

Abstract
A series of 5-alkylsulfamoyl benzimidazole derivatives have been designed and synthesized as novel angiotensin II (Ang II) receptor antagonists. The compounds have been evaluated for in vitro Ang II antagonism and for in vivo antihypertensive activity on isolated rat aortic ring and desoxycortisone acetate induced hypertensive rats, respectively. The activity is found related to size of alkyl group. The maximum activity is observed with a compact and bulky alkyl group like tert-butyl and cyclohexyl. The compounds 4g and 4h have shown promising both in vitro and in vivo activities. A receptor binding model is also proposed on the basis on the basis of structure–activity relationship in this study.
Keywords: Benzimidazole, Angiotensin II antagonists, Sulfamoyl, Antihypertensive, Binding profile
1. Introduction
Angiotensin II (Ang II) AT1 receptor antagonists are widely used in treatment of hypertension and maintenance of homeostasis.1,2 Losartan is the protypical Ang II antagonist and numerous modifications in chemical structure of losartan have generated large artillery of Ang II antagonists3–5 like candesartan, zolasartan, irbesartan, and telmisartan (Fig. 1). In general, all these ‘sartans’ are composed of an appropriately substituted heterocyclic nucleus coupled to an acidic group (carboxylic or tetrazole) bearing biphenyl system through a methylene linker. Candesartan is an Ang II antagonist having benzimidazole nucleus substituted with carboxyl function at 7-position.6,7 Substitution at 6-position of benzimidazole nucleus has also produced derivatives having excellent Ang II antagonistic activity.8 However, the 5-position of benzimidazole nucleus has remained relatively unexplored in search of potent Ang II antagonists. The 5-nitrobenzimidazole derivative 1 (Fig. 1) has been designed and synthesized earlier in our laboratory and it is found more active than candesartan.9 Another series of 5-alkylcarboxamido (−NHCOR) benzimidazole derivatives has also been designed and synthesized in our laboratory10 from which 5-acetamido analog 2 (Fig. 1) is found equipotent to candesartan. Based on these earlier results, a previous study on triazolone based AT1 antagonist 311 and other similar studies12–15 a binding profile was proposed (Fig. 2) in the previous report10 where n-butyl chain interacts with pocket L1 lined by Phe182, Phe171, Ala163, and Ser103 and the biphenyl system interacts with pocket L2 lined by Val108 through van der Waal interactions while N-3 of benzimidazole nucleus and terminal carboxyl group interact through H-bonding with Tyr113 and Tyr184. The nitro group of 1 and acetamido group of 2 can interact with pocket L4 lined by Tyr292, His256, Tyr253, and leu112 through van der Waal and/or H-bonding interactions. However, insignificant increase in activity of 2 with respect to 110 has suggested that interactions of −NO2 group alone with receptor surface are much stronger than those of whole of the −NHCOCH3 group. It indicated that −NO2 group is required for strong binding interaction and it is hypothesized that further extension at nitrogen atom of −NO2 group with substituents (like alkylamino groups) may strengthen the binding interactions through lipophilic and/or H-bonding interactions with pocket L3 or L4. Hence, in continuing endeavors to search novel and potent Ang II antagonists, the present study reports 5-substituted benzimidazole derivatives 4 (Fig. 1) designed by isosteric replacement of −NO2 group with sulfonyl (−SO2) group and extending the latter with alkylamino group (−NHR). The designing of these compounds was based on hypothesis that while the −SO2 mimics the −NO2 group, the −NH– can interact with an H-bond acceptor group and the alkyl group (R) may interact with L3 or L4 to form a stronger drug–receptor complex. The in vitro Ang II antagonism and in vivo antihypertensive activity of the compounds were evaluated using losartan, candesartan and 1 as reference compounds to study the structure–activity relationship.
Figure 1.

Angiotensin II receptor antagonists.
Figure 2.
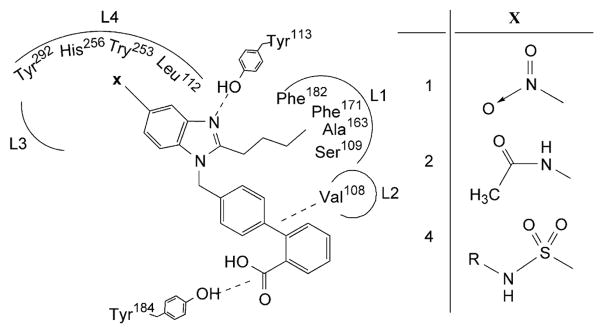
Possible binding profile of 5-substituted benzimidazole derivatives.
2. Results and discussion
2.1. Chemistry
The synthetic scheme employed to synthesize the designed compounds is outlined in Figure 3. The compound I was prepared through a series of reactions as reported earlier9,10 which was then subjected to chlorosulfonation with chlorosulfonic acid at temperature of 10–15 °C. The −SO2Cl was substituted selectively at 5-position of benzimidazole nucleus similar to selective nitration at indole.16 Moreover, the substitution of chlorosulfonyl group was not expected to occur on terminal phenyl ring of biphenyl system due to deactivation of the latter by electron withdrawing −COOH group and the same was also not seemed possible on spacer phenyl ring of biphenyl system due to electron withdrawing effects and steric hindrances of the adjoining benzimidazole nucleus and deactivated terminal phenyl ring. The chlorosulfonyl intermediate II was coupled with varied amines (ammonia or alkylamines) under controlled temperature conditions to form target sulfonamide products 4a–h. The intermediate II was also hydrolyzed with water to give sulfonic acid derivative 5. All compounds were purified by crystallization and purity was ascertained by thin-layer chromatography. Structures of the compounds were established through IR (Table 1), 1H NMR (Table 2) and mass spectral analyses.
Figure 3.
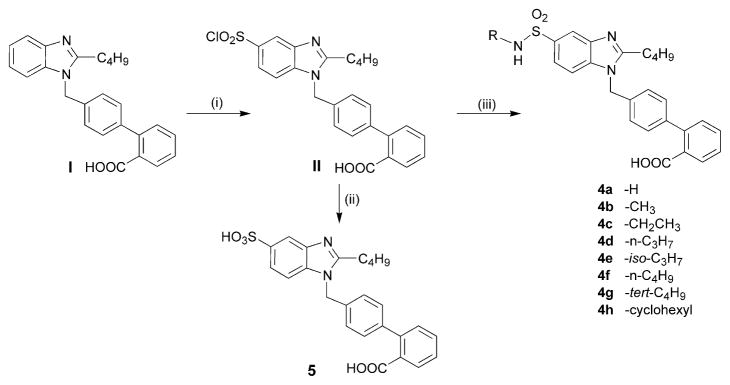
Synthetic scheme (i) chlorosulfonic acid, 10–15 °C stirring 30 min followed by 3.5 h; (ii) 10–15 °C, amine drop wise, continuous stirring, 30 min, dil HCl; (iii) rt, water drop wise, continuous stirring.
Table 1.
IR spectral data of the target compounds.
| Vibrational band | Wave number (cm−1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| 4a | 4b | 4c | 4d | 4e | 4f | 4g | 4h | 5 | |
| Free O–H str. | — | — | 3739 | 3746 | 3750 | 3650 | — | — | — |
| O–H and N–H str. | 3600–2500 | 3700–2200 | 3700–2200 | 3700–2200 | 3700–2200 | 3700–2200 | 3600–2800 | 3600–2800 | 3600–2400 |
| C=O str. | 1711 | 1708 | 1708 | 1710 | 1711 | 1713 | 1713 | 1713 | 1711 |
| S=O str. | 1320, 1160 | 1330, 1123 | 1329, 1126 | 1333, 1132 | 1370, 1161 | 1370, 1170 | 1320, 1160 | 1317, 1170 | 1315, 1172 |
| N–H wagging | 830 | 829 | 826 | 834 | 834 | 846 | 830 | 830 | — |
| C–S str. | 621 | 619 | 918 | 622 | 615 | 630 | 616 | 616 | 614 |
| C–H bend (gem-dimethyl) | — | — | — | — | 1382, 1365 | — | 1395, 1377 | — | — |
| Cyclohexyl bend | — | — | — | — | — | — | — | 1454 | — |
Table 2.
1H NMR spectral data of the target compounds.
| Compound | δ (number of protons, multiplicity, J in Hz, proton assignment) |
|---|---|
| 4a | 8.12–7.92 (2H, m, ArH); 7.87 (1H, m, ArH); 7.68–7.46 (6H, m, ArH, SONH2); 7.35–7.29 (2H, m, ArH); 7.14–7.05 (3H, m, ArH, COOH); 4.01 (2H, s, CH2); 2.97 (2H, t, 7.5, CH2CH2CH2CH3); 1.97 (2H, qv, 7.5, CH2CH2CH2CH3); 1.45 (2H, sx, 7.5, CH2CH2CH2CH3) and 0.97 (3H, t, 7.5, CH2CH2CH2CH3) |
| 4b | 8.03–7.96 (2H, m, ArH); 7.86 (1H, s, br, COOH); 7.76–7.74 (1H, m, ArH); 7.61–7.50 (4H, m, ArH); 7.48–7.42 (2H, m, ArH); 7.36–7.29 (2H, m, ArH); 7.17 (1H, s, br, SONH); 4.04 (2H, s, CH2); 2.58 (3H, s); 2.25 (2H, t, 7.5, CH2CH2CH2CH3); 1.63 (2H, qv, 7.5, CH2CH2CH2CH3); 1.35 (2H, sx, 7.5, CH2CH2CH2CH3) and 0.98 (3H, t, 7.5, CH2CH2CH2CH3) |
| 4c | 8.25–7.70 (5H, m, br, ArH, COOH, SONH); 7.62–7.29 (8H, m, ArH); 4.04 (2H, s, CH2); 3.03 (2H, t, 7.5, CH2CH2CH2CH3); 2.94 (2H, q, 7, CH2CH3); 2.59 (2H, qv, 7.5, CH2CH2CH2CH3); 2.02 (2H, sx, 7.5, CH2CH2CH2CH3); 1.28 (3H, t, 7, CH2CH3) and 0.98 (3H, t, 7.5, CH2CH2CH2CH3) |
| 4d | 7.97–7.70 (5H, m, ArH, COOH, SONH); 7.64–7.54 (4H, m, ArH); 7.52–7.44 (2H, m, ArH); 7.33 (2H, m, ArH); 4.05 (2H, s, CH2); 3.06 (2H, t, 7.5, CH2CH2CH2CH3); 3.03 (2H, t, 7.2, CH2CH2CH3); 2.62 (2H, sx, 7.2, CH2CH2CH3); 1.74 (2H, qv, 7.5, CH2CH2CH2CH3); 1.69 (3H, t, 7.2, CH2CH2CH3); 1.64 (2H, sx, 7.5, CH2CH2CH2CH3) and 0.90 (3H, t, 7.5, CH2CH2CH2CH3) |
| 4e | 8.05–7.90 (3H, br, m, COOH, NH, ArH); 7.76–7.74 (1H, m, ArH); 7.73–7.26 (9H, m, ArH); 4.0 (2H, d, 6, CH2); 3.70 (1H, sp, 7.5, CH(CH3)2); 3.24 (2H, t, 7, CH2CH2CH2CH3); 1.43 (6H, d, 7.5, CH(CH3)2); 1.95 (2H, qv, 7, CH2CH2CH2CH3); 1.45 (2H, sx, 7, CH2CH2CH2CH3) and 0.99 (3H, t, 7, CH2CH2CH2CH3). |
| 4f | 8.06–8.18 (1H, br, s, COOH); 7.75–7.62 (3H, m, ArH and SO2NH); 7.55–7.31 (9H, m, ArH); 4.04 (2H, d, 6, CH2); 3.21 (4H, m, CH2 CH2 CH2 CH3); 1.71 (4H, m, CH2CH2CH2CH3); 1.41 (4H, m, CH2CH2CH2CH3) and 0.96 (6H, m, CH2CH2CH2CH3) |
| 4g | 8.0–7.32 (13H, br, m, COOH, SO2NH and ArH); 4.0 (2H, d, 6, CH2); 3.02 (2H, t, 7, CH2CH2CH2CH3); 2.1 (9H, s, C(CH3)3); 1.93 (2H, qv, 7, CH2CH2CH2CH3); 1.47 (2H, sx, 7, CH2CH2CH2CH3) and 0.99 (3H, t, 7, CH2CH2CH2CH3) |
| 4h | 8.1–7.58 (8H, br, m, COOH, SO2NH and ArH); 7.64–7.31 (5H, br, m, ArH); 4.02 (2H, d, 6, CH2); 4.4 (1H, m, CH); 3.25 (2H, t, 7, CH2CH2CH2CH3); 2.27–1.17 (10H, m, (CH2)5); 1.95 (2H, qv, 7, CH2CH2CH2CH3); 1.43 (2H, sx, 7, CH2CH2CH2CH3) and 0.96 (3H, t, 7, CH2CH2CH2CH3) |
| 5 | 8.12–7.90 (1H, m, ArH); 7.69–7.74 (1H, m, ArH); 7.63–7.58 (2H, m, ArH, COOH); 7.56–7.51 (4H, m, ArH); 7.48–7.39 (3H, m, ArH, SO3H); 7.35–7.28 (2H, m, ArH); 4.25 (2H, s, CH2); 2.95 (2H, t, 7.5, CH2CH2CH2CH3); 1.67 (2H, qv, 7.5, CH2CH2CH2CH3); 1.35 (2H, sx, 7.5, CH2CH2CH2CH3) and 0.97 (3H, t, 7.5, CH2CH2CH2CH3) |
2.2. Pharmacological evaluation
2.2.1. In vitro Ang II receptor antagonism
The target compounds were evaluated for in vitro Ang II antagonism and activity was expressed as pA2 values (Table 3). The pA10 values were also determined to establish the mode of antagonism.17 The unsubstituted sulfonamide derivative 4a was found to be the least active and equipotent to sulfonic acid derivative 5. Both 4a and 5 were significantly less active than the lead compound 1. It indicated that −NO2 group interacts with the receptor more strongly than the isosteric −SO2 group. The activity of alkyl substituted sulfonamide analogs increased with increasing size of alkyl group at the sulfonamide moiety except for compound 4f where activity was found to decrease. The compound 4g emerged as maximally active and even more active than losartan. This varied activity due to different alkyl groups suggested that a compact alkyl group (like tert-butyl and cyclohexyl) at sulfonamide moiety may interact with receptor site by filling L3 or L4 but a long alkyl group (like n-butyl) weakens the binding interactions, probably due to overfilling of the pocket. However, contrary to the hypothesis, activity of the most active derivative 4g was comparable to that of the lead compound 1. It suggested that interactions of the tert-butyl group with receptor site are sufficient to compensate the decreased binding affinity due to replacement of −NO2 with −SO2 group as indicated by decreased activities of 4a and 5. Further, comparison of 3D structures of compounds 2, 3, and 4g in their energy minimized conformations using ChemOffice 6.0 revealed that tert-butyl group of 4g is disposed in an orientation different from positioning of methyl group of 2 but similar to phenyl ring of the benzoic acid moiety of 3. Also, difference in pA2 and pA10 values of the compounds 4a–4f indicated the compounds to be competitive inhibitors while the compounds 4g and 4h were found to be non-competitive ones similar to 1 and candesartan. The changed mode of antagonism of compounds 4a–4f with respect to 1 indicated that binding pattern of −NO2 group is different from that of −SO2 group. It also suggested that the tert-butyl and cyclohexyl groups probably interact with some different sites in the receptor pocket which is responsible for altered mode of antagonism as well as for compensation of decreased binding affinity of −SO2 group. Hence, based on these findings it is proposed that while SO2 interacts with L4, the tert-butyl and cyclohexyl group may be interacting with L3 similar to reported binding profile of 3.11
Table 3.
In vitro and in vivo activities of target and reference compounds.
| Compound | pA2a | pA10a | Mode of antagonism | Maximum decrease in MABPa,b |
|---|---|---|---|---|
| 4a | 6.7 | 6.1 | C | 51.4 ± 6.1 |
| 4b | 6.9 | 6.3 | C | 52.5 ± 3.4 |
| 4c | 7.1 | 6.2 | C | 49.9 ± 4.8 |
| 4d | 7.1 | 6.4 | C | 50.2 ± 5.9 |
| 4e | 7.2 | 6.3 | C | 51.7 ± 3.1 |
| 4f | 7.0 | 6.2 | C | 47.6 ± 6.4 |
| 4g | 8.4 | 5.4 | NC | 62.4 ± 4.5c,d,e |
| 4h | 7.9 | 5.7 | NC | 64.4 ± 3.9c,d,e |
| 5 | 6.6 | 5.8 | C | 44.7 ± 6.1 |
| 1 | 8.5 | 6.7 | NC | 55.9 ± 3.4 |
| Losartan | 8.0 | 7.1 | C | 58.3 ± 5.6 |
| Candesartan | 8.5 | 6.7 | NC | 56.2 ± 6.3 |
n = 6.
Dose is 3.0 mg kg−1 except for losartan and candesartan (5.0 mg kg−1).
p < 0.05 versus 1.
p < 0.05 versus losartan.
p < 0.05 versus candesartan.
2.2.2. In vivo antihypertensive activity
The in vivo antihypertensive activity was evaluated on desoxycortisone acetate (DOCA) induced hypertensive rats18 and activity was expressed as decrease in mean arterial blood pressure (MABP) taking losartan, candesartan, and 1 as reference compounds (Table 3). The MABP in sham control and DOCA induced hypertensive group were 107 ± 4.8 and 187 ± 6.3 mmHg, respectively. The activity was found to increase marginally with increase in alkyl group up to iso-propyl derivative (4e). Thereafter, it decreased for 4f in consonant with in vitro activity data. The decrease in MABP by 4g was significant and this reduction in MABP was even more than all reference compounds. It may be attributed to both optimum binding interactions of the pharmacophoric groups and hydrophilic–lipophilic balance of the molecule. However, in contrast to the in vitro activity trend, the antihypertensive effect of the cyclohexyl substituted analog (4h) was further increased marginally over 4g and it could be attributed probably to increased distribution of the molecule as result of increased lipophilicity of cyclohexyl group.
3. Conclusion
A series of 5-alkylsulfamoyl substituted benzimidazole derivatives are designed and synthesized as Ang II antagonists and are evaluated for in vitro Ang II antagonistic activity as well as in vivo antihypertensive activity to study the structure–activity relationship. The antagonistic activity is found dependent upon size and bulk of alkyl group while antihypertensive activity is found largely corresponding to in vitro activity and also dependent upon the overall lipophilicity of the molecule. The tert-butylsulfamoyl analog 4g has emerged as maximally active compound during in vitro studies but the cyclohexylsulfamoyl analog 4h has maximum decrease in MABP in hypertensive rats. Based on the results a binding profile for the target compounds is proposed where the tert-butyl or cyclohexyl groups can interact with L3 pocket similar to binding profile of non-competitive triazolone based Ang II antagonist.
4. Experimental
4.1. Chemistry
The melting points were recorded in open sulfuric acid bath and uncorrected. 1H NMR spectra were recorded on Bruker AC 300 NMR Spectrometer (300 MHz), mass spectra were recorded on Vg Micro Mass 7070F spectrometer and GC–MS (Gcq) Spectrometer and FT-IR spectra were recorded on FT-IR Perkin-Elmer 1710 series. In 1H NMR, chemical shifts were reported in δ values using tetramethylsilane as internal standard with multiplicities (br, broad; s, singlet; d, doublet; t, triplet; q, quartet; qv, quintet; sx, sextet; sp, septet; m, multiplet; dd, double doublet) and number of protons in DMSO-d6. The coupling constants (J) were expressed in Hz. IR spectra were recorded as KBr pellets. The elemental analyses were performed on Heraus CHN–O rapid elemental analyzer. When necessary, solvents and reagents were dried prior to use over KOH or anhydrous Na2SO4 or fused CaCl2.
4.1.1. 4′-[(2-Butyl-5-chlorosulfonyl-1H-benzimidazol-1-yl)methyl]biphenyl-2-carboxylic acid (II)
Chlorosulfonic acid (1.2 ml, 18 mmol) was cooled to 10–15 °C in a dry two necked 100 ml round bottomed flask and the compound I (1 g, 2.6 mmol) was added with continuous stirring over a period of half an hour. Stirring was further continued for 3–3.5 h maintaining the temperature at 10–15 °C. Formation of sulfonylchloride derivative II was ascertained by TLC. Chloroform/methanol (98:2); Rf 0.78 and cyclohexane/ethylacetate (25:75); Rf 0.58. The reaction mixture was used as such without product isolation to synthesize the target compounds.
4.1.2. General procedure for synthesis of 5-alkylsulfamoyl derivatives (4a–4h)
The amine reactant (45 mmol) was added drop wise to the cooled (10–15 °C) reaction mixture of product II (Caution: vigorous reaction) with continuous stirring. The temperature was not allowed to exceed 15 °C during the addition. After the addition was complete, stirring continued for another 25–30 min at the same temperature. The reaction mixture was acidified with dil HCl and solid was filtered at pump to afford the product as amorphous powder which was recrystallized from ethanol and water.
4.1.2.1. 4′-[(2-Butyl-5-sulfamoyl-1H-benzimidazol-1-yl)methyl] biphenyl-2-carboxylic acid (4a)
Amine reactant: Ammonia solution (25%), Yield 63%, mp 150–152 °C. Anal. Calcd for C25H25 N3O4S: C, 64.59; H, 5.38; N, 9.12. Found: C, 64.78; H, 5.44; N, 9.06. MS: 463 (M).
4.1.2.2. 4′-[(2-Butyl-5-methylsulfamoyl-1H-benzimidazol-1-yl)methyl]biphenyl-2-carboxylic acid (4b)
Amine reactant: Methylamine, Yield 51%, mp 140–142 °C. Anal. Calcd for C26H27 N3O4S: C, 65.08; H, 5.76; N, 8.89. Found: C, 65.39; H, 5.70; N, 8.80. MS: 477 (M).
4.1.2.3. 4′-[(2-Butyl-5-ethylsulfamoyl-1H-benzimidazol-1-yl)methyl]biphenyl-2-carboxylic acid (4c)
Amine reactant: Ethylamine, Yield 53%, mp 146–148 °C. Anal. Calcd for C27H29 N3O4S: C, 65.73; H, 5.89; N, 8.61. Found: C, 65.97; H, 5.95; N, 8.55. MS: 491 (M).
4.1.2.4. 4′-[(2-Butyl-5-propylsulfamoyl-1H-benzimidazol-1-yl)methyl]biphenyl-2-carboxylic acid (4d)
Amine reactant: Propylamine, Yield 42%, mp 141–143 °C. Anal. Calcd for C28H31 N3O4S: C, 66.42; H, 6.26; N, 8.26. Found: C, 66.51; H, 6.18; N, 8.31. MS: 505 (M).
4.1.2.5. 4′-[(2-Butyl-5-isopropylsulfamoyl-1H-benzimidazol-1-yl)methyl]biphenyl-2-carboxylic acid (4e)
Amine reactant: iso-Propylamine, Yield 52%, mp 155–156 °C. Anal. Calcd for C28H31 N3O4S: C, 66.39; H, 6.24; N, 8.37. Found: C, 66.51; H, 6.18; N, 8.31. MS: 505 (M).
4.1.2.6. 4′-[(2-Butyl-5-butylsulfamoyl-1H-benzimidazol-1-yl)methyl]biphenyl-2-carboxylic acid (4f)
Amine reactant: n-Butylamine, Yield 54%, mp 189–191 °C. Anal. Calcd for C29H33 N3O4S: C, 66.88; H, 6.34; N, 7.99. Found: C, 67.03; H, 6.40; N, 8.09. MS: 519 (M).
4.1.2.7. 4′-[(2-Butyl-5-tert-butylsulfamoyl-1H-benzimidazol-1-yl)methyl]biphenyl-2-carboxylic acid (4g)
Amine reactant: tert-Butylamine, Yield 47%, mp 148–150 °C. Anal. Calcd for C29H33 N3O4S: C, 66.92; H, 6.32; N, 8.04. Found: C, 67.05; H, 6.38; N, 8.12. MS: 519 (M).
4.1.2.8. 4′-[(2-Butyl-5-cyclohexylsulfamoyl-1H-benzimidazol-1-yl)methyl]biphenyl-2-carboxylic acid (4h)
Amine reactant: Cyclohexylamine, Yield 43%, mp 170–172 °C. Anal. Calcd for C31H35 N3O4S: C, 67.97; H, 6.51; N, 7.78. Found: C, 68.23; H, 6.46; N, 7.70. MS: 545 (M).
4.1.3. 4′-[(2-Butyl-5-sulfo-1H-benzimidazol-1-yl)methyl]biphenyl-2-carboxylic acid (5)
Cold water (10–15 °C) was added drop wise to the reaction mixture of product II (Caution: vigorous reaction) with continuous stirring. After the addition was complete, stirring continued for another 25–30 min at the same temperature. The reaction mixture was acidified with dil HCl and solid was filtered at pump to afford the product as amorphous powder which was recrystallized from ethanol and water. Yield 56%, mp 156–158 °C. Anal. Calcd for C25H25N3O4S: C, 64.59; H, 5.38; N, 9.12. Found: C, 64.78; H, 5.44; N, 9.06. MS: 464 (M).
4.2. Pharmacological evaluation
The experimental protocol involving use of animals for the study was approved by the duly constituted Institutional Animal Ethical Committee. The animals were taken care of in accordance with the guidelines of Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Ministry of Environment and Forests, Government of India (Reg. No. CPCSEA/107/1999).
4.2.1. In vitro Ang II antagonism
4.2.1.1. Isolated aortic ring preparation
The Ang II antagonism activity of target and reference compounds was evaluated on isolated aortic ring of rats. The rat was sacrificed by cervical dislocation followed by exsanguinations. The thoracic aorta was carefully dissected out, placed in ice-cold aerated Krebs–Henseliet solution (NaCl, 118 mmol; KCl, 4.7 mmol; CaCl2, 5 mmol; MgSO4· 7H2O, 1.2 mmol; NaHCO3, 25 mmol; KH2PO4, 1.2 mmol, and glucose, 11.1 mmol), cleared from the adhered connective tissue and was cut into 3 mm wide segments. Vascular endothelium from each segment was removed by gently rubbing the inside of the ring with tip of a blunt forceps. The ring was mounted in a 10 ml organ bath containing Krebs–Henseleit solution maintained at 37 °C, pH 7.4. The preparation was bubbled with oxygen. The ring was allowed to equilibrate for one and half hour under 1 g tension. The preparation was washed with Krebs–Henseliet solution once in every 15 min. Isometric contractions were recorded using force transducer and BIOPAC four channel recorder (BIOPAC Systems Inc., CA, USA) to plot the cumulative response curve (CRC).
4.2.1.2. Sensitization of rat aortic ring
The ring was sensitized before recording each CRC of Ang II in order to avoid densensitization due to Ang II. KCl (25 mmol) was added in the depolarizing Krebs–Henseleit solution to replace the equivalent isotonic amount of NaCl. The aortic ring was exposed to the resulting depolarizing Krebs–Henseliet solution for 10 min and the preparation was washed for 1 h with Krebs–Henseleit solution relaxation of the preparation to the basal tone.
4.2.1.3. Experimental design
A CRC for Ang II was recorded with increasing concentration of Ang II at an interval of 0.5 log units (1 × 10−10, 3 × 10−10, 1 × 10−9, 3 × 10−9, 1 × 10−8, 3 × 10−8, 1 × 10−7, 3 × 10−7,1 × 10−6, 3 × 10−6, and 1 × 10−5). The preparation was washed repeatedly and it was sensitized before recording the next CRC in presence of losartan. Losartan (1 nmol) was added, preparation was allowed to equilibrate for 15 min and a CRC for Ang II was recorded. Similarly CRC was recorded in absence and in presence of losartan (3 nmol and 10 nmol), candesartan, 1, 4, and 5 (1, 3, and 10 nmol). Results were expressed as percentage of the maximal tension developed in response to Ang II. The pA2 and pA10 value of losartan, candesartan, 1, 4, and 5 were calculated using Schild’s plot.17 Linear regression was employed to plot DRC between developed tension and negative log molar concentration of Ang II. Data were statistically analyzed using paired Student’s t-test. p < 0.05 was considered to be statistically significant.
4.2.2. In vivo antihypertensive activity
4.2.2.1. Experimental hypertension
The rats were uninephroctomized and DOCA (40 mg kg−1, sc) was administered twice a week up to six weeks to produce hypertension. DOCA rats received 1.0% NaCl and 0.2% KCl in their drinking water. Sham rats received tap water. The rats were anaesthetized (sodium thiopental 30 mg kg−1, ip), heparanized (200 IU heparin, ip) and their trachea were cannulated to facilitate respiration. The carotid artery was isolated and cannulated to pressure transducer attached to BIOPAC systems (BIOPAC, CA, USA) for measurement of MABP.
4.2.2.2. Experimental design
The in vivo dose standardization was performed using the lead compound 1. Different doses (0.1, 0.3, 1.0, 3.0, 10.0, and 30.0 mg kg−1, ip) of 1 were administered to rats and MABP was measured after 6 h of administration. It was noted to produce plateau effect at 1 mg kg−1 dose. In light of this, compounds 4 and 5 were administered to rats at doses of 0.1, 1.0, 3.0, and 10.0 mg kg−1 (ip) and MABP was measured after 6 h of their administration. Losartan and candesartan (5 mg kg−1, ip) were used as reference standards in the study. Each value (n = 6) represents mean ± SEM. Data were statistically analyzed by performing one-way ANOVA followed by Tukey’s multiple range test. p < 0.05 was considered to be statistically significant.
References and notes
- 1.Vallotten MB. Trends Pharmacol Sci. 1987;8:69. [Google Scholar]
- 2.Ferrario CM. J Cardiovasc Pharmacol. 1990;15:S1. [PubMed] [Google Scholar]
- 3.Dunica JV, Chiu AT, Carini DJ, Gregory GB, Johnson AL, Timmermans PBMWM. J Med Chem. 1990;33:1312. doi: 10.1021/jm00167a007. [DOI] [PubMed] [Google Scholar]
- 4.Carini DJ, Duncia JV, Aldrich PE, Chiu AT, Johnson AL, Pierc ME, Price WA, Santella JB, Wells J, Wexler R, Wong C, Yoo SE, Timmermans PBMWM. J Med Chem. 1991;34:2525. doi: 10.1021/jm00112a031. [DOI] [PubMed] [Google Scholar]
- 5.Wexler RR, Greenleen WJ, Irvin JD, Goldberg MR, Prendergast K, Smith RD, Timmermans PBMEM. J Med Chem. 1996;39:625. doi: 10.1021/jm9504722. [DOI] [PubMed] [Google Scholar]
- 6.Kubo K, Kohara Y, Imamiya E, Sugiura Y, Inada Y, Furukawa Y, Nishikawa K, Naka T. J Med Chem. 1993;36:2182. doi: 10.1021/jm00067a016. [DOI] [PubMed] [Google Scholar]
- 7.Kubo K, Kohara Y, Yoshimura Y, Inada Y, Shibouta Y, Furukawa Y, Kato T, Nishikawa K, Naka T. J Med Chem. 1993;36:2343. doi: 10.1021/jm00068a011. [DOI] [PubMed] [Google Scholar]
- 8.Ries UJ, Mihm G, Narr B, Hasselbach KM, Wittneben H, Entzeroth M, Van Meel JCA, Wienen W, Kavel NH. J Med Chem. 1993;36:4040. doi: 10.1021/jm00077a007. [DOI] [PubMed] [Google Scholar]
- 9.Bali A, Bansal Y, Sugumaran M, Saggu JS, Balakumar P, Kaur G, Bansal G, Sharma A, Singh M. Bioorg Med Chem Lett. 2005;15:3962. doi: 10.1016/j.bmcl.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 10.Shah DI, Sharma M, Bansal Y, Bansal G, Singh M. Eur J Med Chem. 2008;43:1808. doi: 10.1016/j.ejmech.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 11.Ashton WT, Cantone CL, Chang LL, Hutchins SM, Strelitz RA, Mcross M, Chang RSL, Lotti VJ, Faust KA, Chen T, Bunting P, Schorn TN, Kivlighn SD, Siegl PKS. J Med Chem. 1993;36:591. doi: 10.1021/jm00057a009. [DOI] [PubMed] [Google Scholar]
- 12.Datar PA, Desai PV, Coutinho EC. J Chem Inf Comput Sci. 2004;44:210. doi: 10.1021/ci0341520. [DOI] [PubMed] [Google Scholar]
- 13.Berrllini G, Cruciani G, Mannhold R. J Med Chem. 2005;48:4389. doi: 10.1021/jm049024x. [DOI] [PubMed] [Google Scholar]
- 14.Tuccinardi T, Calderone V, Rapposelli S, Martinelli A. J Med Chem. 2006;49:4305. doi: 10.1021/jm060338p. [DOI] [PubMed] [Google Scholar]
- 15.Zoumpoulakis P, Daliani I, Zervou M, Kyrikou I, Siapi E, Lamprindis G, Mikros E, Mavromoustakos T. Chem Phys Lipids. 2003;125:13. doi: 10.1016/s0009-3084(03)00053-7. [DOI] [PubMed] [Google Scholar]
- 16.Grimmett MR. In: Comprehensive Heterocyclic Chemistry. The Structure, Reactions, Synthesis and Uses of Heterocyclic Compounds. Part 4a. Potts KT, editor. Vol. 5. Pergamon; Oxford: p. 429. [Google Scholar]
- 17.Schild HO. Br J Pharmacol Chemother. 1947;2:189. doi: 10.1111/j.1476-5381.1947.tb00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stanton HC. In: Methods in Pharmacology. Schwartz A, editor. Vol. 1. Appleton-Century-Crofts, Meredith Corporation; New York: 1971. pp. 125–150. [Google Scholar]