14.Wick W, Wick A, Schulz JB, Dichgans J, Rodemann HP, Weller M. Prevention of irradiation-induced glioma cell invasion by temozolomide involves caspase 3 activity and cleavage of focal adhesion kinase. Cancer Res. 2002;62:1915–9. [PubMed] [Google Scholar]
- 15.Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat Rev Cancer. 2005;5:516–25. doi: 10.1038/nrc1650. [DOI] [PubMed] [Google Scholar]
- 16.Madani I, De Neve W, Mareel M. Does ionizing radiation stimulate cancer invasion and metastasis? Bull Cancer. 2008;95:292–300. doi: 10.1684/bdc.2008.0598. [DOI] [PubMed] [Google Scholar]
- 17.Barcellos-Hoff MH. The potential influence of radiation-induced microenvironments in neoplastic progression. J Mammary Gland Biol Neoplasia. 1998;3:165–75. doi: 10.1023/A:1018794806635. [DOI] [PubMed] [Google Scholar]
- 18.Barcellos-Hoff MH, Ravani SA. Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Res. 2000;60:1254–60. [PubMed] [Google Scholar]
- 19.Ohuchida K, Mizumoto K, Murakami M, Qian LW, Sato N, Nagai E, et al. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res. 2004;64:3215–22. doi: 10.1158/0008-5472.CAN-03-2464. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura JL, Haas-Kogan DA, Pieper RO. Glioma invasiveness responds variably to irradiation in a co-culture model. Int J Radiat Oncol Biol Phys. 2007;69:880–6. doi: 10.1016/j.ijrobp.2007.06.052. [DOI] [PubMed] [Google Scholar]
- 21.Park CM, Park MJ, Kwak HJ, Lee HC, Kim MS, Lee SH, et al. Ionizing radiation enhances matrix metalloproteinase-2 secretion and invasion of glioma cells through Src/epidermal growth factor receptor-mediated p38/Akt and phosphatidylinositol 3-kinase/Akt signaling pathways. Cancer Res. 2006;66:8511–9. doi: 10.1158/0008-5472.CAN-05-4340. [DOI] [PubMed] [Google Scholar]
- 22.Badiga AV, Chetty C, Kesanakurti D, Are D, Gujrati M, Klopfenstein JD, et al. MMP-2 siRNA inhibits radiation-enhanced invasiveness in glioma cells. PLoS One. 2011;6:e20614. doi: 10.1371/journal.pone.0020614. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Argyriou AA, Giannopoulou E, Kalofonos HP. Angiogenesis and anti-angiogenic molecularly targeted therapies in malignant gliomas. Oncology. 2009;77:1–11. doi: 10.1159/000218165. [DOI] [PubMed] [Google Scholar]
- 24.Lamszus K, Kunkel P, Westphal M. Invasion as limitation to anti-angiogenic glioma therapy. Acta Neurochir Suppl. 2003;88:169–77. doi: 10.1007/978-3-7091-6090-9_23. [DOI] [PubMed] [Google Scholar]
- 25.Murphy EA, Majeti BK, Barnes LA, Makale M, Weis SM, Lutu-Fuga K, et al. Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proc Natl Acad Sci USA. 2008;105:9343–8. doi: 10.1073/pnas.0803728105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller KD, Soule SE, Calley C, Emerson RE, Hutchins GD, Kopecky K, et al. Randomized phase II trial of the anti-angiogenic potential of doxorubicin and docetaxel; primary chemotherapy as Biomarker Discovery Laboratory. Breast Cancer Res Treat. 2005;89:187–97. doi: 10.1007/s10549-004-2044-y. [DOI] [PubMed] [Google Scholar]
- 27.Gömöri E, Fülöp Z, Mészáros I, Dóczi T, Matolcsy A. Microsatellite analysis of primary and recurrent glial tumors suggests different modalities of clonal evolution of tumor cells. J Neuropathol Exp Neurol. 2002;61:396–402. doi: 10.1093/jnen/61.5.396. [DOI] [PubMed] [Google Scholar]
- 28.Di Nicolantonio F, Mercer SJ, Knight LA, Gabriel FG, Whitehouse PA, Sharma S, et al. Cancer cell adaptation to chemotherapy. BMC Cancer. 2005;5:78. doi: 10.1186/1471-2407-5-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G, et al. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006;13:1238–41. doi: 10.1038/sj.cdd.4401872. [DOI] [PubMed] [Google Scholar]
- 30.Borovski T, De Sousa E Melo F, Vermeulen L, Medema JP. Cancer stem cell niche: the place to be. Cancer Res. 2011;71:634–9. doi: 10.1158/0008-5472.CAN-10-3220. [DOI] [PubMed] [Google Scholar]
- 31.Liu D, Martin V, Fueyo J, Lee OH, Xu J, Cortes-Santiago N, et al. Tie2/TEK modulates the interaction of glioma and brain tumor stem cells with endothelial cells and promotes an invasive phenotype. Oncotarget. 2010;1:700–9. doi: 10.18632/oncotarget.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakano A, Tani E, Miyazaki K, Yamamoto Y, Furuyama J. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in human gliomas. J Neurosurg. 1995;83:298–307. doi: 10.3171/jns.1995.83.2.0298. [DOI] [PubMed] [Google Scholar]
- 33.Chintala SK, Rao JK. Invasion of human glioma: role of extracellular matrix proteins. Front Biosci. 1996;1:d324–39. doi: 10.2741/a135. [DOI] [PubMed] [Google Scholar]
- 34.Rubenstein BM, Kaufman LJ. The role of extracellular matrix in glioma invasion: a cellular Potts model approach. Biophys J. 2008;95:5661–80. doi: 10.1529/biophysj.108.140624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uhm JH, Dooley NP, Villemure JG, Yong VW. Mechanisms of glioma invasion: role of matrix-metalloproteinases. Can J Neurol Sci. 1997;24:3–15. doi: 10.1017/s0317167100021028. [DOI] [PubMed] [Google Scholar]
- 36.Le DM, Besson A, Fogg DK, Choi KS, Waisman DM, Goodyer CG, et al. Exploitation of astrocytes by glioma cells to facilitate invasiveness: a mechanism involving matrix metalloproteinase-2 and the urokinase-type plasminogen activator-plasmin cascade. J Neurosci. 2003;23:4034–43. doi: 10.1523/JNEUROSCI.23-10-04034.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levin VA, Phuphanich S, Yung WK, Forsyth PA, Maestro RD, Perry JR, et al. Randomized, double-blind, placebo-controlled trial of marimastat in glioblastoma multiforme patients following surgery and irradiation. J Neurooncol. 2006;78:295–302. doi: 10.1007/s11060-005-9098-5. [DOI] [PubMed] [Google Scholar]
- 38.Sorbera LA, Castaner J. Prinomastat: oncolytic, matrix metalloproteinase inhibitor. Drugs Future. 2000;25:150–8. doi: 10.1358/dof.2000.025.02.567506. [DOI] [Google Scholar]
- 39.Levitt NC, Eskens FA, O’Byrne KJ, Propper DJ, Denis LJ, Owen SJ, et al. Phase I and pharmacological study of the oral matrix metalloproteinase inhibitor, MMI270 (CGS27023A), in patients with advanced solid cancer. Clin Cancer Res. 2001;7:1912–22. [PubMed] [Google Scholar]
- 40.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–92. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 41.Nakabayashi H, Yawata T, Shimizu K. Anti-invasive and antiangiogenic effects of MMI-166 on malignant glioma cells. BMC Cancer. 2010;10:339. doi: 10.1186/1471-2407-10-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin SG, Jeong YI, Jung S, Ryu HH, Jin YH, Kim IY. The effect of hyaluronic Acid on the invasiveness of malignant glioma cells : comparison of invasion potential at hyaluronic Acid hydrogel and matrigel. J Korean Neurosurg Soc. 2009;46:472–8. doi: 10.3340/jkns.2009.46.5.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Demchik LL, Sameni M, Nelson K, Mikkelsen T, Sloane BF. Cathepsin B and glioma invasion. Int J Dev Neurosci. 1999;17:483–94. doi: 10.1016/S0736-5748(99)00011-8. [DOI] [PubMed] [Google Scholar]
- 44.Bellail AC, Hunter SB, Brat DJ, Tan C, Van Meir EG. Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int J Biochem Cell Biol. 2004;36:1046–69. doi: 10.1016/j.biocel.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 45.Goldbrunner RH, Bernstein JJ, Tonn JC. ECM-mediated glioma cell invasion. Microsc Res Tech. 1998;43:250–7. doi: 10.1002/(SICI)1097-0029(19981101)43:3<250::AID-JEMT7>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 46.Goldbrunner RH, Bernstein JJ, Tonn JC. Cell-extracellular matrix interaction in glioma invasion. Acta Neurochir (Wien) 1999;141(discussion 304-295):295–305, discussion 304-5. doi: 10.1007/s007010050301. [DOI] [PubMed] [Google Scholar]
- 47.Gingras MC, Roussel E, Bruner JM, Branch CD, Moser RP. Comparison of cell adhesion molecule expression between glioblastoma multiforme and autologous normal brain tissue. J Neuroimmunol. 1995;57:143–53. doi: 10.1016/0165-5728(94)00178-Q. [DOI] [PubMed] [Google Scholar]
- 48.Maurer GD, Tritschler I, Adams B, Tabatabai G, Wick W, Stupp R, et al. Cilengitide modulates attachment and viability of human glioma cells, but not sensitivity to irradiation or temozolomide in vitro. Neuro Oncol. 2009;11:747–56. doi: 10.1215/15228517-2009-012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reardon DA, Fink KL, Mikkelsen T, Cloughesy TF, O’Neill A, Plotkin S, et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J Clin Oncol. 2008;26:5610–7. doi: 10.1200/JCO.2008.16.7510. [DOI] [PubMed] [Google Scholar]
- 50.Giese A, Laube B, Zapf S, Mangold U, Westphal M. Glioma cell adhesion and migration on human brain sections. Anticancer Res. 1998;18(4A):2435–47. [PubMed] [Google Scholar]
- 51.Kaczarek E, Zapf S, Bouterfa H, Tonn JC, Westphal M, Giese A. Dissecting glioma invasion: interrelation of adhesion, migration and intercellular contacts determine the invasive phenotype. Int J Dev Neurosci. 1999;17:625–41. doi: 10.1016/S0736-5748(99)00047-7. [DOI] [PubMed] [Google Scholar]
- 52.Gimona M. The microfilament system in the formation of invasive adhesions. Semin Cancer Biol. 2008;18:23–34. doi: 10.1016/j.semcancer.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 53.Hao H, Naomoto Y, Bao X, Watanabe N, Sakurama K, Noma K, et al. Focal adhesion kinase as potential target for cancer therapy (Review) Oncol Rep. 2009;22:973–9. doi: 10.3892/or_00000524. [Review] [DOI] [PubMed] [Google Scholar]
- 54.Parsons JT, Slack-Davis J, Tilghman R, Roberts WG. Focal adhesion kinase: targeting adhesion signaling pathways for therapeutic intervention. Clin Cancer Res. 2008;14:627–32. doi: 10.1158/1078-0432.CCR-07-2220. [DOI] [PubMed] [Google Scholar]
- 55.Slack-Davis JK, Martin KH, Tilghman RW, Iwanicki M, Ung EJ, Autry C, et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. 2007;282:14845–52. doi: 10.1074/jbc.M606695200. [DOI] [PubMed] [Google Scholar]
- 56.Merzak A, Koocheckpour S, Pilkington GJ. CD44 mediates human glioma cell adhesion and invasion in vitro. Cancer Res. 1994;54:3988–92. [PubMed] [Google Scholar]
- 57.Tysnes BB, Mahesparan R. Biological mechanisms of glioma invasion and potential therapeutic targets. J Neurooncol. 2001;53:129–47. doi: 10.1023/A:1012249216117. [DOI] [PubMed] [Google Scholar]
- 58.Yamaguchi H, Oikawa T. Membrane lipids in invadopodia and podosomes: key structures for cancer invasion and metastasis. Oncotarget. 2010;1:320–8. doi: 10.18632/oncotarget.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fenteany G, Zhu S. Small-molecule inhibitors of actin dynamics and cell motility. Curr Top Med Chem. 2003;3:593–616. doi: 10.2174/1568026033452348. [DOI] [PubMed] [Google Scholar]
- 60.Terzis AJ, Thorsen F, Heese O, Visted T, Bjerkvig R, Dahl O, et al. Proliferation, migration and invasion of human glioma cells exposed to paclitaxel (Taxol) in vitro. Br J Cancer. 1997;75:1744–52. doi: 10.1038/bjc.1997.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tonn JC, Haugland HK, Saraste J, Roosen K, Laerum OD. Differential effects of vincristine and phenytoin on the proliferation, migration, and invasion of human glioma cell lines. J Neurosurg. 1995;82:1035–43. doi: 10.3171/jns.1995.82.6.1035. [DOI] [PubMed] [Google Scholar]
- 62.Beadle C, Assanah MC, Monzo P, Vallee R, Rosenfeld SS, Canoll P. The role of myosin II in glioma invasion of the brain. Mol Biol Cell. 2008;19:3357–68. doi: 10.1091/mbc.E08-03-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ivkovic S, Beadle C, Noticewala S, Massey SC, Swanson KR, Toro LN, et al. Direct inhibition of myosin II effectively blocks glioma invasion in the presence of multiple motogens. Mol Biol Cell. 2012;23:533–42. doi: 10.1091/mbc.E11-01-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamazaki D, Kurisu S, Takenawa T. Regulation of cancer cell motility through actin reorganization. Cancer Sci. 2005;96:379–86. doi: 10.1111/j.1349-7006.2005.00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.De Witt Hamer PC. Small molecule kinase inhibitors in glioblastoma: a systematic review of clinical studies. Neuro Oncol. 2010;12:304–16. doi: 10.1093/neuonc/nop068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weber GL, Parat MO, Binder ZA, Gallia GL, Riggins GJ. Abrogation of PIK3CA or PIK3R1 reduces proliferation, migration, and invasion in glioblastoma multiforme cells. Oncotarget. 2011;2:833–49. doi: 10.18632/oncotarget.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoelzinger DB, Demuth T, Berens ME. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J Natl Cancer Inst. 2007;99:1583–93. doi: 10.1093/jnci/djm187. [DOI] [PubMed] [Google Scholar]
- 68.Nico B, Ribatti D. Aquaporins in tumor growth and angiogenesis. Cancer Lett. 2010;294:135–8. doi: 10.1016/j.canlet.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 69.Molenaar RJ. Ion channels in glioblastoma. ISRN Neurol. 2011;2011:590249. doi: 10.5402/2011/590249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Papadopoulos MC, Saadoun S, Verkman AS. Aquaporins and cell migration. Pflugers Arch. 2008;456:693–700. doi: 10.1007/s00424-007-0357-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ding T, Gu F, Fu L, Ma YJ. Aquaporin-4 in glioma invasion and an analysis of molecular mechanisms. J Clin Neurosci. 2010;17:1359–61. doi: 10.1016/j.jocn.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 72.Hayashi Y, Edwards NA, Proescholdt MA, Oldfield EH, Merrill MJ. Regulation and function of aquaporin-1 in glioma cells. Neoplasia. 2007;9:777–87. doi: 10.1593/neo.07454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Patterson LH, McKeown SR. AQ4N: a new approach to hypoxia-activated cancer chemotherapy. Br J Cancer. 2000;83:1589–93. doi: 10.1054/bjoc.2000.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Patterson LH, McKeown SR, Ruparelia K, Double JA, Bibby MC, Cole S, et al. Enhancement of chemotherapy and radiotherapy of murine tumours by AQ4N, a bioreductively activated anti-tumour agent. Br J Cancer. 2000;82:1984–90. doi: 10.1054/bjoc.2000.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Soroceanu L, Manning TJ, Jr., Sontheimer H. Modulation of glioma cell migration and invasion using Cl(-) and K(+) ion channel blockers. J Neurosci. 1999;19:5942–54. doi: 10.1523/JNEUROSCI.19-14-05942.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McFerrin MB, Sontheimer H. A role for ion channels in glioma cell invasion. Neuron Glia Biol. 2006;2:39–49. doi: 10.1017/S1740925X06000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zagzag D, Esencay M, Mendez O, Yee H, Smirnova I, Huang Y, et al. Hypoxia- and vascular endothelial growth factor-induced stromal cell-derived factor-1alpha/CXCR4 expression in glioblastomas: one plausible explanation of Scherer’s structures. Am J Pathol. 2008;173:545–60. doi: 10.2353/ajpath.2008.071197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sciumè G, Santoni A, Bernardini G. Chemokines and glioma: invasion and more. J Neuroimmunol. 2010;224:8–12. doi: 10.1016/j.jneuroim.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 79.Rubin JB, Kung AL, Klein RS, Chan JA, Sun Y, Schmidt K, et al. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc Natl Acad Sci USA. 2003;100:13513–8. doi: 10.1073/pnas.2235846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.De Clercq E. The bicyclam AMD3100 story. Nat Rev Drug Discov. 2003;2:581–7. doi: 10.1038/nrd1134. [DOI] [PubMed] [Google Scholar]
- 81.De Clercq E. Potential clinical applications of the CXCR4 antagonist bicyclam AMD3100. Mini Rev Med Chem. 2005;5:805–24. doi: 10.2174/1389557054867075. [DOI] [PubMed] [Google Scholar]
- 82.Rong Y, Durden DL, Van Meir EG, Brat DJ. 'Pseudopalisading' necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol. 2006;656:529–39. doi: 10.1097/00005072-200606000-00001. [DOI] [PubMed] [Google Scholar]
- 83.Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–31. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Méndez O, Zavadil J, Esencay M, Lukyanov Y, Santovasi D, Wang SC, et al. Knock down of HIF-1alpha in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Mol Cancer. 2010;9:133. doi: 10.1186/1476-4598-9-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chau NM, Rogers P, Aherne W, Carroll V, Collins I, McDonald E, et al. Identification of novel small-molecule inhibitors of hypoxia-inducible factor-1 that differentially block hypoxia-inducible factor-1 activity and hypoxia-inducible factor-1alpha induction in response to hypoxic stress and growth factors. Cancer Res. 2005;65:4918–28. doi: 10.1158/0008-5472.CAN-04-4453. [DOI] [PubMed] [Google Scholar]
- 86.Kong D, Park EJ, Stephen AG, Calvani M, Cardellina JH, Monks A, et al. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005;65:9047–55. doi: 10.1158/0008-5472.CAN-05-1235. [DOI] [PubMed] [Google Scholar]
- 87.Duyndam MC, van Berkel MP, Dorsman JC, Rockx DA, Pinedo HM, Boven E. Cisplatin and doxorubicin repress Vascular Endothelial Growth Factor expression and differentially down-regulate Hypoxia-inducible Factor I activity in human ovarian cancer cells. Biochem Pharmacol. 2007;74:191–201. doi: 10.1016/j.bcp.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 88.Shankar S, Ganapathy S, Hingorani SR, Srivastava RK. EGCG inhibits growth, invasion, angiogenesis and metastasis of pancreatic cancer. Front Biosci. 2008;13:440–52. doi: 10.2741/2691. [DOI] [PubMed] [Google Scholar]
- 89.Zagzag D, Nomura M, Friedlander DR, Blanco CY, Gagner JP, Nomura N, et al. Geldanamycin inhibits migration of glioma cells in vitro: a potential role for hypoxia-inducible factor (HIF-1alpha) in glioma cell invasion. J Cell Physiol. 2003;196:394–402. doi: 10.1002/jcp.10306. [DOI] [PubMed] [Google Scholar]
- 90.Bai X, Cerimele F, Ushio-Fukai M, Waqas M, Campbell PM, Govindarajan B, et al. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J Biol Chem. 2003;278:35501–7. doi: 10.1074/jbc.M302967200. [DOI] [PubMed] [Google Scholar]
- 91.Wolf I, O’Kelly J, Wakimoto N, Nguyen A, Amblard F, Karlan BY, et al. Honokiol, a natural biphenyl, inhibits in vitro and in vivo growth of breast cancer through induction of apoptosis and cell cycle arrest. Int J Oncol. 2007;30:1529–37. [PubMed] [Google Scholar]
- 92.Shigemura K, Arbiser JL, Sun SY, Zayzafoon M, Johnstone PA, Fujisawa M, et al. Honokiol, a natural plant product, inhibits the bone metastatic growth of human prostate cancer cells. Cancer. 2007;109:1279–89. doi: 10.1002/cncr.22551. [DOI] [PubMed] [Google Scholar]
- 93.Ahn KS, Sethi G, Shishodia S, Sung B, Arbiser JL, Aggarwal BB. Honokiol potentiates apoptosis, suppresses osteoclastogenesis, and inhibits invasion through modulation of nuclear factor-kappaB activation pathway. Mol Cancer Res. 2006;4:621–33. doi: 10.1158/1541-7786.MCR-06-0076. [DOI] [PubMed] [Google Scholar]
- 94.Rayet B, Gélinas C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene. 1999;18:6938–47. doi: 10.1038/sj.onc.1203221. [DOI] [PubMed] [Google Scholar]
- 95.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 96.Tertil M, Jozkowicz A, Dulak J. Oxidative stress in tumor angiogenesis- therapeutic targets. Curr Pharm Des. 2010;16:3877–94. doi: 10.2174/138161210794454969. [DOI] [PubMed] [Google Scholar]
- 97.Cooney RV, Pung A, Harwood PJ, Boynton AL, Zhang LX, Hossain MZ, et al. Inhibition of cellular transformation by triphenylmethane: a novel chemopreventive agent. Carcinogenesis. 1992;13:1107–12. doi: 10.1093/carcin/13.7.1107. [DOI] [PubMed] [Google Scholar]
- 98.Salhia B, Rutten F, Nakada M, Beaudry C, Berens M, Kwan A, et al. Inhibition of Rho-kinase affects astrocytoma morphology, motility, and invasion through activation of Rac1. Cancer Res. 2005;65:8792–800. doi: 10.1158/0008-5472.CAN-05-0160. [DOI] [PubMed] [Google Scholar]
- 99.Salhia B, Tran NL, Symons M, Winkles JA, Rutka JT, Berens ME. Molecular pathways triggering glioma cell invasion. Expert Rev Mol Diagn. 2006;6:613–26. doi: 10.1586/14737159.6.4.613. [DOI] [PubMed] [Google Scholar]
- 100.Diaz B, Shani G, Pass I, Anderson D, Quintavalle M, Courtneidge SA. Tks5-dependent, nox-mediated generation of reactive oxygen species is necessary for invadopodia formation. Sci Signal. 2009;2:ra53. doi: 10.1126/scisignal.2000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Munson JM, Fried L, Rowson SA, Bonner MY, Karumbaiah L, Diaz B, et al. Anti-invasive adjuvant therapy with imipramine blue enhances chemotherapeutic efficacy against glioma. Sci Transl Med. 2012;4:27ra36. doi: 10.1126/scitranslmed.3003016. [DOI] [PubMed] [Google Scholar]
- 102.Neilsen PM, Noll JE, Suetani RJ, Schulz RB, Al-Ejeh F, Evdokiou A, et al. Mutant p53 uses p63 as a molecular chaperone to alter gene expression and induce a pro-invasive secretome. Oncotarget. 2011;2:1203–17. doi: 10.18632/oncotarget.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nakamura T, Saito R, Sugiyama S, Sonoda Y, Kumabe T, Tominaga T. Local convection-enhanced delivery of chemotherapeutic agent transiently opens blood-brain barrier and improves efficacy of systemic chemotherapy in intracranial xenograft tumor model. Cancer Lett. 2011;310:77–83. doi: 10.1016/j.canlet.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 104.Gabizon A, Isacson R, Libson E, Kaufman B, Uziely B, Catane R, et al. Clinical studies of liposome-encapsulated doxorubicin. Acta Oncol. 1994;33:779–86. doi: 10.3109/02841869409083948. [DOI] [PubMed] [Google Scholar]
- 105.Siegal T, Horowitz A, Gabizon A. Doxorubicin encapsulated in sterically stabilized liposomes for the treatment of a brain tumor model: biodistribution and therapeutic efficacy. J Neurosurg. 1995;83:1029–37. doi: 10.3171/jns.1995.83.6.1029. [DOI] [PubMed] [Google Scholar]
- 106.Shao J, Zheng D, Jiang Z, Xu H, Hu Y, Li X, et al. Curcumin delivery by methoxy polyethylene glycol-poly(caprolactone) nanoparticles inhibits the growth of C6 glioma cells. Acta Biochim Biophys Sin (Shanghai) 2011;43:267–74. doi: 10.1093/abbs/gmr011. [DOI] [PubMed] [Google Scholar]
- 107.de Boüard S, Herlin P, Christensen JG, Lemoisson E, Gauduchon P, Raymond E, et al. Antiangiogenic and anti-invasive effects of sunitinib on experimental human glioblastoma. Neuro Oncol. 2007;9:412–23. doi: 10.1215/15228517-2007-024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jung YD, Ellis LM. Inhibition of tumour invasion and angiogenesis by epigallocatechin gallate (EGCG), a major component of green tea. Int J Exp Pathol. 2001;82:309–16. doi: 10.1046/j.1365-2613.2001.00205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Roskelley CD, Williams DE, McHardy LM, Leong KG, Troussard A, Karsan A, et al. Inhibition of tumor cell invasion and angiogenesis by motuporamines. Cancer Res. 2001;61:6788–94. [PubMed] [Google Scholar]