

Abstract
The directed migration of mammalian cells is a foundation of development and growth. A variety of processes such as tissue development, wound healing, pathogen recognition/destruction as well as cancer metastasis are the result of regulated or dysregulated cell migration. While the ability to measure a cell's propensity to migrate has clinical relevance in several settings, no universal protocol has been established to measure cell migration. A variety of techniques are currently used to measure migration including manual counting, flow cytometry or Coulter counting, microfluidic devices, computerized spectroscopic methods, or the use of various tracking dyes interfaced with fluorescent or non-fluorescent plate readers. In order to expedite the measurement of migration, we compared several common cytoplasmic and lipophilic cell tracking dyes to determine the best dye for determining migration of rare population of cells. CellVue® Burgundy was found to be superior over calcein AM, Cell Tracker Green CMFDA (chloromethyl fluorescein diacetate), Vybrant CFDA (carboxy fluorescein diacetate succinimidyl ester) in its retention within cells, superior to CellVue® NIR 815, PKH67, and CM DiI with regard to signal to noise ratio, and superior to PKH26 with regard to instrument versatility.
INTRODUCTION
The directed migration of mammalian cells is a foundation of development and growth. A variety of processes such as tissue development, wound healing, pathogen recognition/destruction as well as cancer metastasis are the result of regulated or dysregulated cell migration. Although differentiated cells vary widely with respect to their function, location, and antigen expression the actual process of migration is generally similar among various cell types due to conservation of the underpinning mechanisms.
An important example of cell migration in a clinical setting would be the engraftment of hematopoietic stem/progenitor cells into the bone marrow of a patient undergoing bone marrow transplant. Successful engraftment would depend upon the transplanted cells' ability to home successfully to the bone marrow, principally in a directed fashion towards the chemokine stromal derived factor-1 (SDF-1) [1, 2]. Endothelial cell migration along gradients of locally secreted chemokines has been proposed as a means to repair or create new blood vessels in the process of angiogenesis. And in regard to endothelial health, our laboratory has evaluated the SDF-1 directed migration of endothelial progenitor cells as a potential prognostic indicator of vascular health among different patient populations. While the ability to measure a cell's propensity to migrate has clinical relevance in several settings, no universal protocol has been established to measure cell migration. A variety of techniques are currently used to measure migration including manual counting, flow cytometry or Coulter counting, microfluidic devices, computerized spectroscopic methods, or the use of various tracking dyes interfaced with fluorescent or non-fluorescent plate readers. For the measurement of migration to have clinical value, factors such as assay speed, reduced examiner bias, economy, and compatibility with electronic data bases are important considerations in assay development and many of the current methods used are tedious and time consuming. In order to expedite the measurement of migration, we tested several common cytoplasmic and lipophilic cell tracking dyes to determine the best dye for determining migration of rare population of cells.
METHODS
Cell culture and isolation
This study was approved by the IRB of the University of Florida. Human CD34+ BMDAC were harvested from peripheral blood of normal volunteers and from patients from a variety of different out-patient clinics at UF upon acquisition of signed informed consent. Blood was collected by routine venipuncture into CPT tubes containing heparin (BD Biosciences, Franklin Lakes, NJ). After centrifugation at room temperature in a swinging bucket rotor (Eppendorf USA, Westbury, NY) for 20 min at 1,500g, the mononuclear cells were diluted with PBS supplemented with 2 mM EDTA (PBS-E). The cells were centrifuged for 15 min at 300g and the cell pellet washed; this procedure was repeated once. A concentration of 3.3 × 107 peripheral blood mononuclear cells was resuspended in 100 μl PBS-E, to which 33 μl of FcR-blocking reagent and 33 μl of magnetic microbeads conjugated with an anti-CD34 antibody (CD34 Isolation Kit, Miltenyi Biotec, Auburn, CA) were added. After incubation for 30 min at 4°C, the cells were diluted in 2 mls of PBS-E. The CD34+ BMDAC were positively selected using an automated magnetic selection column autoMACS (Miltenyi Biotec). The depleted peripheral blood mononuclear cells (PBMC) from these patient samples were used in some experiments to compare dye retention propensities relative to the progenitor cell populations.
Jurkat cells, a human T cell lymphoma line (ATCC, Manassas, VA), were cultured in RPMI supplemented with 10% fetal bovine serum and 100 ug/ml penicillin and streptomycin. Cells were harvested on the day of the assay and centrifuged at 500g for 5 minutes in a swinging bucket rotor, washed and resuspended in RPMI without supplements.
Cell dyes
Calcein-AM, Cell Tracker Green CMFDA (chloromethyl fluorescein diacetate), Vybrant CFDA (carboxy fluorescein diacetate succinimidyl ester) and CM-DiI were purchased from Invitrogen, Eugene, OR. Visible lipophilic membrane dyes PKH26 and PKH67 were purchased in kit form from Sigma Aldrich, St Louis MO. Near infrared lipophilic membrane dyes CellVue Burgundy (CVB) and CellVue NIR815 (CV815) were purchased in kit form from Molecular Targeting Technologies, West Chester, PA.
Cell Labeling
Each dye was used according to manufacturer's recommendations. Concentration and excitation and emission wavelength are listed in TABLE 1.
Table1.
Dye Properties and Cell Labeling Conditions.
| Dye | Solvent | Final concentration | Excitation | Emission |
|---|---|---|---|---|
| Calcein-AM | DMSO | 12 μM | 495 | 515 |
| CMFDA | DMSO | 10 μM | 485 | 538 |
| CFDA | DMSO | 10 μM | 492 | 517 |
| CM-Dil | DMSO | 4 μM | 553 | 570 |
| PKH26 | Diluent C | 2 μM | 551 | 567 |
| PKH67 | Diluent C | 2 μM | 490 | 502 |
| CellVue Burgundy | Diluent C | 2 μM | 683 | 707 |
| CellVue NIR815 | Diluent C | 2 μM | 786 | 814 |
Cytoplasmic dyes Calcein-AM CMFDA and CFDA
Jurkat cells or PBMCs were used routinely as models to measure the behavior or suitability of different dyes. Jurkat or PBMC cells were centrifuged at 500g for five minutes and supernatants were aspirated. After precipitation the cells were resuspended in PBS and loaded with dye at the concentration listed in Table 1. The cells were incubated for 30 mins and then the cells were precipitated and washed several times in PBS before being resuspended in RPMI migration medium without supplements.
After isolation, CD34+ BMDAC used in dye suitability or migration experiments were centrifuged at 300g for 15 minutes and washed again in PBS and precipitated by centrifuging at 300g for 15 minutes.
CMDil
A 4μM solution of CM DiI in DMSO was added to approximately 1×105 PBS-washed cells and incubated for 15 mins in a humidified CO2 incubator at 37°C and then incubated for an additional 15 mins at 4°C before being pelleted, washed, and resuspended in RPMI.
Membrane Dyes: PKH26, PKH67, CVB, and CV815 cell membrane labeling kits
Lipophilic dyes partition more efficiently in the lipid bilayer when cells are suspended in a low-salt iso-osmotic buffer. All lipophilic dyes were loaded in a similar fashion. Approximately 100,000 cells previously washed in PBS or PBS-E were pelleted at 500g for 5 minutes and resuspended in 250 μl of an iso-osmotic aqueous solvent Diluent C provided as part of the cell membrane labeling kits (Sigma Chemicals, St Louis MO). PKH67 was tested at 2 μM and 6 μM concentrations. PKH26 was tested at 4 μM concentrations. Cellvue Dyes were tested at 2 μM concentration routinely. Otherwise loading conditions were identical. An equal volume of 2× dye solution was added to the cells and incubated in the dark for 5 minutes. Two volumes of complete media (RPMI plus 10% fetal bovine serum) were added to the cells to complex with the residual dye for removal. Cells were then precipitated and washed two times in RPMI-FBS before being resuspended in RPMI migration medium.
In order to determine the signal strength of PKH67 or the CV dyes, two fold serial dilutions of dye-loaded Jurkat cells were made and the fluorescent intensities were determined in an FL600 (PKH67) or a Li-COR Odyssey NIR Imager (CV dyes).
Chemotaxis assay
Migration towards the chemokine SDF-1 was measured in a modified Boyden chamber (Neuroprobe, Gaithersburg, MD). Twenty-five μl aliquots of tenfold dilutions of SDF-1 in RPMI were added to the lower wells of a Boyden chamber which was overlaid with an 8 μm pore polycarbonate membrane (Neuroprobe) coated with 10% Bovine collagen. Fifty μl volumes of CVB labeled Jurkat or CD34+ BMDAC at concentrations of between 2000 to 10,000 cells per well were added to the top wells of the apparatus before incubating the Boyden chamber at 37° degrees for 4 hours in a CO2 incubator. At assay completion, the non-migrated material in the upper wells was aspirated and discarded. The chamber was then disassembled and 20 μl aliquots from the lower wells were removed and dispensed in wells of a 384 well black glass bottom plate (Matrical, Dot Scientific, Burton, MI). In addition, 20 μl serially diluted samples of the CVB labeled stock cell suspensions (counted on hemacytometer) were added to the 384 well plate to serve as a standard curve, and were used to estimate the migrated cell number.
Migration of CVB stained cells was quantified using a Li-COR Odyssey Near Infrared Imaging System (Li-Cor, Lincoln, NE), with image derived integrated data or relative fluorescence (RF). Migration was routinely expressed as the percent change relative to the 0 nM SDF-1 control.
Relative fluorescence could be transformed into an approximate number of migrated cells from the accompanying standard curve and migration could also be expressed as the number of cells migrating.
Flow cytometry
CVB has a fluorescent excitation/emission maximum of 683/707nm which allows the dye to be maximally detected in the FL4 detector of a FACSCalibur flow cytometer. Therefore, the migration of samples measured by the Odyssey NIR imager were compared by counting the total number of events by flow cytometry. Material from the lower wells was removed after fluorescence measured in an Odyssey Imager. The material was diluted in 100 μl of flow cytometry buffer (PBS, 0.05% NaN3 and 2% paraformaldehyde) and total events in 60 seconds were collected for each sample in the FL4 detector of a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ).
In addition, samples of CVB stained Jurkat cells were counted in a FACSCalibur after 72 h and 30 days in order to evaluate the suitability of CVB for studying cell proliferation by measuring the partitioning of dye into daughter cells.
Dye retention and limits of detection
In order to measure dye retention, cells were loaded with several different cytoplasmic dyes as well as lipophilic dye CVB in separate experiments. Cells were incubated at 37°C in a humidified CO2 incubator for approximately 3 hours. At periodic intervals, aliquots from the culture were removed, fluorescent measurement was taken and then cells were precipitated. The supernatant was removed and the cell pellet was washed in PBS. The equivalent volume of buffer was added back to the residual pellet. Equivalent volumes of supernatant and washed cell pellets were measured again to determine the percent of dye still retained in the cell compared to that which was extruded from the cells.
In parallel experiments, two fold serial dilutions were performed using dye-loaded cells in order to determine the limits of sensitivity and detection of each of the most promising dyes. Signal-to-noise ratios were determined by measuring the specific signal of dye-loaded cells at a given cell concentration compared to the signal of the same number of cells which had not been loaded with dye.
In some cases, cells were incubated for 4 hours in titrated quantities of the chemokine stromal derived factor in order to evaluate the effect of this chemokine on the signal amplification and retention of dye. Aside from the addition of SDF-1, these experiments were performed as described above.
Dye diffusion
In the course of experiments, we observed an unexpected efflux of soluble fluorescent calcein from CD34+ BMDAC within minutes after loading, which would obfuscate the interpretation of cellular migration. To measure the rate of diffusion of soluble dye across an 8 μm pore polycarbonate membrane, we performed a mock migration by placing a 10 μM solution the dye Rhodamine 110 in the top wells of a Boyden chamber. At 10 minute intervals the material in the top wells was removed by aspiration, the well washed with PBS and then the material in the lower wells was removed by puncturing the membrane and removing 20 μl from the lower well. Contents were read on a 384 well black plate at 485 nm excitation/530 nm emission.
RESULTS
Calcein extrusion
The cell permeant dye calcein- acetoxymethylester has commonly been used as a measure of cell viability because as a non-fluorescent ester it is moves freely across cell membranes and is hydrolyzed by intracellular esterases producing a fluorescent calcein anion. The fluorescence is considered to be an indication of an intact membrane and cell viability. In the course of measuring cell migration, we noted that hydrolyzed calcein-AM was not retained well in CD34+ bone marrow derived angiogenic cells (BMDAC). Although calcein provided a strong signal in a fluorescent plate reader with an apparent sensitivity to detect as few as 250 cells (Figure 1A), its use in quantitating cells was complicated by the fact that the dye appeared to be extruded from the cells rapidly, almost immediately in some cases. CD34+ BMDAC were found to extrude fluorescent calcein rapidly after cells were loaded with the colorless AM-esterified dye. As shown in Figure 1B washed calcein-AM loaded CD34+ BMDAC extruded fluorescent calcein shortly after the initial incubation period and as the RFU of calcein dye increased in the cell supernatant, the RFU in the cell pellet decreased. After 4 hours of incubation, the percent of calcein still retained inside the CD34+ BMDACs ranged from as low as 1.8% up to a high of 12.6% of total available calcein signal measured by the fluorescent plate reader in CD34+ BMDAC.
Figure 1.

A. Fluorescent measurements (485ex/530em) of increasing concentration of CD34+ BMDAC cells labeled with calcein-AM.
B. The level of calcein extrusion from ( ) and calcein retention within (
) and calcein retention within ( ) the cell pellet of CD34+ BMDAC was measured by RFU over time. Percent calcein retained in the cell pellet (▲) was determined by measuring total fluorescence, media and cells, then pelleting the cells and measuring the fluorescence of the washed cell pellet resuspended in vehicle. The results shown are a representative sample of 4 separate experiments.
) the cell pellet of CD34+ BMDAC was measured by RFU over time. Percent calcein retained in the cell pellet (▲) was determined by measuring total fluorescence, media and cells, then pelleting the cells and measuring the fluorescence of the washed cell pellet resuspended in vehicle. The results shown are a representative sample of 4 separate experiments.
C. Calcein retention in CD34+ BMDAC from healthy volunteers verse subjects with risk factors for cardiovascular disease (Endothelial at risk subjects). Results represent the average of 4 or more subjects per cohort. Calcein retention within CD34+ BMDAC was determined after 4 hours incubation.
D. The level of calcein retention in CD34+ BMDAC (■) versus PBMC ( ), from the same subject over time, is graphed over the time period shown. Shown is a representative of 4 similar experiments.
), from the same subject over time, is graphed over the time period shown. Shown is a representative of 4 similar experiments.
Calcein retention by CD34+ BMDAC varied between subject samples. Interestingly, the percent of calcein retained in CD34+ BMDAC from healthy subjects was higher (8%) after 4 hours incubation than calcein retention in CD34+ BMDAC from patients with endothelial risk factors (diabetes, end-stage kidney disease, and hypertension) (3.5%) (Figure 1C).
While dye retention was variable between patient samples, peripheral blood mononuclear cells (PBMC) from the same patient always retained a greater percent of calcein than did the CD34+ BMDAC, regardless of the origin of the sample. Figure 1D is a representative efflux of calcein over time from CD34+ BMDAC and PBMC from the same patient over the course of 4 hours. After 0.5 hour incubation, only 50% of available calcein was detected in the CD34+ BMDAC pellet (Figure 1D). By comparison over 80% of the calcein was retained in the PBMC at the same time point. Calcein retention dropped consistently throughout the assay for both cell types. After 4 hours, less than 10% of calcein was retained within the CD34+ BMDAC and only about 21% within the PBMC.
The extrusion rate of the dye appeared to be influenced by the concentration of other reactive substances such as when the chemokine SDF-1 was added to the migration medium. It was theorized that the concentration of SDF-1 could actually obfuscate the interpretation of a migration assay by causing the cells to extrude or retain calcein in an SDF-1 dependent fashion. In order to address this issue, CD34+ BMDAC were exposed to varying concentrations of SDF-1 in a microfuge tube for 4 hours before determining the amount of calcein retained within the cells. As shown in Figure 2A, there was considerable variation in the amount of calcein retained in CD34+ BMDAC from different patients and this variation had some SDF-1 dependence. In addition, PBMC also demonstrated a SDF-1 dependent calcein extrusion, Figure 2B. This SDF-1 dependent calcein retention in CD34+ BMDAC was significantly different at different SDF-1 concentrations (Figure 2C).
Figure 2.

A. SDF-1 influence on calcein retention was measured in subjects with a variety of clinical syndromes. CD34+ BMDAC were exposed to increasing concentrations of SDF-1 and the calcein retention was measured as described in Methods after 4 hours. Subjects were healthy smokers ( ), hypertensive African American women (□), female subjects ischemic heart disease (■), and a subject that had received a kidney transplant (
), hypertensive African American women (□), female subjects ischemic heart disease (■), and a subject that had received a kidney transplant ( ).
).
B. Effect of increasing concentrations of SDF-1 on calcein retention in PBMC (■) and CD34+ BMDAC ( ), averaged from 7 different healthy controls.
), averaged from 7 different healthy controls.
C. Differences in calcein retention in CD34+ BMDAC exposed to increasing concentration of SDF-1. Expressed as the percent change compared to the 0 nM SDF-1 control.
D. Amount of calcein extruded (■) verse retained (◇) after labeling an equal number of PBMC and CD34+ BMDAC with 12 μM calcein-AM and incubating for 4 hours.
Although PBMC retained calcein better than CD34+ BMDAC, it was possible that differences were due to differences in cell number. Thus we suspended equal numbers of PBMC & CD34+ BMDAC and added 12 μM calcein. After 4 hours of incubation, the RFU of the total cell suspensions and the washed pellets were measured. As shown in Figure 2D the total counts elaborated by CD34+ BMDAC were nearly twice that of an equal number of PBMC, with the amount of calcein retained by PBMC being 60× higher than CD34+ BMDAC. This suggested to us that cell metabolism may influence the extrusion rate of calcein into the medium.
Since soluble calcein extruded by non-migrated cells is likely to be able to penetrate the 8 μm pores in the polycarbonate membrane of the Boyden chamber, extrusion of calcein could cause misinterpretation of migration assays when it is used as a cell marker. Using a 10 μM solution of Rhodamine 110 in a mock migration (Figure 3A), this dye quickly penetrated the 8 μm polycarbonate pores with nearly 80% of the dye equilibrated on both sides of the polycarbonate membrane of a Boyden chamber by 45 minutes. Extruded calcein dye is likely to penetrate the membrane pore as rapidly as Rhodamine 110, strongly suggesting that calcein is not a suitable dye for measuring the migration of BMDAC.
Figure 3.

A. The ability of the soluble fluorescent dye Rhodamine 110 (485ex/530em) to diffuse across an 8 μm pore polycarbonate membrane was determined by adding Rhodamine to the top of a Boyden chamber and at the indicated time point determining the level of fluorescent dye that was in the lower chamber.
B. Level of retained calcein-AM within Jurkat cells at 30 and 180 minutes after labeling.
C. Level of calcein retention by Jurkat cells after a 4 hour exposure to increasing concentrations of SDF-1.
We also examined the ability of calcein to label Jurkat cells, which are frequently used as a model to study cell migration. In these cells, calcein efflux was robust, with dye retention considerably lower than that recorded in PBMC. As shown in Figure 3B after 180 minutes of incubation only ~9% of calcein was retained within the cell pellet. Calcein retention in Jurkat cells also demonstrated an SDF-1 dependent relationship (Figure 3C). In this case, SDF-1 appeared to cause the cells to retain calcein relative to the 0 nM SDF-1 control.
In an effort to find a dye better retained by highly metabolic cells, we compared dye retention in Jurkat cells using 3 cytoplasmic dyes: calcein-AM, Cell Tracker Green CMFDA, and Vybrant CFDA. As shown in Figure 4A, dye retention was generally 20% or less with all three dyes after 4 hours of incubation. Results were similar when these 3 dyes were compared in PBMC (data not shown). As a result of these experiments and because the cytoplasmic dyes were extruded from cells and in an SDF-1 dependent fashion, we elected to evaluate lipophilic dyes for suitability for the migration assay.
Figure 4.
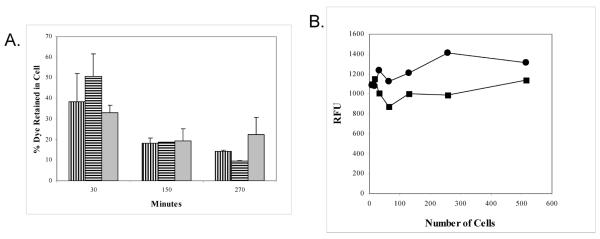
A. Level of dye retention of Cell Tracker ( ), Vybrant (
), Vybrant ( ), and Calcein-AM (
), and Calcein-AM ( ) labeled Jurkat cells over time.
) labeled Jurkat cells over time.
B. Level of dye retention of Jurkat cells labeled with 2 μM (■) or 6 μM (●) concentrations of PKH67. Two fold dilutions of cells were performed and fluorescence was measured in a FL600 at 485ex/530em to determine the sensitivity of the signal.
Lipophilic dyes
Lipophilic dyes possess polar headgroups and long aliphatic hydrocarbon tails, partition into the lipid bilayer and are essentially retained irreversibly in the plasma membrane via non-covalent hydrophobic interactions. They have been used for in vivo cell tracking studies for periods up to several months. We compared 5 commercially available lipophilic dyes with long hydrocarbon tails for suitability in the migration assay: CM-DiI (ex/em 553/570), PKH26 (551/567), PKH67 (490/502), CVB(683/707) and CV815 (786/814). Table 1 shows the concentrations used, application solvent and spectral characteristics of these dyes.
PKH26, PKH67 and CM DiI
PKH26, PKH67 and CMDiI dyes are attractive in that they are amenable to use in a fluorescent plate reader format with multiple well plates since they excite and emit in the visible region of the spectrum. However, we found that very narrow Stokes shift between the excitation and emission signal of PKH26 made it unsuitable for use with routine filters. CMDiI did not provide a strong enough signal to detect CD34+ BMDAC, which are found in low numbers in peripheral blood, in a migration assay. PKH67 possesses similar excitation and emission wavelengths as calcein, making it versatile for use in a fluorescent plate reader, fluorescent microscope, and flow cytometer. Using Jurkat cells as a model, PKH67 signal strength was measured at 6 μM and 2 μM concentrations. However the resolution of cells at the lower limit of detection by PKH67 was not sufficiently strong to detect the migration of a rare population of cells such as peripheral CD34+ BMDAC, Figure 4B.
CellVue Dyes
CellVue dyes are a relatively new family of lipophilic dyes and are available with a variety of fluorescence ex/em properties ranging from the long wavelength UV into the near infrared. Similar to the PKH dyes, they are designed for rapid uniform labeling of live cell membranes with minimal transfer of dye between cells. The fluorescence properties of two near infrared CellVue dyes, CVB and CV815, are well matched with the excitation lines and emission detectors of the Odyssey near infrared imaging system (LI-COR Biosciences, Lincoln, NE). Therefore these dyes were studied to determine their suitability for labeling CD34+ BMDAC and then to determine the lower limits of detection of cells labeled with these dyes using the Odyssey Imaging system.
A single culture of Jurkat cells was diluted to 1,000 cells per μl and labeled with either CVB or CV815. Two fold serial dilutions of labeled cells were loaded onto a black glass bottom plate and imaged using the Odyssey a near infrared imager (Figure 5A). As shown in Figure 5B, CVB signal plateaued above 5,000 total cells, whereas the standard curve of cells loaded with CV815 was still linear to 250,000 cells. However the CVB was much more sensitive, able to distinguish between 50 and 100 cells whereas the sensitivity of the CV815 dye, was only in the 300 to 400 cell range (Figure 5C).
Figure 5.

A. Sensitivity of detection of Jurkat cells labeled with a 2 μM solution of CVB dye (◆) or CV815 (■). Two fold serial dilutions were performed and the integrated fluorescent intensities of the images from the Odyssey software were plotted to produce a standard curve.
B. Expanded view of cell number of the lower concentration of cells verse fluorescent intensity of Jurkat cells labeled with a 2 μM solution of CVB dye (◆) or CV815 (■).
In order to determine the optimal dye concentration of CVB, Jurkat cells were stained with two fold serial dilutions of dye, starting with 4 μM or twice the manufacturer's recommended concentration. Readings of diluted cells at each dye concentration were taken at 2h and 16h. As shown in figure 6A, there was a linear relationship between cell number and relative fluorescence at each concentration. Signal was sufficiently strong to distinguish between 60 and 30 cells at concentrations of CVB as low as 0.25 μM although the strongest signal was produced at 4 μM. However, greater standard errors were associated with the higher concentration. When CVB signal strength was measured after 16h of incubation, it was found that the signal actually increased at all concentrations except 4 μM (Figure 6B). Importantly, CVB signal was retained within the cell (Figure 6C) and SDF-1 appeared to have limited effect on CVB dye retention (Figure 6D). We observed some variability in CVB retention depending on SDF-1 exposure, but it was negligible compared to the differences observed with calcein.
Figure 6.

A. Effect of CVB concentration on the ability to label Jurkat cells. Jurkat cells were stained with 0.25 μM (X), 0.5 μM (◯), 1 μM (▲), 2 μM (■), 4 μM (◆) of CVB and diluted aliquots of stained cells were imaged after 2 hours.
B. Stability of CVB labeling over-time. The signal strength of 500 Jurkat cells labeled at the indicated concentration of CVB was measured at 2h (■) and 16h ( ).
).
C. Retention of CVB(■) or calcein ( ) within Jurkat cells over time.
) within Jurkat cells over time.
D. Ability of Jurkat cells to retain CVB (■) or calcein ( ) in the face of exposure to increasing concentrations of SDF-1 for 4 hours.
) in the face of exposure to increasing concentrations of SDF-1 for 4 hours.
Chemotaxis assay
Ultimately, we were evaluating dyes for the use in measuring the migration of rare endothelial progenitor cells isolated from human peripheral blood. When comparing different dye properties, both CV dyes were found to provide increased sensitivity over any other lipophilic dye previously studied in our lab with good signal to noise ratios as shown in Table 2. By contrast, PKH26 yielded low signal to noise ratios due to incompatible instrument filter sets, which was also true for CM DiI (data not shown). PKH67 and calcein had similar signal to noise ratios but PKH67 was insufficiently sensitive to be suitable in the CD34+ BMDAC migration assay. Because CVB provided the greatest sensitivity in measuring a small number of cells with respectable signal to noise ratio and excellent dye stability, we evaluated its usefulness to measure cell migration and initial migration experiments were conducted using human Jurkat cells.
Table 2.
Signal to Noise Ratios for Detection of Dyes
| Dye | Device | Filters | S/N |
|---|---|---|---|
| PKH 26 | Fluorescent Plate Reader | 551/567 | 1.64 |
| PKH67 | Fluorescent Plate Reader | 490/530 | 16.2 |
| Calcein | Fluorescent Plate Reader | 490/530 | 16.9 |
| CellVue Burgundy | Odyssey NIR Imager | 683/707 | 174 |
| CellVue 815 | Odyssey NIR Imager | 786/814 | 3850 |
As seen in Figure 7A, migrated Jurkat cells provided a strong signal using CVB dyes as measured in the LiCor Odyssey. A typical migration of Jurkat cells to SDF-1 demonstrates a 140% increase in migration compared to the background control at 0 nM SDF. When migrated cells were subsequently counted in the FL4 detector of a FACSCalibur, the flow events provided similar results corroborating the digital data from the Odyssey with an r value of 0.96 (Figure 7A).
Figure 7.
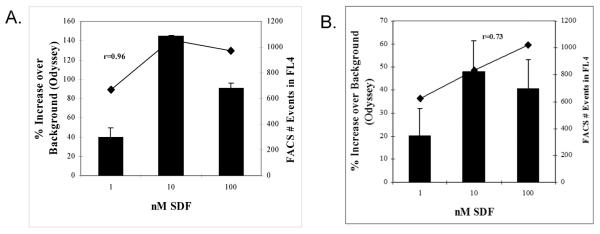
A. The ability of CVB labeled Jurkat cells to migrate was determined. Jurkat cells were labeled with CVB dye and migrated to 0 nM, 1 nM, 10 nM, and 100 nM SDF-1. The fluorescent intensity was imaged and the cells that migrated at 1, 10 and 100 nM are expressed as percent change from the 0 nM well (■). After determining the fluorescent intensity the number of events was determined by flow cytometry (◆). Each migration was run in triplicate.
B. The ability of CVB labeled human CD34+ BMDAC to migrate was determined. Human CD34+ BMDAC were labeled with CVB dye and migrated to 0 nM, 1 nM, 10 nM, and 100 nM SDF-1, fluorescent intensity was imaged and the cells that migrated at 1, 10 and 100 nM are expressed as percent change from the 0 nM well (■). After determining the fluorescent intensity the number of events was determined by flow cytometry (◆). Each migration was run in triplicate
Migration of CD34+ BMDAC
CVB dye was found to provide a strong signal with acceptable signal-to-noise ratios which leant itself well to the measurement of Jurkat cell migration in the Boyden chamber system. However, it was necessary to determine whether this dye was sufficiently sensitive to detect CD34+ BMDAC in a similar format, since the total number of primary CD34+ BMDAC available for migration is typically between 3,000 and 10,000 cells per well and the actual number of cells successfully penetrating an 8 μm pore towards the chemokine would be a fraction of that total. Using CVB we were routinely able to measure CD34 migration using 2000 cells per well. Results of a typical CD34+ BMDAC migration are shown in Figure 7B with migration relative to background control (0 nM SDF) calculated from integrated intensity data.
In order to corroborate our CVB based migration, we compared the integrated intensity values generated by the Odyssey with events per minute generated by flow cytometry as described above. Migrated CD34+ BMDAC aspirated from the lower well of the Boyden chamber were first read in the Odyssey imager and then analyzed by flow cytometry. Results of the flow cytometry confirmed the near infrared plate reader results (Figure 7B) with an r value of 0.73.
Cell proliferation studies
CVB is detectable in the FL4 detector of the FACSCalibur flow cytometer, making it potentially valuable in cell proliferation studies as well as tracking cells for migration. CVB signal is very durable, when protected from light and was not found to deteriorate even 30 days after the initial reading when stored at room temperature in a 384 well plate. However, the signal strength per cell in dividing, viable cells was unknown. After 96 hours of culture, CVB labeled Jurkat cells still emitted a total signal strength which was 90% or above that of the initial reading as measured by the Odyssey imager. Moreover, the standard 2 μM concentration of CVB dye was not found to be toxic, in that these cells multiplied at the same rate as those which were treated only with vehicle Diluent C (data not shown). However, the Odyssey only measures total signal, not the signal strength per cell, being unable to monitor the partitioning of the dye into daughter cells. In order to evaluate CVB usefulness in proliferation assays, Jurkat cells underwent flow cytometry 0, 48, and 96 hours after labeling with 2 μM CVB. As shown in the Figure 8A, as the time for cell division increased, the signal intensity per cell decreased proportionally. At zero hours, the concentration of labeled Jurkat cells was 3 ×105 (Figure 8B) and the Odyssey fluorescent intensity (measuring total available signal) was 10 and the flow geometric mean was 32. By 96 h the cell concentration had increased to 1.6 × 106 and the Odyssey fluorescent intensity was stable at 13, indicating that the total signal had not degraded. While a 5.3-fold increase in cells the mean fluorescent intensity appropriately decreased 8-fold (Figure 8B). The similarity between the two generated ratios suggests that CVB might prove to be a useful agent in proliferation studies.
Figure 8.

A. Jurkat cells were labeled with 2 μM CVB and cultured in a humidified 5% CO2 incubator for 0h ( ), 48 h (
), 48 h ( ), and 96 h (
), and 96 h ( ) after which time the samples were removed and 100,000 events were evaluated by flow cytometry. Inset shows, for comparison, 0h unlabeled cells (
) after which time the samples were removed and 100,000 events were evaluated by flow cytometry. Inset shows, for comparison, 0h unlabeled cells ( ) verse 48h CVB labeled cells (
) verse 48h CVB labeled cells ( ).
).
B. The signal ratios between 0h and 96h CVB labeled Jurkat cells were calculated to compare the change in cell number over time and flow cytometry signal intensity over the same time period. The ratio of cells/ml between 96h and 0h was 5.3; the ratio of flow cytometry signal intensity geometric mean between the 0h and 96h was 8.0.
Discussion
In 1997 Asahara et al published landmark findings of the existence of stem cells in the circulation which could give rise to endothelial cells [3]. Since then a number of papers have been published demonstrating the therapeutic value of these endothelial progenitor cells in repairing damaged vasculature (For a review, see [4]). In addition, it has been proposed that the chemotactic behavior of human BMDAC in response to SDF-1 may be a key component in the mechanism of vascular repair [5]. The number and migratory propensity of an individual's BMDAC may reflect an innate ability to repair damaged endothelium. We and others have postulated that the chemotactic response of CD34+ BMDAC towards SDF-1 may be perturbed (suppressed or even exaggerated) in compromised patient populations whose vasculature is at risk [6]. Therefore, the ability of CD34+ BMDAC to migrate towards the chemokine SDF-1 is a potential measure of vascular health and may provide a prognostic indicator of vascular health or disease. If this measurement is to have clinical value, then the method used to measure BMDAC migration should be simple, amenable to electronic data-basing and high throughput technologies, objective, and inexpensive.
In developing a test to measure BMDAC chemotactic response, we compared a variety of techniques to calculate migration, including manual enumeration of cells by light microscopy, Coulter counting, and flow cytometry. Each of these methods is cumbersome or labor intensive making them impractical for a clinical setting. In contrast, the use of a tracking dye measurable by spectroscopy is an attractive alternative for reasons of speed, high throughput, reduction of bias and compatibility with data-basing.
A large variety of dyes are available for use in labeling and tracking eukaryotic cells, each with characteristic strengths and weaknesses. Because we were monitoring the migration of a rare population of cells, the dye's signal strength, low signal to noise ratio, and applicability in a 384 well format were of particular importance.
For a number of years, the cytoplasmic dye calcein-AM has received much attention for its ability to differentiate between viable and dead cells. The acetomethoxyester (AM) derivative of calcein prevents fluorescence of the molecule because the calcium binding site is blocked on the molecule by the AM ester. Calcein-AM passively diffuses across an intact cell membrane and non-specific internal cell esterases cleave the AM group allowing fluorescence to develop, resulting in a powerful green signal, measurable in a typical fluorescent plate reader with fairly low background. Other cytoplasmic dyes (CFDA, CMFDA) behave in a similar fashion. Each of these dyes is able to passively diffuse across the cell membrane as a colorless reagent. Once internalized, cellular esterases (calcein or CFDA) or thiol reactivity (CMFDA) alter the dyes rendering them fluorescent throughout the cytoplasm. All three dyes fluoresce at 530 nM which is compatible with a variety of different instruments: Fluorescent plate reader, fluorescent microscopes, and flow cytometry, making them attractive and versatile tracking agents. However, we discovered that all three cytoplasmic dyes were extruded rapidly from live BMDAC, even though the dyes have been promoted as a means to distinguish being living and dead cells.
Calcein efflux has received a considerable amount of attention in the literature, particularly with cancerous cells. In 1998, Essodaigui M, et al. reported that tumor cells extruded calcein which was attributable to the presence of the multidrug resistance protein (MRP) and the MDR1 encoded P-glycoprotein in the plasma membrane [7]. These proteins are believed to be responsible for the resistance of cancer cells to some chemotherapeutic agents, are part of an ATP binding cassette family of transport proteins, dependent upon ATP hydrolysis for the extrusion of the substrates across the membrane. P-glycoprotein is normally expressed in capillary endothelial cells among a number of other normal cells types, such as renal proximal tubule cells and those cells lining the intestine [8, 9].
It is possible that cytoplasmic dyes are treated as toxins by certain cells and that the rate of extrusion is related to the metabolic state of the cell. In fact, we noted a variation in calcein extrusion from CD34+ BMDAC and Jurkat cells which appeared to be dependent on the concentration of the chemokine SDF-1. It is interesting to note that calcein efflux was also rapid in the Jurkat cell line, a human T-cell lymphoma cell line. By contrast, calcein was fairly well retained in human peripheral blood mononuclear cells. This suggests that the expression of ATP binding cassette transporter proteins is greater in BMDAC than in more fully mature white blood cells. Other cytoplasmic dyes such as Cell Tracker Green CMFDA (chloromethyl fluorescein diacetate), Vybrant CFDA (carboxy fluorescein diacetate succinimidyl ester) were also extruded from Jurkat cells and PBMC at rates similar to calcein, making them equally unsuitable for use in the measurement of BMDAC migration.
After recognizing the limitations of cytoplasmic dyes we evaluated the suitability of lipophilic dyes such as CM DiI, PKH26, and PKH67 with excitation and emission fluorescence in the visible region of the spectrum. Each of these dyes possesses long aliphatic hydrocarbon tails and polar fluorescent head groups which allow them to insert into the lipid bilayers of cells, reducing the likelihood of extrusion. However, due to the spectral characteristics of these dyes, none was able to provide a strong enough signal for tracking the migration of the limited number of BMDAC cells isolated from 24 mls of peripheral blood samples.
Finally we evaluated the suitability of two recently commercialized near infra-red emitting lipophilic dyes CVB and CV815 for use in BMDAC migration. Like their visible emitting counterparts, both dyes possess polar heads and long aliphatic tails, allowing them to rapidly intercalate into the lipid bilayer of the cell and be retained by hydrophobic interactions. Since these dyes emit in the near infra-red, a stronger fluorescence signal is expected because background autofluorescence from physiological compounds is reduced in this region of the spectrum.
Initially, we established a range of sensitivity for each of these dyes, using Jurkat cells as a model. The sensitivity of CVB was considerably stronger than CV815, even though CV815 emitted further into the infrared region. When two-fold dilutions of Jurkat cells were measured in the Odyssey NIR Imager, we were able to detect between 50 and 100 cells using CVB with a signal to noise ratio of 174. By comparison, although the signal to noise ratio for CV815 was considerably higher than CVB, due to its longer wavelength emission, its signal strength was not sufficient to allow for the detection of a rare population of cells such as BMDAC. This suggests however that CV815, by virtual of its strong signal to noise ratio, might be a valuable tracking dye in vivo using a larger population of cells.
CVB is also detectable by flow cytometry in the FL4 detector of a FACSCalibur and fluorescence microscopy if the microscope is equipped with a custom filter set, a Xenon light source and a CCD camera able to detect long wavelengths.
Because the strength of the CVB signal allowed us to measure as few as 50 to 100 cells, we found that it was most appropriate for use in the CD34+ BMDAC migration assay in which often as few as 2,000 to 10,000 cells are available for migration and only a fraction of those cells migrate into the lower chamber containing the chemokine SDF-1. Furthermore, we were able to compare cell migration data obtained from the Odyssey imager with data by flow cytometry and found that both techniques yielded similar values for quantification of migration. In addition, because over 90% of CVB was well retained in the lipid bilayer for at least 96 hours, results were not confounded by dye extrusion, and this provides a broad time frame for assays in which cell monitoring is necessary. Moreover, this dye might be appropriate for use in measuring cell proliferation, owing to the partitioning of the dye into daughter cell populations, which could be monitored by flow cytometry.
Table 3 summarizes the characteristics of the dyes tested with regard to suitability for rare cell migration. The CVB dye was found to be superior to any other tracking dye tested by virtue of its signal strength and high signal to noise ratio allowing detection of small numbers of cells in a Boyden migration assay. Because detection was measurable in the Odyssey Near Infrared Imager, with a high throughput format, large amounts of data could be rapidly generated. Furthermore, the fluorescence excitation and emission profile of the dye make it suitable for use in flow cytometry and also in fluorescence microscopy with some microscope modifications. Lastly, CVB was found to be extremely stable, nontoxic, and well retained in two cell types tested making it attractive not only for use in migration assays but also for cell proliferation studies.
Table 3.
Summary Of Characteristics Of Dyes With Regard To Suitability For Rare Cell Migration
| Dye | PKH26 | Calcein AM | Cell Tracker Green CMFDA | Vybrant CFDA | PKH67 | CMDil | CellVue® NIR 815 | CellVue® Burgundy |
|---|---|---|---|---|---|---|---|---|
| Class | Visible lipophilic membrane dyes | Visible lipophilic membrane dyes | Visible lipophilic membrane dyes | Visible lipophilic membrane dyes | Visible lipophilic membrane dyes | Visible lipophilic membrane dyes | Near infrared lipophilic membrane dyes | Near infrared lipophilic membrane dye |
| Standard Filter Suitability | Poor | Excellent | Excellent | Excellent | Excellent | Excellent | Excellent | Excellent |
| Cell Retention | Poor | Poor | Poor | Excellent | ||||
| Cell Detection Sensitivity | Poor | Poor | Poor | Excellent |
References
- 1.Aiuti A, Webb IJ, Bleul C, Springer T, Gutierrez-Ramos JC. The Chemokine SDF-1 Is a Chemoattractant for Human CD34+ Hematopoietic Progenitor Cells and Provides a New Mechanism to Explain the Mobilization of CD34+ Progenitors to Peripheral Blood. J Exp Med. 1997;185:111–120. doi: 10.1084/jem.185.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peled A, Grabovsky V, Habler L, Sandbank J, Arenzana-Seisdedos F, Petit I, Ben-Hur H, Lapidot T, Alon R. The chemokine SDF-1 stimulates integrin-mediated arrest of CD34(+) cells on vascular endothelium under shear flow. J Clin Invest. 1999;104:1199–1211. doi: 10.1172/JCI7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, Kearne M, Magner M, Isner JM. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85:221–8. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 4.Dimmeler S, Zeiher AM. Vascular repair by circulating endothelial progenitor cells: the missing link in atherosclerosis? J Mol Med. 2004;82:671–7. doi: 10.1007/s00109-004-0580-x. [DOI] [PubMed] [Google Scholar]
- 5.Yamaguchi J, Kusano KF, Masuo O, et al. Stromal cell-derived factor-1 effects on ex vivo expanded endothelial progenitor cell recruitment for ischemic neovascularization. Circulation. 2003;107:1322–1328. doi: 10.1161/01.cir.0000055313.77510.22. [DOI] [PubMed] [Google Scholar]
- 6.Segal MS, Shah R, Afzal A, et al. Nitric oxide cytoskeletal-induced alterations reverse the endothelial progenitor cell migratory defect associated with diabetes. Diabetes. 2006;55:102–9. [PubMed] [Google Scholar]
- 7.Essodaigui M, Broxterman HJ, Garnier-Suillerot A. Kinetic analysis of calcein and calcein-acetoxymethylester efflux mediated by the multidrug resistance protein and P-glycoprotein. Biochemistry. 1998;37:2243–2250. doi: 10.1021/bi9718043. [DOI] [PubMed] [Google Scholar]
- 8.Hauser IA, Koziolek M, Hopfer U, Thévenod F. Therapeutic concentrations of cyclosporine A, but not FK506, increase P-glycoprotein expression in endothelial and renal tubule cells. Kidney Int. 1998;54:1139–1149. doi: 10.1046/j.1523-1755.1998.00095.x. [DOI] [PubMed] [Google Scholar]
- 9.Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci USA. 1987;84:7735–7738. doi: 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]