

Abstract
HIC1 (hypermethylated in cancer 1) is a tumor suppressor gene, expression of which is frequently suppressed in human cancers. Very little is known about the molecular basis of HIC1 in antagonizing oncogenic pathways. Here, we report that HIC1 forms complexes with the signal transducers and activators of transcription 3 (STAT3) and attenuates STAT3-mediated transcription. STAT3 was identified as a HIC1-interacting protein by affinity capture and followed by mass spectrometry analysis. Overexpression or depletion of HIC1 resulted in decreased or increased levels of interleukin-6 (IL-6)/oncostatin M (OSM)-induced STAT3-mediated reporter activity and expression of target genes such as VEGF and c-Myc, respectively. Furthermore, HIC1 suppressing the VEGF and c-Myc promoter activity and the colony formation of MDA-MB 231 cells were STAT3-dependent. Further studies showed that HIC1 interacts with the DNA binding domain of STAT3 and suppresses the binding of STAT3 to its target gene promoters. Domain mapping study revealed that HIC1 C-terminal domain binds to STAT3. HIC1 mutant defective in STAT3 interaction reduced its repressive effect on STAT3 DNA binding activity, the reporter activity and gene expression of the VEGF and c-Myc genes, and cell growth in MDA-MB 231 cells. Altogether, our findings not only provide a novel role of HIC1 in antagonizing STAT3-mediated activation of VEGF and c-Myc gene expression and cell growth, but also elucidate a molecular basis underlying the inhibitory effect of HIC1 on STAT3 transcriptional potential.
Keywords: HIC1, STAT3, protein interaction, transcriptional regulation, DNA binding
Introduction
Hypermethylated in cancer 1 (HIC1) is first identified as a candidate tumor suppressor gene located at chromosome 17p13.3, a region frequently deleted and epigenetically silenced in a variety of human cancers.1 HIC1 protein is a sequence-specific transcriptional repressor composed of 2 autonomous repression domains, an N-terminal BTB/POZ domain, and a central region, followed by 5 C2H2 zinc fingers in the C terminus. The BTB/POZ domain forms a transcriptional repression complex with the class III histone deacetylase SIRT1 and also recruits the PRC2 repression complexes to suppress the transcription of HIC1 target genes.2 The central region can recruit transcriptional co-repressor CtBP or MTA1 through the GLDLSKK motif3,4 or via SUMO modification at a consensus motif,5 respectively, for its repression activity. The C-terminal zinc fingers 2–5 are responsible for DNA binding activity in recognizing HIC1-responsive element (HiRE).6 To date only 11 direct target genes of HIC1 have been described, which are associated with the regulation of several cellular pathways including DNA damage response.7,8 In addition to the DNA binding-mediated target gene regulation, HIC1 could modulate other gene expression via protein-protein interactions. For instance, HIC1 interacts with TCF4 and attenuates TCF4/β-catenin-mediated transcription in Wnt-stimulated cells.9 Given that the role of HIC1 in regulating cellular pathways is not limited by its direct target gene expression, identification of novel HIC1-associated factors would broaden our understanding in HIC1 function.
The signal transducers and activators of transcription (STAT) family proteins, consisting of at least 7 members STAT1–4, STAT5A, STAT5B, and STAT6, play important roles in regulating fundamental cellular processes, including cell growth, survival and differentiation, inflammation, angiogenesis, and metastasis.10-12 Among this protein family, STAT3 is an oncogenic transcription factor involved in cell transformation. Activated STAT3 can mediate oncogenic transformation in cultured cells and tumor formation in nude mice.12,13 Furthermore, STAT3 is frequently overexpressed or persistently activated in a broad range of human cancers and promotes tumor progression.11,14-17 STAT3-mediated tumorigenesis was thought to be associated with its potential in activating genes encoding anti-apoptotic proteins (Mcl-1, Bcl-2, and survivin), cell cycle regulators (cyclinD1 and c-Myc), and angiogenic factor (VEGF).16,18-20 STAT3 generally resides in the cytoplasm and can be activated through phosphorylation of tyrosine 705 by Janus kinases (JAKs) in response to various cytokines such as Interleukin-6, IL-10, IL-21, G-CSF, leukemia inhibitory factor, and oncostatin M (OSM) or by receptor tyrosine kinases upon ligand activation such as EGF and HGF.21,22 Tyrosine 705 phosphorylation facilitates STAT3 to form either homodimers or heterodimers with other STAT proteins, then translocate into the nucleus where they bind to specific DNA elements for gene activation.23,24 In addition, lysine 685 acetylation stimulates STAT3 DNA binding activity and transactivation potential.25,26 On the other hand, STAT activities can be negatively regulated by phosphotyrosine phosphatases (e.g., SHPs, CD45, PTP1B/TC-PTP),27-30 the suppressor of cytokine signaling 3 (SOCS3) that interacts with JAKs and inhibits their activities for STAT3 phosphorylation,31 factors that inhibit STAT3 DNA binding activity (e.g., the protein inhibitor of activated STAT3 [PIAS3], Daxx, Duplin, and ARHI),32-36 or proteins that prevent STAT3 phosphorylation and nuclear translocation (e.g., KAP1 and LMW-DSP2).37,38 Whether other factors can also modulate STAT3 activity remains to be further explored.
Here, we report that STAT3 is one of HIC1-interacting proteins. HIC1 could suppress STAT3-mediated reporter activity and target gene expression, such as VEGF and c-Myc. Furthermore, the effect of HIC1 on cell growth inhibition was attenuated in STAT3-depleted cells, suggesting the important role of HIC1 in antagonizing STAT3-mediated cell growth. Further study showed that HIC1 associates with STAT3 and inhibits STAT3 DNA binding activity via its C-terminal domain. Deletion of this C-terminal domain resulted in a loss of HIC1-repressive effect on the promoter activity and expression levels of VEGF and c-Myc genes and cell growth in MDA-MB 231 cells. Collectively, our findings provide a novel mechanism of HIC1 in repressing cell growth by inhibiting STAT3 DNA binding potential.
Results
HIC1 interacts with STAT3
We sought to identify HIC1-interacting proteins to explore HIC1 cellular function. Flag-tagged HIC1 protein expressed in 293T cells were subjected to immunoprecipitation experiments. Among the proteins precipitated by HIC1, we isolated a distinct band with a molecular mass around 90 kDa (Fig. 1A, arrow). Mass spectrometry analysis suggested that this band represented STAT3 protein. We further confirmed the HIC1-STAT3 interaction in HeLa cells transiently expressing Flag-tagged HIC1 and HA-tagged STAT3 proteins. Reciprocal immunoprecipitation experiments showed that HIC and STAT3 formed complexes in cells (Fig. 1B). To further substantiate the HIC1-STAT3 interaction at an endogenous level, the nuclear extracts of WI-38 normal lung embryonic fibroblast cell line were subjected to immunoprecipitation experiments using anti-STAT3 antibody. Endogenous HIC1 could be precipitated by STAT3 (Fig. 1C, left panel). We attempted to perform a reciprocal experiment, but the anti-HIC1 antibody was inefficient for immunoprecipitation (data not shown). Notably, treatment with OSM, a well-known cytokine in activating STAT3 signaling pathway, which could induce STAT3 nuclear translocation (right panel, lanes 1 and 2), increased the HIC1 levels precipitated by anti-STAT3 (lane 5 vs. 6). Moreover, GST pull-down assays showed that in vitro-synthesized HIC1 protein was precipitated by recombinant GST-STAT3 but not GST protein (Fig. 1D). These results suggest that HIC1 interacts with STAT3.

Figure 1. HIC1 interacts with STAT3 in vivo and in vitro. (A) SYPRO Ruby gel shows STAT3 pulled down from the cell lysates of 293T cells expressing Flag-tagged HIC1 by immunoprecipitation with anti-Flag antibody. STAT3 peptides detected by mass spectrometry are indicated. (B) Western blots show complex formation of Flag-HIC1 and HA-STAT3 in HeLa cells by immunoprecipitation with indicated antibodies. IP, immunoprecipitation; WCL, whole cell lysates; WB, western blotting (C) Immunoblotting shows immunoprecipitated endogenous HIC1 and STAT3 from the nuclear extracts of WI38 cells (left panel) or WI38 cells starved for 24 h and followed by treatment of 20 ng/ml OSM for 30 min (right panel). Input represents the 10% amount of nuclear extracts subjected to immunoprecipitation. (D) Immunoblotting analysis of in vitro-synthesized HIC1 protein pulled down by recombinant GST-STAT3 fusion proteins (top panel). Input represents 5% of the reticulocyte lysates containing synthesized HIC1 protein subjected to binding assays. Coomassie blue staining showing the levels of GST and GST-STAT3 fusion proteins used in each sample (bottom). The asterisks depict GST and GST-STAT3 proteins.
HIC1 suppresses the STAT3-mediated transcriptional activity and cell growth
Because HIC1 and STAT3 function as a tumor suppressor and an oncogenic protein in transcriptional regulation, respectively, the HIC1-STAT3 interaction raised a possibility that HIC1 might affect STAT3 transcriptional potential. To this end, we first tested whether STAT3-mediated reporter gene activity is affected by HIC1. Reporter activity was analyzed from 293T cells transfected with a STAT3 reporter (3× Ly6E-Luc) and a construct expressing HA-tagged HIC1. As a control, IL-6 treatment induced STAT3 tyrosine 705 phosphorylation and STAT3-mediated reporter activity (Fig. 2A, lane 2). Interestingly, increasing amounts of HIC1 expression rendered a dose-dependent repression of IL-6-induced STAT3 reporter activity (lanes 3–5). These results suggest that HIC1 suppresses STAT3-mediated transcriptional potential. In line with this notion, the reporter activity transactivated by the STAT3-CA, a constitutively active form of STAT3 which could activate reporter gene activity independent to ligand stimulation, was repressed by HIC1 in a dose-dependent fashion (Fig. 2B, lanes 5–8). It should be noted that HIC1 repression is STAT3-dependent, because overexpression of HIC1 did not alter the basal promoter activity of this reporter (Fig. 2B, lanes 1–4).

Figure 2. HIC1 suppresses IL-6/STAT3-activated reporter activity. (A and B) Reporter gene activity of 293T cells transfected with 3xLy6E-Luc reporter and increasing amounts of HA-tagged HIC1 expression construct and then treated with IL-6 (20 ng/ml) for 16 h (A) or along with Flag-tagged STAT3-CA, a constitutively active form of STAT3, transfection for 36 h (B). Error bars represent the mean ± SD (n = 3)
To further substantiate the negative effect of HIC1 on STAT3-mediated transactivation, MDA-MB231, a breast cancer cell line with a constitutive active form of STAT3, and no HIC1 expression, was subjected to analyses of STAT3 target gene promoter activity and expression. STAT3 target genes such as VEGF and c-Myc were selected for this study. Reporter gene analysis showed that HIC1 inhibited the reporter gene activity driven by VEGF or c-Myc promoter (Fig. 3A, lanes 2 and 3 in each panel). If HIC1 repressed both VEGF and c-Myc promoter activities via STAT3, then STAT3 knockdown should attenuate HIC1’s effect on both reporter activities. As expected, HIC1 expression failed to effectively suppress both reporter gene activities in STAT3-depleted cells (lanes 4–6). These results suggest that HIC1-elicited repression of VEGF and c-Myc promoter activities are via STAT3 activity inhibition.

Figure 3. HIC1 represses the promoter activity and expression of VEGF and c-Myc genes and cell colony formation. (A) Reporter gene activity of MDA-MB 231 cells transfected with indicated shRNAs for 24 h and then transfected again with VEGF-Luc or c-Myc-Luc reporter and increasing amounts of pcDNA3-Flag-HIC1 for additional 36 h. Western blots show transfected Flag-HIC1 and endogenous STAT3 levels of each sample. Error bars represent the mean ± SD (n = 3) (B) Real-time qPCR analysis of endogenous STAT3 target gene VEGF and c-Myc mRNAs from HIC1 lentivirus infected MDA-MB 231 cells. Bar graph shows the relative expression of VEGF and c-Myc normalized to GAPDH (the sample with empty vector was taken as 1). Error bars represent the mean ± SD (n = 3) (C) Real-time qPCR analysis of VEGF and c-Myc mRNAs from WI-38 cells infected with lentivirus expressing HIC1 shRNAs for 24 h, serum starved for additional 24 h and then treated with OSM (20 ng/ml) for 2 h. Error bars represent the mean ± SD (n = 3). (D) Colony formation analysis of MDA-MB 231 cells infected with lentivirus expressing Flag-HIC1 and/or indicated shRNAs for 14 d culture with puromycin (2 μg/ml) selection and followed by crystal violet staining (top panel). Bar graph shows the quantification of colony formation of the MDA-MB-231 cells infected with indicated lentivirus. Error bars represent the mean ± SD (n = 3) (bottom panel).
In line with HIC1 in the regulation of VEGF and c-Myc promoter activities, HIC1 overexpression reduced VEGF and c-Myc mRNA levels in MDA-MB231 cells (Fig. 3B). We further substantiated the role of HIC1 in downregulation of VEGF and c-Myc mRNA levels at an endogenous level. The efficacies of two independent shRNA clones (shHIC1-1 and shHIC1-2) were tested their against endogenous HIC1 expression in WI-38 cells (Fig. 3C, western blot). We then examined VEGF and c-Myc mRNA levels in HIC1-depleted WI-38 cells treated with OSM. Real-time quantitative PCR analyses showed that the OSM treatment increased VEGF and c-Myc mRNA levels, while HIC1 mRNA levels remained comparable in both treated and non-treated cells (Fig. 3C, lanes 1 and 2 in each bar graph). Notably, HIC1 knockdown further increased OSM-activated VEGF and c-Myc mRNA levels (lanes 4 and 6 vs. 2). Along with above reporter gene analysis, these results suggest that HIC1 negatively regulates STAT3 target gene promoter activity and expression.
Given that STAT3-activated genes including c-Myc and VEGF are associated with cell growth, our findings of HIC1 repressing the promoter activity and expression of these 2 genes raised a possibility whether HIC1 suppressing cell growth is in part via STAT3 inactivation. Notably, HIC1 expression resulted in a reduction of cell colony formation in MDA-MB 231 cells (Fig. 3D, lane 1 vs. 2). Such inhibition of cell colony formation by HIC1 was significantly abrogated in STAT3-depleted MDA-MB 231 cells (lane 3 vs. 4), suggesting a role of HIC1 in downregulation of cell growth via a STAT3-dependent pathway. Altogether, these results further support the negative effect of HIC1 on STAT3 transcriptional potential.
HIC1 attenuates the DNA binding activity of STAT3
We next explored the molecular basis of HIC1 in downregulating STAT3 transcriptional activity. It is possible that HIC1 modulates STAT3 protein level, phosphorylation, acetylation, nuclear translocation, DNA binding activity, and/or transactivation ability. Overexpression of HIC1 did not significantly alter STAT3 protein steady-state level, the phosphorylation status of STAT3 at Tyr705 and Ser727 or STAT3 Lys685 acetylation in 293T or MDA-MB 231 cells (Figs. 2A and 4A). We further analyzed the effect of HIC1 expression on STAT3 nuclear translocation. Upon OSM stimulation, the levels of STAT3, phospho-Y705, and acetylated K685 STAT3 were substantially increased in the nuclear fractions of MCF7 cells (Fig. 4B, lane 6). However, HIC1 overexpression failed to alter the level of STAT3 nuclear translocation induced by OSM treatment (lane 8 vs. 6). These data suggest that HIC1 does not affect STAT3 phosphorylation, acetylation, or nuclear translocation modulated by cellular signaling(s).

Figure 4. HIC1 attenuates the DNA binding activity of STAT3. (A) Western blot analysis of STAT3 phosphorylation in MDA-MB 231 cells infected with lentivirus expressing Flag-tagged HIC1. (B) Western blotting shows STAT3 phosphorylation, acetylation, and nuclear translocation in MCF7 cells transfected with Flag-HIC1, serum starved for 24 h and then treated with OSM (20 ng/ml; 30 min). Tubulin and lamin A/C were used as loading controls for cytosolic and nuclear fractions, respectively. (C) Phosphoimager analysis of the EMSA gel showing 32P-labeled SIE oligonucleotide with nuclear extracts from 293T cells transfected with HA-STAT3 along with or without increasing amounts of Flag-HIC1 and treated with OSM (20 ng/ml; 30 min) or incubated with anti-STAT3 for supershift as indicated. Western blots show the levels of indicated proteins in the nuclear extracts of each sample for EMSA (bottom). The intensity of STAT3/SIE complex was quantified by densitometry and the relative binding of STAT3 to SIE is indicated after normalization to the sample with OSM treatment but without Flag-HIC1 transfection taken as 1. (D) ChIP semi-quantitative and real-time qPCR analyses of MDA-MB 231 cells infected with lentivirus expressing Flag-tagged HIC1. ChIP assays were performed using indicated antibodies, and PCR was performed with primers specific for a region encompassed the STAT3 or Sp1 binding site in the promoter regions of c-Myc and VEGF genes. The semi-quantitative PCR products were resolved on 1.5% agarose gel (top panel). Bar graph shows the ChIP real-time qPCR data with anti-STAT3 or control antibody (bottom panel). Error bars, standard deviation; n = 3. “Input” represents 1% of the chromatin used for the immunoprecipitation with indicated antibodies. (E) ChIP real-time qPCR analyses of WI38 cells infected with lentivirus expressing shHIC1 for 24 h, serum starved for additional 24 h and then treated with OSM (20 ng/ml; 30 min). ChIP assays were performed using anti-STAT3 antibody and qPCR was performed with primers specific for a region encompassed the STAT3 binding site in the promoter regions of c-Myc and VEGF genes or for the exon region of GAPDH as a negative control. Error bars, standard deviation; n = 3. “Input” represents 1% of the chromatin used for the immunoprecipitation with anti-STAT3 antibody.
We further tested whether HIC1 affects STAT3 DNA binding activity. EMSAs were performed using nuclear extracts from OSM-treated 293T cells with or without Flag-tagged HIC1 overexpression. OSM treatment induced a DNA/protein complex (Fig. 4C, lane 3, arrow), which could be supershifted by adding anti-STAT3 antibody (lane 6). HIC1 attenuated the STAT3-DNA complex formation in a dosage-dependent manner (lanes 3–5). These results suggest that HIC1 may interfere with STAT3 DNA binding activity. To further test whether HIC1 reduces STAT3 DNA binding in cells, MDA-MB 231 cells infected with lentivirus expressing HIC1 were subjected to chromatin immunoprecipitation for analysis of endogenous STAT3 bound to its target gene promoter. HIC1 overexpression significantly attenuated endogenous STAT3 recruitment to the promoter regions of VEGF and c-Myc (Fig. 4D, top panel, lanes 5 and 6). It should be noted that the effect of HIC1 on STAT3 inhibition is specific, because Sp1 recruitment to both promoter regions was not significantly altered by HIC1 expression (lanes 11 and 12). We further quantified the effect of HIC1 on STAT3 recruitment to both promoter regions by real-time quantitative PCR analysis (Fig. 4D, bottom panel). In contrast, depletion of endogenous HIC1 in WI-38 cells increased OSM-elicited STAT3 binding to VEGF and c-Myc promoter regions (Fig. 4E). Altogether, these results suggest that HIC1 represses STAT3 transcriptional activity by reducing STAT3 binding to its target DNA sequences.
HIC1 interacts with STAT3 DNA binding domain
The results of HIC1 interrupting the STAT3 DNA binding capacity raised a possibility that HIC1 binds to the DNA binding domain of STAT3. To test this possibility, a series of STAT3 truncation mutants were generated and tested for its interaction with HIC1 (Fig. 5A). The results of immunoprecipitation experiments revealed that STAT3 fragments (1–490), (318–770), but not (1–317), could precipitate HIC1 like WT (Fig. 5A, lanes 2–5), suggesting that the DNA binding domain (318–490) is required for STAT3 binding to HIC1. As expected, STAT3 (318–490) itself was sufficient for HIC1 interaction (lane 6). These results further suggest that HIC1 binds to STAT3 DNA binding domain, thereby attenuating STAT3 DNA binding ability.

Figure 5. Interaction domain mapping of HIC1 and STAT3 (A) Schematic presentation of STAT3 domains and deletion mutants used in this study. The interaction of each STAT3 deletion mutant with HIC1 is indicated (top panel). Western blots show immunoprecipitation of 293T cells expressing HA-tagged STAT3 deletion mutants and Flag-tagged HIC1 with anti-HA antibody (bottom panel). (B) Schematic presentation of HIC1 domains and deletion mutants used in this study. The interaction of each HIC1 deletion mutant with STAT3 is indicated. BTB, BTB-POZ domain; CR, central region; ZF, zinc finger domain. Western blots show the immunoprecipitation of 293T cells expressing HA-tagged HIC1 deletion mutants and STAT3 by anti-HA or STAT3 antibody. (C) Phosphoimager analysis of the EMSA gel showing 32P-labeled SIE oligonucleotide with nuclear extracts from 293T cells transfected with HA-STAT3 and treated with OSM (20 ng/ml; 30 min) incubated with in vitro-synthesized HIC1 wild-type or (1–420) mutant fragment proteins or anti-STAT3 for supershift as indicated. The western blot shows the levels of indicated proteins added to the nuclear extracts for EMSA (input). The intensity of STAT3/SIE complex was quantified by densitometry and the relative binding of STAT3 to SIE is indicated after normalization to the sample with OSM treatment taken as 1.
HIC1 C-terminal domain is essential for STAT3 interaction and activity regulation
If HIC1 binding to STAT3 is important for HIC1 in suppressing STAT3 activity, HIC1 mutant defective in STAT3 interaction would attenuate its repressive effect on STAT3 DNA binding activity, target gene expression and cell growth. We then mapped the domain of HIC1 involved in STAT3 interaction. Several HIC1 deletion mutants including fragment (1–140), (141–714), (420–714), and (1–420) were generated and tested for their capacities to associate with endogenous STAT3 in 293T cells (Fig. 5B). The results of immunoprecipitation experiments showed that HIC1 truncation mutants (141–714) and (420–714), like WT, could precipitate endogenous STAT3, while HIC1 mutants (1–140) and (1–420) failed to do so (Fig. 5B). These results suggest that HIC1 C-terminal fragment (420–714) containing the zinc finger domain is responsible for STAT3 interaction.
We next tested whether the C-terminal domain deletion mutant (1–420) reduces HIC1 effect on STAT3 DNA binding activity. HIC1 wild-type and (1–420) fragment proteins synthesized by in vitro transcription and translation reactions were subjected to EMSA with the nuclear extracts of STAT3-transfected 293T cells with OSM treatment. Notably, HIC1 (1–420) fragment protein failed to inhibit STAT3/SIE complex formation as compared with wild-type (Fig. 5C, lanes 2–4). These results further support the notion that HIC1 attenuates STAT3 DNA binding activity via its interaction with STAT3. In line with EMSA study, reporter gene assays revealed that HIC1 (1–420) fragment could not suppress the reporter activity of the VEGF-Luc or c-Myc-Luc in MDA-MB 231 cells, compared with wild-type HIC1 (Fig. 6A). Accordingly, real-time quantitative PCR analyses showed that this HIC1 mutant was not effective in downregulating VEGF and c-Myc expressions compared with WT (Fig. 6B, left panel). Similar results were made by western analysis in detecting c-Myc and VEGF protein levels (right panel). Consistent with these observations, expression of HIC1 mutant (1–420) protein reduced the inhibitory effect of HIC1 on MDA-MB 231 cell growth (Fig. 6C). Collectively, these results provide a nice correlation of HIC1 in STAT3 interaction and activity regulation.
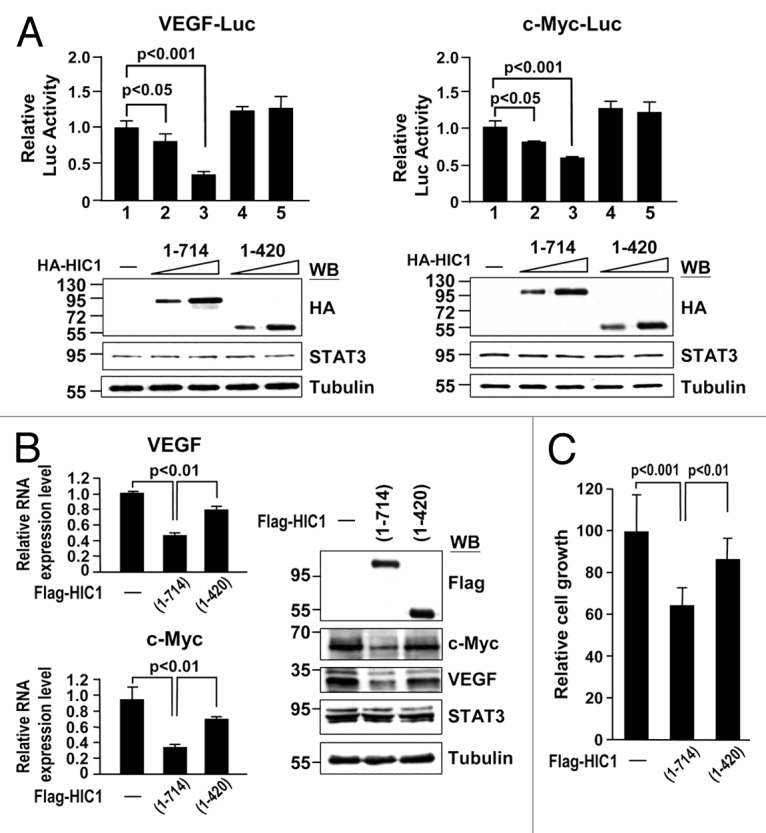
Figure 6. HIC1 C-terminal domain is important to suppress VEGF and c-Myc gene expression and cell growth (A) Reporter gene activity of MDA-MB 231 cells transfected with indicated reporter along with increasing amounts of HIC1 WT or (1–420) mutant. Error bars represent the mean ± SD (n = 3). Western blots show the expression levels of HIC1 WT and (1–420) mutant in samples. (B) Real-time qPCR and western analyses of VEGF and c-Myc mRNAs and protein levels from MDA-MB 231 cells infected with lentivirus expressing Flag-tagged HIC1 WT or (1–420) mutant. Error bars represent the mean ± SD (n = 3). (C) Cell growth analysis of MDA-MB 231 cells infected with indicated lentivirus expression constructs for 24 h and incubated for 5 d and followed by MTT assays. Error bars represent the mean ± SD (n = 3).
Discussion
While HIC1 was identified as a tumor-suppressor gene, the inhibitory mechanisms of tumor progression are largely unclear. The molecular basis of HIC1 in suppressing tumor progression was thought to be through its target gene regulation and/or protein-protein interactions. Previous reports showed that HIC1 downregulates target genes including SIRT1, ephrin-A1, EphA2, cyclin D1, P57KIP2, CXCR7, and ADRB2, which are associated with tumor growth and apoptosis as well as cell migration and invasion.7,39-43 Re-expression of HIC1 target genes such as ephrin-A1, EphA2, or ADRB2 partially rescued the negative effects on tumor growth, cell migration, and invasion induced by HIC1 overexpression.39,40,43 These observations suggest that other HIC1 regulated genes and/or pathways are also important for HIC1-mediated downregulation of cell proliferation, migration, and invasion. In this study, we identified STAT3 as one of HIC1-interacting proteins and demonstrated HIC1 in downregulation of STAT3-mediated VEGF and c-Myc expressions and promoter activities as well as cell growth. Previous studies showed that STAT3 is crucial for IL-6-induced VEGF expression in promoting tumor growth and angiogenesis,44 and OSM-induced gene expression including c-Myc in cell proliferation.26 Our findings that HIC1 suppresses VEGF and c-Myc expression, and cell growth via inhibition of STAT3 activity provide an additional molecular basis associated with HIC1 tumor suppressor activity.
Several lines of evidence indicate that STAT3 activation by cytokines such as OSM is not required for HIC1-STAT3 interaction. First, in vitro-synthesized HIC1 protein could bind to STAT3 recombinant protein (Fig. 1D). Second, the STAT3 DNA binding domain (318–490), lacking of other activation modifications such as Y705 phosphorylation, S727 phosphorylation, and K685 acetylation, was sufficient to associate with HIC1 (Fig. 5A). Third, a parallel increment of HIC1 precipitated by increased levels of STAT3 from the nuclear extracts of OSM-treated cells was observed in the immunoprecipitation experiments (Fig. 1C, right panel, lane 5 vs. 6), suggesting that OSM stimulation simply affects the STAT3 nuclear translocation for HIC1 interaction, instead of HIC1-STAT3 interaction per se. In addition, while HIC1 and STAT3 could form complexes in vitro (Fig. 1D), we cannot exclude the possibility that some other factor(s) may stabilize or facilitate the complex formation of HIC1 and STAT3 in cells.
Our data that HIC1 interacts with STAT3 and inhibits STAT3 DNA binding activity resembles the scenario that HIC1 binds to TCF4 and prevents TCF4 binding to its target gene promoters in cells. Although the loss of both factors in association with target gene promoters is similar, the molecular mechanisms underlying these two cases are distinct. Domain mapping studies showed that the BTB/POZ and C-terminal domains are responsible for HIC1 binding to TCF49 and STAT3 (Fig. 5B), respectively. Accordingly, the BTB/POZ or C-terminal domain deletion mutant conferred reduced levels of HIC1 in downregulation of TCF4- or STAT3-mediated reporter activity, respectively. While the BTB/POZ domain is crucial for HIC1 oligomerization to form nuclear bodies and sequester TCF4 into the HIC1 bodies, this domain is dispensable for STAT3 interaction (Fig. 5B) and not required for STAT3 activity inhibition as evidenced by data that the HIC1 C-terminal domain (420–714) able to bind STAT3 (Fig. 5B) is sufficient to repress STAT3-mediated promoter activity (unpublished data). Because HIC1 (420–714) cannot form HIC1 nuclear bodies, these findings suggest that HIC1 attenuated STAT3 DNA binding activity is distinct from the molecular mechanism of TCF4 sequestration by HIC1 bodies.
Previous study of microarray analysis from HIC1-overexpressed cells revealed that HIC1 downregulated the expression of leukemia inhibitory factor (LIF) in U2OS osteosarcoma cells.42 Since LIF, one of the IL-6-type cytokines, can activate JAK/STAT3 pathway,45 this finding led to a possibility that HIC1 overexpression can reduce STAT3 Tyr 705 phosphorylation activated by LIF. However, we observed that HIC1 mainly altered STAT3 DNA binding activity but not STAT3 Tyr 705 phosphorylation in all the experimental conditions. Although we cannot exclude the possibility that HIC1 downregulates STAT3 pathway via inhibition of LIF expression, the data that HIC1 could attenuate the transactivation potential of the constitutively active form STAT3-CA (Fig. 2B) support the molecular basis of HIC1 in attenuating STAT3-mediated pathway via inhibition of STAT3 DNA binding activity.
STAT3 is frequently activated in a broad range of human cancers.11,14-17 According to our current finding, the loss of HIC1 expression in the majority of cancers by hypermethylation on HIC1 promoter may result in an enhancement of the STAT3 activity and thereby promote oncogenesis. Thus, increasing HIC1 expression and/or HIC1-STAT3 interaction may provide a potential therapeutic strategy in antagonizing STAT3-associated human cancers. In addition, while HIC1 could attenuate STAT3 target gene promoter activity and gene expression, our findings that HIC1 C-terminal DNA binding domain can interact with STAT3 also raised a possibility that STAT3 may modulate HIC1 DNA binding activity and target gene expression. Whether such interaction and regulation contribute to STAT3 oncogenic potential requires further study.
In summary, we identified STAT3 as a novel HIC1-interacting protein and showed that HIC1 negatively regulates STAT3-dependent transcription, VEGF and c-Myc gene expression, and cell growth through decreasing STAT3 DNA binding potential. These findings provide a possible mechanism that HIC1 acts as a tumor suppressor to counteract STAT3 oncogenic potential and also a molecular basis underlying the inhibitory effect of HIC1 on STAT3-dependent transcription.
Materials and Methods
Plasmids, antibodies, and reagents
pcDNA3-Flag-HIC1, 3X Ly6E-Luc, VEGF-Luc, and c-Myc-Luc reporter constructs were described previously.46-49 For the lentivirus construct expressing Flag-HIC1, HIC1 cDNA was subcloned into pLKO AS3W-puro (RNAi consortium at Academia Sinica). The cDNAs coding HIC1 WT, (1–140), (141–714), (420–714), and (1–420) were generated by PCR and inserted into pcDNA3-HA vector for expressing corresponding HA-tagged HIC1 WT or truncated proteins. The lentivirus expressing shHIC1-1 or shHIC1-2, both 21-nucleotide sequences corresponding to coding sequences 1473–1493 and 74–94 of HIC1, respectively, inserted into pLKO.1-puro and the pLKO.1-Luc obtained from the RNAi consortium at Academia Sinica. The 21 nucleotides of human STAT3 sequence, 5′-GCACAATCTACGAAGAATCAA-3′, corresponding to coding sequence 240–260, were subcloned into pLKO.1-puro vector to generate the lentivirus expressing shSTAT3. pcDNA3-HA-STAT3 (1–770), (1–317), (318–770) constructs were kindly provided by Dr Jeou-Yuan Chen at Academia Sinica, while pcDNA3-HA-STAT3 (1–490) and (318–490) were generated by inserting the corresponding coding cDNAs into pcDNA3-HA vector. pGEX-4T-STAT3 was made by inserting STAT3 cDNA into pGEX-4T-2 vector. All constructs were verified by DNA sequencing analyses. The antibodies and cytokines are indicated as following: anti-Flag M2, anti-tubulin, and anti-HIC1 (Sigma), anti-HA (Covance), anti-STAT3, anti-Sp1, and control rabbit IgG (Santa Cruz Biotechnology), anti-phospho-STAT3 (Y705), anti-phospho-STAT3 (S727), and anti-acetyl STAT3 (K685) (Cell Signaling), IL-6 and Oncostatin M (PeproTech).
Cell culture, transfection, reporter assays, and real-time qPCR
293T and MCF7 cells were maintained in Dulbecco modified Eagle’s medium (DMEM) and MDA-MB 231 and WI-38 cells were maintained in RPMI1640 medium and minimum essential medium (MEM) supplemented with 1 mM sodium pyruvate, 1× nonessential amino acids, 2 mM L-glutamine, 10% FBS, and 100 μg/ml streptomycin/penicillin, respectively. Transfections were performed by using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. For reporter gene assays, 5 × 104 cells plated in 24-well were transfected with indicated expression vectors along with 250 ng of 3× Ly6E-Luc, VEGF-Luc, or c-Myc-Luc reporter construct and 50 ng of pRL-TK as an indicator for normalization of transfection efficiency. The luciferase activities were measured using Dual-Glo Luciferase Assay System (Promega). For RT-PCR, 2 × 106 MDA-MB 231 cells were infected with HIC1 lentivirus for 24 h and followed by puromycin (2 μg/ml) selection for 3 d, or 5 × 105 WI-38 cells were infected with shHIC1 lentivirus for 24 h, serum starved for additional 24 h, and with or without 20 ng/ml OSM treatment for 2 h. Total cellular RNAs from these cells were extracted using TRIzol reagent (Invitrogen), and 2.5 μg total RNA of each sample was then reverse transcribed by ThermoScript RT-PCR system (Invitrogen). Real-time qPCR analysis was performed using SYBR Green PCR master mix (Applied Biosystems) and ABI 7500 sequence detection system (Applied Biosystems). The following specific primers were used for PCR amplification: c-Myc, forward: 5′-TGA AAG GCT CTC CTT GCA GC-3′ and reverse: 5′-GCT GGT AGA AGT TCT CCT CC-3′; VEGF, forward: 5′-CGC AAG AAA TCC CGG TAT AA-3′; and reverse: 5′-TCT CCG CTC TGA GCA AGG-3′ and GAPDH, forward: 5′-TCT TTT GCG TCG CCA GCC GAG-3 and reverse: 5′-TGA CCA GGC GCC CAA TAC GAC-3′.
Colony-formation assay and cell growth analysis
For colony-formation assay, 2 × 106 MDA-MB 231 cells were infected with lentivirus empty vector or expressing shSTAT3 for 24 h and followed by another infection with lentivirus empty vector or expressing Flag-HIC1 for additional 24 h. Five hundred lentivirus-infected MDA-MB231 cells from each set were seeded in 6 cm cultured dish with 2 μg/ml puromycin selection. Cells were allowed to grow for 14 d to form colonies, which were then stained with crystal violet (Sigma) and counted. For cell growth analysis, 2 × 106 MDA-MB 231 cells were infected with lentivirus expressing HIC1 wild-type or mutant for 24 h. The resulting cells were re-plated in a 96-well plate at 1000 cells/well and further incubated for 5 d. The cell growth was evaluated by the MTT assay. Briefly, cells were washed twice with PBS and 50 μl of 1 mg/ml MTT (Sigma) solution was added to each well for 4 h incubation at 37 °C. The formazan crystals were dissolved using 150 μl of DMSO and absorbance was measured at 595 nm on a multiwell plate reader (Bio-Rad).
Immunoprecipitation, western blot, and GST pull-down analyses-
Transfected or infected cells were lysed with a modified RIPA buffer (50 mM Tris pH7.8, 150 mM NaCl, 5 mM EDTA, 0.5% Triton X-100, 0.5% Nonidet P-40, and 0.1% sodium deoxycholate) supplemented with protease inhibitor mixture (Roche). Cell lysates were subjected to immunoprecipitation and western analyses as described previously.50 GST pull-down assays were performed using 3 µg recombinant GST or GST-STAT3 fusion proteins immobilized to glutathione agarose beads (Pierce) in binding buffer (10 mM Hepes pH 7.5, 0.5 mM EDTA, 50 mM NaCl, 0.1% NP-40, and 0.5 mM DTT) along with in vitro-synthesized HIC1 proteins for 2 h. After binding and washing, bound proteins were fractionated by SDS-PAGE and analyzed by western blot. For identification of STAT3 by LC-MS/MS analysis, the procedures were performed as described previously.51 In brief, 293T cells transfected with Flag-HIC1 were lysed with a lysis buffer (50 mM Hepes pH7.5, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 1 mM DTT, protease inhibitor) and immunoprecipitated with anti-Flag conjugated agarose beads for 2 h at 4 °C. The resulting bound proteins were resolved by SDS-PAGE and followed by SYPRO-RUBY staining. The gel piece around 90 kDa was excised and subjected to in-gel digestion with trypsin. The resulting digested peptides were desalted using C18 Zip-Tip (Millipore) and eluted with 50% acetonitrile (ACN)/0.1% trifluoroacetic acid (TFA) and further analyzed by LC-MS/MS.
Electrophoretic mobility shift assay
Human high affinity sis inducible element (hSIE) (5′-AGC TTC ATT TCC CGT AAA TCC CTA AAG CT-3′) that derived from the c-fos gene promoter52 was synthesized and annealed to form a double strand. EMSA was performed as described with slight modifications.53 Five μg nuclear extract were incubated with 32P-labeled oligonucleotide probe in binding buffer (10 mM HEPES, pH7.9, 80 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5 μg poly-dIdC, and 1 mM DTT) for 30 min at room temperature. For HIC1 (1–420) mutant defective in reducing STAT3/SIE complex formation experiments, HIC1 wild-type, and (1–420) fragment proteins separately synthesized by TNT® Quick Coupled transcription/Translation system (Promega). Equal amount of synthesized WT and (1–420) proteins in TNT lysates was added to the indicated nuclear extracts for the incubation of DNA-protein complex binding reaction. For supershift assay, the nuclear extracts were preincubated with anti-STAT3 antibody for 20 min before addition of SIE probe. The DNA–protein complexes were resolved by electrophoresis on 5% non-denaturing polyacrylamide gel in 0.25× TBE buffer at 180 V for 6 h. Gels were dried, exposed, and analyzed by PhosphorImager (Molecular Dynamics).
Chromatin immunoprecipitation
ChIP experiments were performed as described.54 5 × 106 HIC1-lentivirus infected MDA-MB231 or 5 × 107 shHIC1-1 lentivirus infected WI-38 cells were subjected to ChIP procedure using anti-STAT3, anti-Sp1 or a control anti-IgG antibody for chromatin fragment precipitation. The following primers were used for PCR amplification: STAT3 binding site on c-Myc promoter, forward: 5′-AAA AGG GGA AAG AGG ACC TGG-3′ and reverse: 5′-TAA AAG GGG CAA GTG GAG AGC-3′ ; STAT3 binding site on VEGF promoter, forward: 5′-CTG GCC TGC AGA CAT CAA AGT GAG-3′ and reverse: 5′-CTT CCC GTT CTC AGC TCC ACA AAC-3′; Sp1 binding site on c-Myc promoter, forward: 5′-CTT GGC GGG AAA AAG AAC-3′ and reverse: 5′-GCT GTA GTA ATT CCA GCG AG-3′ ; Sp1 binding site on VEGF promoter, forward: 5′-GGT CGA GCT TCC CCT TCA-3′ and reverse: 5′- TGC CCC AAG CCT CCG CGA TCC TC-3′ and GAPDH exon region, forward: 5′-CTA CTG GCG CTG CCA AGG CT-3′ and reverse: 5′-ACG TTG GCA GTG GGG ACA CG-3′. The ChIP product was used for semi-quantitative PCR and real-time qPCR analyses. For semi-quantitative PCR analysis, the PCR products were then resolved by 1.5% agarose gel containing ethidium bromide and visualized under UBV light. The real-time qPCR was performed using the Applied Biosystem PRISM 7500 Real-Time qPCR System with SYBR green dye (Applied Biosystems) for detection. For each sample, the average threshold (Ct) value was resulted from triplicate assays. A fraction (1%) of the sonicated chromatin was set aside as input control before the antibody affinity manipulations. Percent input was calculated by 100 × 2^(Ctadjusted Input - CtIP).
Statistical analysis
Data from reporter gene assays, cell growth, ChIP-qPCR, and real-time qPCR analyses in this study are expressed as the mean ± standard deviation (SD). Statistical significance between different experimental groups was ascertained using Student t test.
Acknowledgments
We thank Dr Jeou-Yuan Chen for STAT3 WT and deletion mutant constructs. This work was supported by NSC Frontier Grant NSC101-2321-B-001-029 and an Academia Sinica Investigator Award to H-M Shih.
Glossary
Abbreviations:
- HIC1
hypermethylated in cancer 1
- STAT3
signal transducers and activators of transcription 3
- OSM
oncostatin M
- TCF
T-cell factor
- ChIP
chromatin immunoprecipitation
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25365
References
- 1.Wales MM, Biel MA, el Deiry W, Nelkin BD, Issa JP, Cavenee WK, et al. p53 activates expression of HIC-1, a new candidate tumour suppressor gene on 17p13.3. Nat Med. 1995;1:570–7. doi: 10.1038/nm0695-570. [DOI] [PubMed] [Google Scholar]
- 2.Boulay G, Dubuissez M, Van Rechem C, Forget A, Helin K, Ayrault O, et al. Hypermethylated in cancer 1 (HIC1) recruits polycomb repressive complex 2 (PRC2) to a subset of its target genes through interaction with human polycomb-like (hPCL) proteins. J Biol Chem. 2012;287:10509–24. doi: 10.1074/jbc.M111.320234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deltour S, Pinte S, Guerardel C, Wasylyk B, Leprince D. The human candidate tumor suppressor gene HIC1 recruits CtBP through a degenerate GLDLSKK motif. Mol Cell Biol. 2002;22:4890–901. doi: 10.1128/MCB.22.13.4890-4901.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stankovic-Valentin N, Verger A, Deltour-Balerdi S, Quinlan KG, Crossley M, Leprince DA. A L225A substitution in the human tumour suppressor HIC1 abolishes its interaction with the corepressor CtBP. FEBS J. 2006;273:2879–90. doi: 10.1111/j.1742-4658.2006.05301.x. [DOI] [PubMed] [Google Scholar]
- 5.Van Rechem C, Boulay G, Pinte S, Stankovic-Valentin N, Guérardel C, Leprince D. Differential regulation of HIC1 target genes by CtBP and NuRD, via an acetylation/SUMOylation switch, in quiescent versus proliferating cells. Mol Cell Biol. 2010;30:4045–59. doi: 10.1128/MCB.00582-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinte S, Stankovic-Valentin N, Deltour S, Rood BR, Guérardel C, Leprince D. The tumor suppressor gene HIC1 (hypermethylated in cancer 1) is a sequence-specific transcriptional repressor: definition of its consensus binding sequence and analysis of its DNA binding and repressive properties. J Biol Chem. 2004;279:38313–24. doi: 10.1074/jbc.M401610200. [DOI] [PubMed] [Google Scholar]
- 7.Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123:437–48. doi: 10.1016/j.cell.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 8.Dehennaut V, Loison I, Dubuissez M, Nassour J, Abbadie C, Leprince D. DNA double-strand breaks lead to activation of hypermethylated in cancer 1 (HIC1) by SUMOylation to regulate DNA repair. J Biol Chem. 2013;288:10254–64. doi: 10.1074/jbc.M112.421610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valenta T, Lukas J, Doubravska L, Fafilek B, Korinek V. HIC1 attenuates Wnt signaling by recruitment of TCF-4 and beta-catenin to the nuclear bodies. EMBO J. 2006;25:2326–37. doi: 10.1038/sj.emboj.7601147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jove R. Preface: STAT signaling. Oncogene. 2000;19:2466–7. doi: 10.1038/sj.onc.1203549. [DOI] [PubMed] [Google Scholar]
- 11.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levy DE, Inghirami G. STAT3: a multifaceted oncogene. Proc Natl Acad Sci USA. 2006;103:10151–2. doi: 10.1073/pnas.0604042103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/S0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 14.Darnell JE., Jr. Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2:740–9. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 15.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 16.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–88. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 17.Calò V, Migliavacca M, Bazan V, Macaluso M, Buscemi M, Gebbia N, et al. STAT proteins: from normal control of cellular events to tumorigenesis. J Cell Physiol. 2003;197:157–68. doi: 10.1002/jcp.10364. [DOI] [PubMed] [Google Scholar]
- 18.Bowman T, Broome MA, Sinibaldi D, Wharton W, Pledger WJ, Sedivy JM, et al. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc Natl Acad Sci USA. 2001;98:7319–24. doi: 10.1073/pnas.131568898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grandis JR, Drenning SD, Zeng Q, Watkins SC, Melhem MF, Endo S, et al. Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc Natl Acad Sci USA. 2000;97:4227–32. doi: 10.1073/pnas.97.8.4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21:2000–8. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- 21.Shao H, Cheng HY, Cook RG, Tweardy DJ. Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer Res. 2003;63:3923–30. [PubMed] [Google Scholar]
- 22.Kermorgant S, Parker PJ. Receptor trafficking controls weak signal delivery: a strategy used by c-Met for STAT3 nuclear accumulation. J Cell Biol. 2008;182:855–63. doi: 10.1083/jcb.200806076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Darnell JE., Jr. STATs and gene regulation. Science. 1997;277:1630–5. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 24.Schindler C, Darnell JE., Jr. Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annu Rev Biochem. 1995;64:621–51. doi: 10.1146/annurev.bi.64.070195.003201. [DOI] [PubMed] [Google Scholar]
- 25.Wang R, Cherukuri P, Luo J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J Biol Chem. 2005;280:11528–34. doi: 10.1074/jbc.M413930200. [DOI] [PubMed] [Google Scholar]
- 26.Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269–73. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 27.Wormald S, Hilton DJ. Inhibitors of cytokine signal transduction. J Biol Chem. 2004;279:821–4. doi: 10.1074/jbc.R300030200. [DOI] [PubMed] [Google Scholar]
- 28.Irie-Sasaki J, Sasaki T, Matsumoto W, Opavsky A, Cheng M, Welstead G, et al. CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature. 2001;409:349–54. doi: 10.1038/35053086. [DOI] [PubMed] [Google Scholar]
- 29.Myers MP, Andersen JN, Cheng A, Tremblay ML, Horvath CM, Parisien JP, et al. TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J Biol Chem. 2001;276:47771–4. doi: 10.1074/jbc.C100583200. [DOI] [PubMed] [Google Scholar]
- 30.ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, Zhu W, Tremblay M, David M, et al. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol. 2002;22:5662–8. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Starr R, Hilton DJ. Negative regulation of the JAK/STAT pathway. Bioessays. 1999;21:47–52. doi: 10.1002/(SICI)1521-1878(199901)21:1<47::AID-BIES6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 32.Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, et al. Specific inhibition of Stat3 signal transduction by PIAS3. Science. 1997;278:1803–5. doi: 10.1126/science.278.5344.1803. [DOI] [PubMed] [Google Scholar]
- 33.Muromoto R, Nakao K, Watanabe T, Sato N, Sekine Y, Sugiyama K, et al. Physical and functional interactions between Daxx and STAT3. Oncogene. 2006;25:2131–6. doi: 10.1038/sj.onc.1209235. [DOI] [PubMed] [Google Scholar]
- 34.Muromoto R, Kuroda M, Togi S, Sekine Y, Nanbo A, Shimoda K, et al. Functional involvement of Daxx in gp130-mediated cell growth and survival in BaF3 cells. Eur J Immunol. 2010 doi: 10.1002/eji.201040688. [DOI] [PubMed] [Google Scholar]
- 35.Yamashina K, Yamamoto H, Chayama K, Nakajima K, Kikuchi A. Suppression of STAT3 activity by Duplin, which is a negative regulator of the Wnt signal. J Biochem. 2006;139:305–14. doi: 10.1093/jb/mvj033. [DOI] [PubMed] [Google Scholar]
- 36.Nishimoto A, Yu Y, Lu Z, Mao X, Ren Z, Watowich SS, et al. A Ras homologue member I directly inhibits signal transducers and activators of transcription 3 translocation and activity in human breast and ovarian cancer cells. Cancer Res. 2005;65:6701–10. doi: 10.1158/0008-5472.CAN-05-0130. [DOI] [PubMed] [Google Scholar]
- 37.Tsuruma R, Ohbayashi N, Kamitani S, Ikeda O, Sato N, Muromoto R, et al. Physical and functional interactions between STAT3 and KAP1. Oncogene. 2008;27:3054–9. doi: 10.1038/sj.onc.1210952. [DOI] [PubMed] [Google Scholar]
- 38.Sekine Y, Tsuji S, Ikeda O, Sato N, Aoki N, Aoyama K, et al. Regulation of STAT3-mediated signaling by LMW-DSP2. Oncogene. 2006;25:5801–6. doi: 10.1038/sj.onc.1209578. [DOI] [PubMed] [Google Scholar]
- 39.Zhang W, Zeng X, Briggs KJ, Beaty R, Simons B, Chiu Yen RW, et al. A potential tumor suppressor role for Hic1 in breast cancer through transcriptional repression of ephrin-A1. Oncogene. 2010;29:2467–76. doi: 10.1038/onc.2010.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Foveau B, Boulay G, Pinte S, Van Rechem C, Rood BR, Leprince D. The receptor tyrosine kinase EphA2 is a direct target gene of hypermethylated in cancer 1 (HIC1) J Biol Chem. 2012;287:5366–78. doi: 10.1074/jbc.M111.329466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Rechem C, Boulay G, Pinte S, Stankovic-Valentin N, Guérardel C, Leprince D. Differential regulation of HIC1 target genes by CtBP and NuRD, via an acetylation/SUMOylation switch, in quiescent versus proliferating cells. Mol Cell Biol. 2010;30:4045–59. doi: 10.1128/MCB.00582-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Rechem C, Rood BR, Touka M, Pinte S, Jenal M, Guérardel C, et al. Scavenger chemokine (CXC motif) receptor 7 (CXCR7) is a direct target gene of HIC1 (hypermethylated in cancer 1) J Biol Chem. 2009;284:20927–35. doi: 10.1074/jbc.M109.022350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boulay G, Malaquin N, Loison I, Foveau B, Van Rechem C, Rood BR, et al. Loss of Hypermethylated in Cancer 1 (HIC1) in breast cancer cells contributes to stress-induced migration and invasion through β-2 adrenergic receptor (ADRB2) misregulation. J Biol Chem. 2012;287:5379–89. doi: 10.1074/jbc.M111.304287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu Q, Briggs J, Park S, Niu G, Kortylewski M, Zhang S, et al. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene. 2005;24:5552–60. doi: 10.1038/sj.onc.1208719. [DOI] [PubMed] [Google Scholar]
- 45.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stankovic-Valentin N, Deltour S, Seeler J, Pinte S, Vergoten G, Guérardel C, et al. An acetylation/deacetylation-SUMOylation switch through a phylogenetically conserved psiKXEP motif in the tumor suppressor HIC1 regulates transcriptional repression activity. Mol Cell Biol. 2007;27:2661–75. doi: 10.1128/MCB.01098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wen Z, Zhong Z, Darnell JE., Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–50. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 48.Hsieh YL, Kuo HY, Chang CC, Naik MT, Liao PH, Ho CC, et al. Ubc9 acetylation modulates distinct SUMO target modification and hypoxia response. EMBO J. 2013;32:791–804. doi: 10.1038/emboj.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chao TT, Chang CC, Shih HM. SUMO modification modulates the transrepression activity of PLZF. Biochem Biophys Res Commun. 2007;358:475–82. doi: 10.1016/j.bbrc.2007.04.157. [DOI] [PubMed] [Google Scholar]
- 50.Lin DY, Lai MZ, Ann DK, Shih HM. Promyelocytic leukemia protein (PML) functions as a glucocorticoid receptor co-activator by sequestering Daxx to the PML oncogenic domains (PODs) to enhance its transactivation potential. J Biol Chem. 2003;278:15958–65. doi: 10.1074/jbc.M300387200. [DOI] [PubMed] [Google Scholar]
- 51.Chang CC, Naik MT, Huang YS, Jeng JC, Liao PH, Kuo HY, et al. Structural and functional roles of Daxx SIM phosphorylation in SUMO paralog-selective binding and apoptosis modulation. Mol Cell. 2011;42:62–74. doi: 10.1016/j.molcel.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 52.Wagner BJ, Hayes TE, Hoban CJ, Cochran BH. The SIF binding element confers sis/PDGF inducibility onto the c-fos promoter. EMBO J. 1990;9:4477–84. doi: 10.1002/j.1460-2075.1990.tb07898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen KY, Huang LM, Kung HJ, Ann DK, Shih HM. The role of tyrosine kinase Etk/Bmx in EGF-induced apoptosis of MDA-MB-468 breast cancer cells. Oncogene. 2004;23:1854–62. doi: 10.1038/sj.onc.1207308. [DOI] [PubMed] [Google Scholar]
- 54.Huang YS, Chang CC, Huang TC, Hsieh YL, Shih HM. Daxx interacts with and modulates the activity of CREB. Cell Cycle. 2012;11:99–108. doi: 10.4161/cc.11.1.18430. [DOI] [PubMed] [Google Scholar]