

Abstract
The aim of study was to develop self-nanoemulsifying pellets (SNEP) for oral delivery of poorly water soluble drug, repaglinide (RPG). Solubility of RPG in oily phases and surfactants was determined to identify components of self-nanoemulsifying drug delivery system (SNEDDS). The surfactants and cosurfactants were screened for their ability to emulsify oily phase. Ternary phase diagrams were constructed to identify nanoemulsification area for the selected systems. SNEDDS formulations with globule size less than 100 nm were evaluated for in vivo anti-hyperglycemic activity in neonatal streptozotocin rat model. A significant reduction in glucose levels was produced by optimized SNEDDS formulation in comparison to the control group. The optimized SNEDDS formulations were pelletized via extrusion/spheronization technique using microcrystalline cellulose and lactose. SNEP were characterized by X-ray powder diffraction and scanning electron microscopy. X-ray diffraction study indicated loss of crystallinity of RPG in SNEP. The SNEP exhibited good flow properties, mechanical strength and formed nanoemulsion with globule size less than 200 nm. SNEP showed in vitro release of more than 80% RPG in 10 min which was significantly higher than RPG containing reference pellets. In conclusion, our studies illustrated that RPG, a poorly water soluble drug can be successfully formulated into SNEP which can serve as a promising system for the delivery of poorly water soluble drugs.
Key words: anti-hyperglycemic activity, extrusion/spheronization, repaglinide, self-nanoemulsifying pellets
INTRODUCTION
Poor aqueous solubility is considered to lead to low oral bioavailability. The high intra- and inter-subject variability is also documented to be the result of poor solubility. The rate of absorption of poorly soluble compounds from gastrointestinal lumen is governed by dissolution. A number of formulation options such as micronization, salt formation, solid dispersions, cyclodextrin complexation, micellar solutions are very well documented in literature to enhance oral absorption of these candidates. Lipid formulations are reported to reduce the inherent limitations of slow and incomplete dissolution of poorly water soluble drugs by facilitating the formation of solubilized phases which promote absorption (1).
Repaglinide (RPG), a member from meglitinide class, is a post prandial glucose regulator used in management of type II diabetes. It stimulates the release of insulin by binding to a receptor that is distinct from sulfonylurea receptor (2,3). RPG is practically insoluble in water with a solubility of 34 μg/mL (4). It is reported to possess low and variable bioavailability of 50% with high inter-individual variability in plasma concentrations during clinical trials (5,6). Various formulation approaches such as preparation of nanostructured particles (7), complexation with cyclodextrins (8), solid dispersions (9–11), micellar solubilization (12), ultra-rapid freezing technique (13) have been employed to improve the solubility of repaglinide. However, there is no study reported in literature on the application of self- nanoemulsifying drug delivery system to improve the solubility and oral absorption of repaglinide.
SNEDDS are systems comprising of oil, surfactant, cosurfactant and drug which form spontaneous oil-in-water emulsion upon gentle mixing. The agitation essential for in vivo emulsification is provided by the digestive motility of stomach and intestine. SNEDDS present drug in nanosized droplets offering large interfacial area for drug diffusion and absorption (14,15). The possible mechanisms involved in improved absorption include increasing membrane fluidity to facilitate transcellular absorption, opening tight junction to allow paracellular transport, inhibiting P-gp, and/or CYP450 to increase intracellular concentration and residence time by surfactants and stimulating lipoprotein/chylomicron production by lipid (16). The most important advantage of SNEDDS over other novel delivery systems, such as solid dispersions, liposomes, and nanoparticles is their ease of manufacture, scale-up, and excellent stability (17). In addition, the marketed products such as Neoral® (Cyclosporine A), Fortovase (Saquinavir), and Norvir (Ritonavir) represent the commercial viability of the self-emulsifying system to deliver the drug.
There is a concern encapsulating these liquid systems in capsules as there is a possibility of interaction with the capsule shell leading to brittleness or softness and drug leakage (18,19). Adsorbing liquid SNEDDS onto powder would overcome these limitations. In addition, solid SNEDDS would combine the advantages of liquid SNEDDS with those of solid dosage form. Solid self-emulsifying dosage forms like self-emulsifying tablets (20), self-nanoemulsifying granules (21), spray dried powder (22) and pellets (23–26) which can finally be incorporated into hard gelatin capsules are very well documented in literature. Pellets being multiple unit dosage form possess advantages, such as flexibility of manufacture and reduction of intra- and inter-subject variability of plasma profiles thus improving drug safety and efficacy (26). A study conducted by Tuleu et al. have shown equivalent bioavailability of progesterone self-emulsifying formulations presented in a pellet and liquid form (23).
In this study, SNEDDS containing RPG were formulated with the objective of improving solubility and reducing variations in the bioavailability. The optimized SNEDDS formulations were evaluated for in vivo efficacy and converted into self-nanoemulsifying pellets (SNEP) formulation. Pellets were fabricated using lactose and microcrystalline cellulose (MCC) by extrusion/spheronization technique. SNEP were characterized for size distribution, friability, flow properties, X-ray diffraction, SEM, and in vitro release profile.
EXPERIMENTAL
Materials and Methods
Materials
RPG was a generous gift from USV India Pvt. Ltd. (Mumbai, India). Solutol HS-15 (SHS-15), Cremophor EL (CR) (BASF, Mumbai, India), Plurol Oleique, Capryol 90 (CP), Lauroglycol 90, Lauroglycol FCC, Labrafac CC, Labrasol, Labrafac lipophile WL1349, Maisine 35-1 (Colorcon Asia Pvt. Ltd., Mumbai, India), Akoline-MCM (Ak-MCM), Akomed E, Akomed R (Karlshamns AB, Sweden), Imwitor 742 (S. Zaveri & Co., Mumbai, India), Sefsol 228 (Nikko Chemical Co. Ltd., Japan), Acconon MC8 (Ac) (Abitec Corporation, USA) were obtained as gift samples. Hard gelatin capsules were gifted by Associated Capsules Ltd. (Mumbai, India). PEG 400, Propylene glycol, Tween 80 and Tween 20 were purchased from S.D. Fine Chemicals (Mumbai, India). All other reagents and solvents used were of analytical grade. Double-distilled water was prepared freshly whenever required. Neonatal streptozotocin induced Wistar rats were procured from Glenmark Pharmaceuticals Ltd. (Mumbai, India).
Methods
Method of Analysis
High-performance liquid chromatography (HPLC) was used for analysis of RPG in various vehicles and formulations. The HPLC system consisted of PU 2080 pump (Jasco Corporation, Tokyo, Japan) with UV/VIS detector (UV 2075 plus) (Jasco Corporation, Tokyo, Japan) in the range of 200–400 nm. A reverse phase C18 column (250 × 4.6 mm, 5 μm) (HiQ Sil, KyaTech Corporation, Japan) with acetonitrile- phosphate buffer pH 2.5 (45:55) as mobile phase was used for RPG separation. The mobile phase was maintained at a flow rate of 1 mL/min and the detector was fixed at a wavelength of 242 nm.
Solubility Studies
The solubility of RPG in different oils, surfactants and cosurfactants was determined using shake flask method. To each vial containing 1 g of vehicle, i.e., oil, surfactant, or cosurfactant, an excess quantity of RPG was added. The vial was sealed and mixture was vortexed using a CM101, cyclomixer (Remi, Mumbai, India) for 10 min to aid proper mixing of RPG with the vehicles. The mixtures were shaken for 48 h in a water bath shaker (Remi Motors, Mumbai, India) maintained at room temperature followed by centrifugation at 5,000 rpm for 5 min. The supernatant was separated and diluted appropriately with methanol. HPLC method as described under “Method of Analysis” was used to quantify dissolved RPG. The concentration of RPG was determined at detection wavelength of 242 nm. The experiment was performed in triplicate and results were represented as mean value (in milligram per gram) ± SD.
Preliminary Screening of Surfactants
The surfactants were screened for emulsification ability as per method reported in literature (27). Oil was mixed with each surfactant in 1:1 ratio and vortexed for 5 min to ensure proper mixing. This mixture, 50 mg, was weighed and diluted to 50 mL with double-distilled water to obtain emulsion. The emulsions were observed for physical appearance, clarity and phase separation. The emulsions were left undisturbed for 2 h and analyzed for transmittance at 638.2 nm on UV-160A double beam spectrophotometer (Jasco Corporation, Japan) using double distilled water as blank. The experiment was performed in duplicate.
Preliminary Screening of Cosurfactants
The turbidimetric method described under preliminary screening of surfactants was used to screen the cosurfactants. During screening, oil/surfactant/cosurfactant ratio was kept constant as 3:2:1. The experiment was performed in duplicate.
Construction of Ternary Phase Diagrams
Ternary mixtures were prepared by mixing oil, surfactant, and cosurfactant. Various combinations of surfactant, cosurfactant and oil were used and their concentrations were varied in each formulation. The concentration of surfactant and cosurfactant was varied from 14 to 60% (w/w) for construction of phase diagram. Twenty five experiments were performed for a particular combination of oil, surfactant, and cosurfactant. Each formulation, 200 mg was diluted to 10 mL with distilled water and observed for nanoemulsion (NE) formation. The mean globule size of stable NEs was recorded by Photon Correlation Spectroscopy (Beckmann N5 coulter, Miami, FL, USA). The formulations with mean globule size of 150 nm or less were considered desirable. Phase diagrams were plotted for each combination of oil, surfactant, and cosurfactant and the area of NE formation was determined. The emulsions with desired globule size determined the area of NE formation for each system.
Preparation of SNEDDS
The required quantity of oil, surfactant, and cosurfactant were mixed thoroughly by stirring on a CM101, cyclomixer (Remi, Mumbai, India). The requisite quantity of RPG was weighed accurately and dissolved in the mixture by stirring. The mixtures were equilibrated for 12 h to observe for any signs of phase separation. SNEDDS were filled in size “5” hard gelatin capsules until further evaluation.
Effect of pH on Stability and NE Globule Size
SNEDDS, 200 mg was diluted to 10 mL with distilled water, pH 1.2 and pH 5.0 buffer separately. Visual observations were made immediately for assessment of self-nanoemulsification efficiency, appearance, phase separation and drug precipitation. The mean globule size was determined using photon correlation spectroscopy. The physical stability of NE was assessed by allowing the dispersions to stand for 12 h at room temperature.
Effect of Dilution and pH of Dilution
The optimized formulations were evaluated for robustness to dilution and the effect of pH of dilution media. The formulations were diluted 50-, 100-, and 1000-fold with water, simulated gastric fluid (0.1 N HCl) and pH 5.0 buffer. The emulsions were monitored immediately and after 6 h of dilution for any physical changes such as precipitation or phase separation.
Dispersion Test
SNEDDS filled in hard gelatin capsules were evaluated for dispersion in pH 5.0 buffer, 500 mL at 75 rpm using USP type II apparatus (Electrolab, Mumbai, India). Dispersion and solubilization of RPG in the dispersion medium was determined by HPLC with detection wavelength of 242 nm.
Stability study
SNEDDS formulations, F4 and F6 were stored in sealed amber glass vials under various storage conditions, namely 5 ± 3°C, 25 ± 2°C/65 ± 5% RH, 40 ± 2°C/75 ± 5% RH for a period of 2 months and evaluated for stability. The physical stability was determined by monitoring any drug precipitation from the formulations. The chemical stability was assessed by determining RPG content in SNEDDS using HPLC.
Pharmacodynamic Activity in Rats
The animal study protocol was approved by Institutional Animals Ethics Committee IAEC/CPCSEA, Mumbai. Neonatal streptozotocin induced rat model of type 2 diabetes mellitus was used for the study. Wistar rats in weight range of 150–170 g, 6–8 weeks old were used. The animals were housed in cages in a randomized order in groups of six at a constant temperature of 23 ± 2°C and 55 ± 5% RH humidity with a light/dark cycle of 12 h. All the animals were given food (pellet diet supplied by Hindustan Lever Ltd) and water ad libitum. Rats were fasted overnight (18 h) and not fed during the study, although they had free access to water during night and during the experimental protocol. Oral glucose tolerance test (OGTT) was performed on neonatal streptozotocin rats to evaluate the anti-hyperglycemic action. Blood was withdrawn from overnight fasted rats followed by oral administration of test samples (RPG and SNEDDS) under ketamine anesthesia. Glucose, 2 g/kg was administered 60 min post treatment to test samples. Blood samples were collected via retro-orbital plexus under ketamine anesthesia at 30, 60, 90, and 120 min after glucose administration. The serum glucose levels were determined using glucose estimation kits (Erba diagnostics, India). Statistical analysis was performed to determine the difference in glucose levels of control and treatment groups using one-way analysis of variance followed by Bonferroni’s Multiple Comparison test. The results were considered to be significant at p < 0.05.
Formulation and Optimization of Self-Nanoemulsifying Pellets of RPG
SNEP were prepared by extrusion/spheronization technique using microcrystalline cellulose PH101 (MCC) and lactose in 2:1 ratio. Kollidon® CL was accurately weighed and added to MCC, lactose blend. SNEDDS formulation was added slowly to the powder blend with thorough mixing. Polyvinyl pyrrolidone, 2% (w/v) was added in small proportion until a mass suitable for extrusion was obtained. The wet mass was extruded through 1 mm screen (B. S. S. 18 mesh) to obtain extrudates. Spheronizer (Fuji Paudal Q-230, Japan) fitted with a cross-hatch frictional plate of dimensions 1 mm × 1 mm × 1 mm was used to obtain spheres. The extrudates were spheronized at 690 rpm for 2 min and dried in an oven at 45°C until constant weight was obtained. SNEP containing RPG equivalent to 1 mg were filled in size “3” hard gelatin capsules manually.
Size Distribution of SNEP
Pellets, 10 g were manually shaken on a nest of British standard sieves (1,000, 710, 425, 250, and 180 μm) for 5 min. The pellet fraction retained on each sieve was weighed and a frequency distribution curve was constructed. The arithmetic mean value was determined from the cumulative frequency oversize curve.
Measurement of Flow Property and Friability
The flow characteristics of SNEP were evaluated by determining angle of repose. The values for Hausner’s ratio and Carr’s index were also calculated. The friability of SNEP was assessed using Roche friabilator. SNEP, 5 g were placed in the drum and rotated for 4 min at 25 rpm. The weight of pellets was determined and friability was calculated.
X-Ray Powder Diffraction (XRPD)
XRPD patterns were recorded on a Philips PW 17291 powder X-ray diffractometer using Ni-filtered, Cu Kα radiation, a voltage of 40 kV and a 25 mA current. The scanning rate employed was 1°/ min over 10–40° 2θ angle. The XRPD patterns of RPG, RPG reference pellets and SNEP were recorded.
Morphological Analysis of SNEP
The surface morphology of SNEP was characterized using scanning electron microscope (Jeol JSM-840, USA). Gold sputter coating of all the samples was done to make the surface of particles electroconductive.
Globule Size Determination of SNEP
SNEP, 100 mg were dispersed in 5 mL distilled water using cyclomixer to determine the mean globule size distribution. The mixtures were left undisturbed for 15 min to allow the solid content to sediment. The supernatant was filtered through a coarse filter (Whatman filter No.1, 90 mm diameter) and the filtrate was used to determine globule size distribution using photon correlation spectroscopy.
In Vitro Drug Release Study
USP type II apparatus (Electrolab, Mumbai, India) was used to study the release of RPG from SNEP capsules. In vitro release study was performed in 500 mL, pH 5.0 buffer at 75 rpm as indicated in United States Pharmacopeia. The samples were withdrawn at predetermined time intervals and analyzed using HPLC with a detection wavelength of 242.0 nm. The dissolution studies were conducted in triplicate and the average RPG release ±SD was calculated.
RESULTS
Method of Analysis
The mean calibration curve for RPG was given by the equation y = 75799x − 8942.3, where y represents area under the curve and x is the concentration in microgram per milliliter. The method was found to be linear in the range of 0.5-50 μg/mL with a coefficient of correlation of 0.9999.
Solubility Studies
Solubility studies were undertaken to recognize oil, surfactant/cosurfactant that exhibit maximum solubility for RPG. The solubility of RPG in different oils and various surfactants and cosurfactants is presented in Figures 1 and 2. respectively. CP showed maximum solubility for RPG as compared to other oils. Hence, CP was used as oily phase for further studies. The surfactants and cosurfactants were selected based on two parameters, ability to solubilize RPG and their emulsification efficiency.
Fig. 1.

Solubility of RPG in various oily phases. Data expressed as mean ± SD, n = 3
Fig. 2.

Solubility of RPG in various surfactants and cosurfactants. Data expressed as mean ± SD, n = 3
Screening of Surfactants for Emulsifying Ability
Turbidimetry studies were performed to evaluate the ability of various surfactants to emulsify the selected oily phase, CP. For oil–surfactant mixture to be used in SNEDDS formulation, it was essential to determine whether it could disperse efficiently to form spontaneous NE. It has been reported that well formulated SNEDDS get dispersed within seconds under gentle stirring conditions (28). CR, SHS-15, Tween 20, and Tween 80 were selected for emulsification study as they showed good solubility potential for RPG. The percent transmittance values of various dispersions are quoted in Table I. CR and SHS-15 were found to emulsify the dispersions in a short time as compared to Tween 20 and Tween 80. The percent transmittance value for all dispersions was found to be more than 90%. This suggests good emulsification ability of all the four surfactants for the selected oily phase, CP. Hence, all four surfactants were used for further investigation.
Table I.
Emulsification Efficiency of Various Surfactants
| Surfactant | Transmittancea (%) |
|---|---|
| Cremophor EL | 94.6 |
| Solutol HS 15 | 93.3 |
| Tween 20 | 99.4 |
| Tween 80 | 97.4 |
aData expressed as mean (n = 2)
Screening of Cosurfactants
Addition of a cosurfactant to surfactant containing formulation is reported to improve dispersibility and drug absorption from the formulation (29). As the ratio of cosurfactant to surfactant was kept constant, the turbidity of resulting NEs helped in assessing the relative efficacy of the cosurfactants to improve the nanoemulsification ability of surfactants. The percent transmittance values depicted in Table II indicate maximum emulsification ability of PEG 400 and Ac amongst the cosurfactants tried.
Table II.
Emulsification Studies on Surfactant/ Cosurfactant Combinations
| Cosurfactant | Transmittancea (%) | |||
|---|---|---|---|---|
| Cremophor EL | Solutol HS 15 | Tween 20 | Tween 80 | |
| PEG 400 | 99.55 | 92.7 | 86.57 | 92.01 |
| Acconon MC8 | 98.37 | 97.52 | 72.1 | 77.70 |
| Capmul MCM L | 92.60 | 87.48 | 44.97 | 66.45 |
aData expressed as mean, n = 2
Based on the results of preliminary screening, two systems were selected:
System A: Consisted of CP as oily phase/ CR as surfactant/ PEG 400 as cosurfactant
System B: Consisted of CP as oily phase/ SHS-15 as surfactant/ Ac as cosurfactant
Construction of Phase Diagrams
Ternary phase diagram was plotted for system A (CP, CR, PEG 400) and system B (CP, SHS-15, Ac) to identify the nanoemulsification region and to determine the optimum concentrations of oil, surfactant, and cosurfactant to form a stable self-nanoemulsifying formulation. The outer region in the phase diagram represents the area studied for locating nanoemulsification region and the shaded region represent the formulations with desired globule size. In the current investigation, system A gave a wider nanoemulsification region (Fig. 3) as compared to system B (Fig. 4). This shows better self- nanoemulsification property of the former system. A minimum surfactant concentration of 25% for system A and 33.33% for system B was required to form clear and stable nanoemulsions. It was observed that more than 14% (w/w) oil was essential to keep RPG in solubilized state after conversion into nanoemulsion.
Fig. 3.

Ternary phase diagram of CP, CR and PEG 400 system
Fig. 4.

Ternary phase diagram of CP, SHS-15, Ac system
Effect of pH on Stability and NE Globule Size
The composition of optimized SNEDDS formulations is indicated in Table III. The optimized formulations were selected based on their globule size and stability upon dilution after 12 h. F1, F2, F3, F4, F5, and F6 did not show any signs of drug precipitation or phase separation after 12 h. The effect of pH was studied to determine the influence of gastrointestinal tract pH on the mean globule size and stability of emulsion. SNEDDS were diluted with different buffer solutions and the mean globule size was recorded as given in Table IV. None of the formulations showed any signs of phase separation or drug precipitation at different pH conditions. The mean globule size of the formulations was in the range of 47–180 nm. However, an increase in globule size was observed when pH 5.0 buffer was used as dilution medium but the mean size remained below 200 nm. Formulations F4 and F6 showed lowest globule sizes hence were selected for further studies. The globule size of F4 and F6 was found to be 65.1 nm ± 2.33 and 56.2 nm ± 1.27 respectively when evaluated after 12 h.
Table III.
Optimized Formulae of SNEDDS
| Ingredients | Quantity (mg) | |||||
|---|---|---|---|---|---|---|
| F1 | F2 | F3 | F4 | F5 | F6 | |
| RPG | 1 | 1 | 1 | 1 | 1 | 1 |
| CP | 10 | 25 | 16.66 | 18.75 | 12.5 | 10 |
| CR | 20 | 8.33 | 25 | 18.75 | – | – |
| SHS-15 | – | – | – | – | 25 | 30 |
| PEG 400 | 20 | 16.66 | 8.33 | 12.5 | – | – |
| Ac | – | – | – | – | 12.5 | 10 |
Table IV.
Mean Globule Size of RPG SNEDDS and their Respective SNEP in Different pH Media
| Medium | Distilled water | pH 1.2 | pH 5.0 |
|---|---|---|---|
| SNEDDS | |||
| F1 | 117 ± 12.7 | 105 ± 3.7 | 132.6 ± 4.38 |
| F2 | 145.8 ± 13.7 | 150 ± 1.21 | 180 ± 7.65 |
| F3 | 88.65 ± 1.20 | 83.1 ± 4.7 | 103.8 ± 5.9 |
| F4 | 63.3 ± 0.03 | 47.3 ± 0.58 | 117 ± 0.66 |
| F5 | 107.1 ± 6.22 | 110 ± 4.66 | 180.1 ± 3.88 |
| F6 | 55.3 ± 1.21 | 66.8 ± 0.82 | 130.3 ± 2.17 |
| SNEP | |||
| F4 | 80.2 ± 6.97 | 78.1 ± 3.13 | 122.8 ± 2.87 |
| F6 | 107.9 ± 1.93 | 97.1 ± 5.64 | 155.1 ± 5.18 |
Each value is mean ± SD of two determinations
Effect of Dilution and pH of Dilution
The effect of dilution and pH of dilution is an important parameter for evaluation since the delivery system is subjected to dilution by body fluids after administration. F4 and F6 SNEDDS dispersed well upon dilution and formed NE without any precipitation of drug. The resulting NEs were robust to all dilutions in different media with no signs of phase separation or drug precipitation after 6 h.
Dispersion Test
The optimized SNEDDS, F4 and F6 exhibited good dispersibility and formed a stable emulsion in the dispersion medium. F4 and F6 SNEDDS were found to disperse easily within 5 min from the capsule. The dispersion profile of F4 showed 94% release of RPG whereas F6 released 95% RPG in 5 min. Both the formulations gave 100% release of RPG in 10 min.
Stability Study
F4 and F6 were chemically stable when stored at 5°C and at ambient conditions for a period of 2 months (data not shown). However, the content of RPG was reduced by 10% when stored at 40°C (data not shown). This suggests the formulations should be stored at ambient and refrigerated conditions. The formulations were physically stable with no drug precipitation.
Pharmacodynamic Activity in Rats
F4 and F6 were evaluated for anti-hyperglycemic activity using OGTT. Blood glucose levels (Fig. 5) of all groups reached a peak at 60 min post glucose administration and thereafter a gradual decrease was observed. SNEDDS treated groups significantly (p < 0.01) restricted the increase in blood glucose levels at 60 min post glucose administration when compared to control group. No significant difference was observed in blood glucose levels of RPG suspension treated group and control group, 60 min post glucose load. The blood glucose levels of RPG suspension treated group was found to be 340 ± 24.29 mg/dl, 60 min post glucose administration. On the other hand, the blood glucose levels for F4 and F6 SNEDDS treated groups were found to be 233 ± 7.11 mg/dl and 220 ± 2.98 mg/ml respectively, 60 min post glucose load. The formulations showed a significant (p < 0.05) reduction in glucose levels of rats when compared with standard RPG suspension, 60 min post glucose administration. This confirms the superiority of SNEDDS treated groups in inhibiting the increase in blood glucose levels. F4 and F6 offered a significant reduction (p < 0.05 and p < 0.01 respectively) compared to RPG suspension at 180 min. The reduction in glucose levels offered by SNEDDS formulation is due to increase in the dispersion of RPG leading to faster and better absorption.
Fig. 5.

Effect of SNEDDS formulation on oral glucose tolerance test in rats. Value is expressed as mean ± SEM (n = 5), *significant with respect to control group, #significant with respect to RPG suspension treated group
Preparation and Optimization of SNEP Formulation
F4 and F6 SNEDDS were fabricated into SNEP formulations by extrusion spheronization technique. Preliminary studies were carried out to identify the excipients suitable for development of SNEP. Trials were conducted using different proportions of lactose: MCC and MCC alone to assess the possibility of obtaining SNEP. The prepared pellets were assessed for size and shape in order to arrive at an optimum ratio of excipients. The spheronization speed was optimized to 690 rpm. From the preliminary studies it could be concluded that MCC: lactose when used in 2:1 proportion gave SNEP with good sphericity. Initially, the proportion of excipients to SNEDDS was identified to completely adsorb SNEDDS. When one part of solid (lactose and MCC) was used to adsorb one part of SNEDDS, the pellets obtained were found to stick to each other during the spheronization process. This suggests incomplete adsorption of SNEDDS on the surface of excipients. It was observed that two parts of solid (lactose-MCC) effectively adsorbed one part of liquid SNEDDS. Polyvinylpyrrolidone, 2% (w/v) was identified as an appropriate binding agent that imparted good mechanical strength to SNEP. Table V gives the composition of SNEP formulations.
Table V.
Formulae of Optimized SNEP Formulations
| Ingredients (% w/w) | F4 SNEP | F6 SNEP | Reference RPG pellets |
|---|---|---|---|
| RPG | – | – | 0.93 |
| SNEDDS | 33.15 | 33.15 | – |
| MCC | 43.22 | 43.36 | 62.57 |
| Lactose | 21.58 | 21.65 | 31.23 |
| Kollidon CL | 1.29 | 1.3 | 1.87 |
| PVP | 0.84 | 0.52 | 3.37 |
Size Distribution of SNEP
Details of the percentage of F4 SNEP and F6 SNEP retained on various sieve fractions are presented in Table VI. SNEP in the size range of 710–1,000 μm were considered appropriate and reported as pellet yield.
Table VI.
Percentage of the Optimized SNEP in Each Size Range
| Size range (μm) | Percentage of pellets (%) | |
|---|---|---|
| F4 SNEP | F6 SNEP | |
| 1,200–1,000 | 27.956 | 15.315 |
| 1,000–710 | 51.843 | 65.09 |
| 710–425 | 15.821 | 17.267 |
| 425–250 | 3.609 | 1.576 |
| 250–180 | 0.768 | 0.75 |
Determination of Flow Property and Friability
The angle repose for F4 SNEP and F6 SNEP was found to be 20.36° and 25.97°, respectively. The Hausner’s ratio for F4 SNEP and F6 SNEP was 1.135 and 1.145, respectively, thus indicating good flow ability of the self-nanoemulsifying pellets. The value for Carr’s index was less than 15 thus confirming excellent flow property of SNEP. F4 SNEP and F6 SNEP exhibited friability values less than 1%, indicating good mechanical strength.
X-Ray Powder Diffraction
Figure 6 shows the XRPD patterns for RPG, reference pellets containing RPG, F4 SNEP and F6 SNEP. The XRPD pattern of RPG showed sharp peaks at a diffraction angle (2θ) of 7.55, 10.01, 20.19, 16.58, 22.85, 23.93 and 18.53 which indicates presence of crystalline RPG. Drug crystallinity peaks were also detectable in XRPD pattern of reference RPG pellets. XRPD patterns of SNEP were characterized by diffuse spectra and the characteristic peaks of RPG were not observed. This indicates there was no precipitation of RPG while converting SNEDDS into SNEP.
Fig. 6.

XRD spectra of (a) F4 SNEP, (b) F6 SNEP, (c) reference RPG pellets, (d) RPG
Morphological Analysis of SNEP
The surface morphology of SNEP is depicted in Fig. 7. F4 SNEP and F6 SNEP showed a regular circular shape with no RPG crystals evident on the smooth pellet surface. This indicates presence of RPG in a solubilized state in the self-nanoemulsifying pellet formulations.
Fig. 7.
Scanning electron micrographs of optimized SNEP
Globule Size Determination of SNEP
The droplet size determination following self-nanoemulsification is a critical factor in evaluation of SNEDDS since droplet size is reported to have an effect on drug absorption. The smaller is the droplet size; the larger is the interfacial surface area available for drug absorption. The mean globule size of NEs produced on mixing SNEP with aqueous media is given in Table IV. SNEP were converted to stable NEs after mixing with the dilution media, however an increase in the mean globule size was observed. The increase could be attributed to the time taken by surfactant and cosurfactant to release from the pellet matrix. Although, an increase in globule size was observed, the mean size was below 200 nm.
In Vitro Drug Release Study
Reference pellets comprising of RPG, microcrystalline cellulose, lactose, Kollidon CL and PVP K30 (binder) were prepared. The content of RPG in F4 SNEP, F6 SNEP and reference pellets was determined. Table VII indicates the content of RPG in the prepared formulations. In vitro dissolution profile of F4 SNEP, F6 SNEP and reference pellets is shown in Fig. 8. The capsules containing F4 SNEP exhibited a release of 79% RPG whereas F6 SNEP capsules released 85% RPG in 10 min. On the other hand, capsules containing reference pellets released only 5% RPG in 10 min followed by a cumulative release of 17% RPG at the end of 1 h.
Table VII.
RPG Content in SNEP (in milligram) and Percent RPG Dissolved in 10 min from reference Pellets and SNEP
| Parameters | Reference RPG pellets | F4 SNEP | F6 SNEP |
|---|---|---|---|
| RPG content (mg) | 1.01 ± 0.21 | 0.98 ± 0.02 | 0.99 ± 0.02 |
| %DP10 | 5.11 ± 1.06 | 79.05 ± 3.07 | 86.12 ± 0.66 |
Fig. 8.
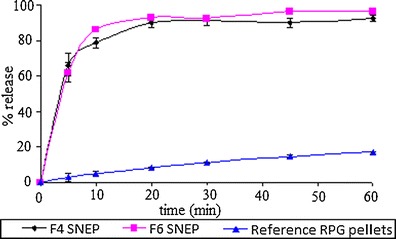
Comparison of dissolution profile of capsules filled with SNEP and reference RPG pellets
DISCUSSION
Self-nanoemulsifying drug delivery systems have gained tremendous popularity due to their ability to form spontaneous oil-in-water emulsion. These systems not only improve drug solubilization but also enhance the release and absorption properties due to the already dissolved form of drug in the formulation (30). Several researchers have formulated these systems into solid form, i.e., solid SNEDDS to overcome the stability and compatibility issues that arise upon incorporation in capsule. The idea of combining the advantages of self-emulsifying system with pellets using extrusion/spheronization technique was introduced by Newton et al. (31). In the current investigation, self-nanoemulsifying pellet formulation of RPG was designed to surmount its low solubility and bioavailability.
It is known that only a very specific combination of pharmaceutical excipients lead to formation of an efficient self-emulsifying system. Therefore, selection of appropriate excipients possessing good solubilization capacity for drug is desirable for development of SNEDDS. The solubility of drug in oily phase is critical as it would determine the drug loading capacity. CP showed highest solubilization for RPG when compared to other oily phases. If the surfactant or cosurfactant is contributing to drug solubilization, there could be a risk of precipitation upon dilution in in vivo environment (15). Non-ionic surfactants are generally considered to be non-toxic than ionic surfactants. The efficiency of self-emulsification is related to the HLB of surfactants. Surfactants with HLB values greater than 10 are suitable for formation of o/w emulsion. Hence, all the surfactants screened possessed HLB higher than 10. CR, SHS-15, Tween 20 and Tween 80 were chosen as surfactants as they showed good solubilizing potential for RPG and possessed good emulsification ability. PEG 40 and Ac as cosurfactants were effective in improving the nanoemulsification ability of the surfactants, CR and SHS-15.
The ternary phase diagram plot for system B showed a smaller nanoemulsification area when compared to system A. In case of system A, an increase in surfactant concentration was found to decrease the globule size of the formed nanoemulsions. The decrease in droplet size is a result of more surfactant being available to stabilize oil–water interface. In case of system B, high surfactant concentration was required to form a stable interfacial film in order to stabilize the oil droplets when compared to system A. The number of stable nanoemulsions obtained after dilution were also found to be less as compared to system A. Both the systems, A & B required more than 14% oil to keep RPG in solubilized state after conversion into nanoemulsion. However, it was also observed that systems containing less than 20% oil phase caused RPG to precipitate out of solution when observed after 12 h. This could be due to insufficient concentration of oil to keep the drug in solubilized state in the formulation. Therefore, it is very essential to assess the stability of formulations after dilution.
The influence of emulsion droplet size on bioavailability was clearly demonstrated by the commercial products, Neoral and Sandimmune formulations of cyclosporine. Neoral with droplet size of approximately 30 nm showed improved and less variable bioavailability compared to the earlier Sandimmune formulation, which formed a coarse emulsion with droplet size of 2 to 5 μm (32). The globule sizes of prepared SNEDDS formulations were in the range of 47–180 nm. It is necessary to assess the solubilization capacity of SNEDDS in different media as it would give an idea about its in vivo performance. An increase in globule size of all the optimized formulations was observed in pH 5.0 buffer. There was no precipitation of drug or phase separation in different pH media. F4 and F6 SNEDDS showed no significant change in globule size on evaluation after 12 h, thus indicating stability of the formulations. F4 and F6 SNEDDS were robust to dilution and showed good stability in water, pH 1.2 and pH 5.0 buffer even after 6 h of dilution.
The SNEDDS formulations easily dispersed in dissolution media with 100% release of RPG in 10 min. The stability study results revealed chemical stability of F4 and F6 SNEDDS formulations when stored at ambient and refrigerated conditions for a period of 2 months.
Repaglinide is a post- prandial glucose regulator used in the treatment of type II diabetes mellitus. The ability of SNEDDS formulations to reduce blood glucose levels was studied in neonatal streptozotocin induced rat model. SNEDDS were effective and superior in inhibiting the increase in blood glucose levels when compared to RPG suspension group and animal control group. The reduction in the glucose levels could be due to effective solubilization of RPG in SNEDDS leading to faster and complete absorption.
It is desirable to convert liquid SNEDDS into solid form to overcome the limitations associated with liquid SNEDDS. The selection of a particular process to prepare solid SNEDDS would depend on the content of oily excipients in the formulation, properties of API like solubility, heat stability, and compatibility with other ingredients. F4 and F6 SNEDDS formulations were converted to pellet dosage form by using extrusion–spheronization technique. The identification of appropriate excipients and the ratio of excipients to liquid SNEDDS are very important in the fabrication of SNEP. The ratio of excipients to liquid SNEDDS needs to be tailored in such a manner that there is effective adsorption without any loss of drug during the process. Microcrystalline cellulose: lactose in 2:1 proportion was used to prepare SNEP. Two parts of solid (MCC-lactose) was required to adsorb one part of liquid SNEDDS.
The compressibility index and friability values indicated good flow property and mechanical strength for F4 SNEP and F6 SNEP. Solid SNEDDS after mixing with the dilution media should be able to regain their self-nanoemulsifying properties and form stable NE. F4 and F6 SNEP were converted to stable NEs after mixing with the dilution media. An increase in the mean globule size was observed but it was below 200 nm. This could be attributed to the time taken by surfactant and cosurfactant to release from pellet matrix. The physical state of RPG in SNEP formulation was investigated as it would have an important influence on its in vitro and in vivo release characteristics (22). X-ray diffraction spectra of SNEP revealed there was no drug precipitation during conversion of SNEDDS to SNEP. SEM study also indicated absence of any drug crystals on the pellet surface thus confirming presence of RPG in solubilized state in the self-nanoemulsifying pellet formulation. The in vitro drug release profiles of reference pellets showed a release of only 16.9% RPG at the end of one hour whereas F4 and F6 SNEP gave a release of more than 80% RPG in 10 min. The faster drug release observed from SNEP could be due to solubilizing ability of surfactants and cosurfactants and lower globule size of the formed nanoemulsion. The faster dissolution from SNEPs may be ascribed to the fact that in these formulations the drug is in solubilized form and upon exposure to dissolution medium form small droplets that dissolve rapidly in dissolution medium.
CONCLUSIONS
In this study, SNEDDS of RPG were designed which showed good release profiles and exhibited a rapid rate of emulsification. The optimized SNEDDS formulations showed a significant reduction in blood glucose levels in comparison to RPG suspension. The present investigation indicates that SNEDDS could be successfully developed into pellets by extrusion/spheronization technique. The resulting SNEP possessed good mechanical strength with preserved self-nanoemulsifying properties. The superior dissolution profile of SNEP capsule dosage form confirms the use of this system as a potential delivery system for water-insoluble drugs.
Acknowledgments
The authors are thankful to USV India Pvt. Limited, India for providing gift sample of repaglinide. We are thankful to Colorcon Asia Pvt. Ltd., India; Gattefosse India Pvt Ltd., India; BASF India Ltd., India; Karlshamns AB., Sweden; S. Zaveri & Co., India; for providing gift samples of oils and surfactants. The authors are also thankful to Associated Capsules, India, for gift sample of hard gelatin capsules. The authors wish to thank Mr. Nilesh Kulkarni, Tata Institute of Fundamental Research (TIFR), Mumbai, India for carrying out X-ray diffraction studies. The authors are thankful to the Department of Biotechnology (DBT), New Delhi, for providing fellowship.
References
- 1.Humberstone AJ, Charman WN. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv Drug Deliv Rev. 1997;25:103–128. doi: 10.1016/S0169-409X(96)00494-2. [DOI] [Google Scholar]
- 2.Culy C, Jarvis B. Repaglinide, a review of its therapeutic use in type 2 diabetes mellitus. Drugs. 2001;61:1625–1660. doi: 10.2165/00003495-200161110-00008. [DOI] [PubMed] [Google Scholar]
- 3.Malaisse W. Repaglinide, a new oral antidiabetic agent: a review of recent preclinical studies. Eur J Clin Invest. 1999;29:21–29. doi: 10.1046/j.1365-2362.1999.00001.x. [DOI] [PubMed] [Google Scholar]
- 4.Mandic Z, Gabelica V. Ionization, lipophilicity and solubility properties of repaglinide. J Pharm Biomed Anal. 2006;41(3):866–871. doi: 10.1016/j.jpba.2006.01.056. [DOI] [PubMed] [Google Scholar]
- 5.Marbury TC, Ruckle JL, Hatorp V, Andersen MP, Nielsen KK, Huang WC, Strange P. Pharmacokinetics of repaglinide in subjects with renal impairment. Clin Pharmacol Ther. 2000;67:7–15. doi: 10.1067/mcp.2000.103973. [DOI] [PubMed] [Google Scholar]
- 6.Davis SN, Granner DK. Insulin, oral hypoglycemic agents and the pharmacology of the endocrine pancreas. In: Hardman JG, Limbrid LE, editors. Goodman and Gilman’s the pharmacological basis of therapeutics. USA: McGraw-Hill; 2001. pp. 1704–1705. [Google Scholar]
- 7.Sinswat P, Matteucci ME, Johnston KP, Williams RO., III Dissolution rates and supersaturation behavior of amorphous repaglinide particles produced by controlled precipitation. J Biomed Nanotechnol. 2007;3:18–27. doi: 10.1166/jbn.2007.001. [DOI] [Google Scholar]
- 8.Desai NS, Bramhane DM, Nagarsenker MS. Repaglinide–cyclodextrin complexes: preparation, characterization and in vivo evaluation of antihyperglycemic activity. J Incl Phenom Macrocycl Chem. 2011;70(1–2):217–225. doi: 10.1007/s10847-010-9895-0. [DOI] [Google Scholar]
- 9.Zawar LR, Bari SB. Preparation, characterization and in vivo evaluation of antihyperglycemic activity of microwave generated repaglinide solid dispersion. Chem Pharm Bull. 2012;60(4):482–487. doi: 10.1248/cpb.60.482. [DOI] [PubMed] [Google Scholar]
- 10.Kavitha R, Sathali AAH. Enhancement of solubility of repaglinide by solid dispersion technique. Int J Chem Sci. 2012;10(1):377–390. [Google Scholar]
- 11.Patel M, Pandya N, Bhaskar VH. Preparation, characterization and in vitro evaluation of repaglinide binary solid dispersions with hydrophilic polymers. Int J Drug Dev & Res. 2011;3(2):107–117. [Google Scholar]
- 12.Seedher N, Kanojia M. Micellar solubilization of some poorly soluble antidiabetic drugs: a technical note. AAPS PharmSciTech. 2008;9(2):431–436. doi: 10.1208/s12249-008-9057-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Purvis T, Mattucci ME, Todd MC, Johnston KP, Williams RO., III Rapidly dissolving repaglinide powders produced by the ultra-rapid freezing process. AAPS PharmSciTech. 2007;8(3):E52–E60. doi: 10.1208/pt0803058. [DOI] [PubMed] [Google Scholar]
- 14.Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58:173–182. doi: 10.1016/j.biopha.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Lawrence MJ, Rees GD. Microemulsion-based media as novel drug delivery systems. Adv Drug Deliv Rev. 2000;45:89–121. doi: 10.1016/S0169-409X(00)00103-4. [DOI] [PubMed] [Google Scholar]
- 16.Wei W, Yang W, Li Q. Enhanced bioavailability of silymarin by self-microemulsifying drug delivery system. Eur J Pharm Biopharm. 2006;63:288–294. doi: 10.1016/j.ejpb.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Date A, Desai N, Dixit R, Nagarsenker M. Self-nanoemulsifying drug delivery systems: formulation insights, applications and advances. Nanomedicine. 2010;5(10):1595–1616. doi: 10.2217/nnm.10.126. [DOI] [PubMed] [Google Scholar]
- 18.Chang RK, Raghavan KS, Hussain MA. A study on gelatin capsule brittleness: moisture transfer between the capsule shell and its content. J Pharm Sci. 1998;87:556–558. doi: 10.1021/js9704238. [DOI] [PubMed] [Google Scholar]
- 19.Mei X, Etzler FM, Wang Z. Use of texture analyzer to study hydrophilic solvent effects on the mechanical properties of hard gelatin capsules. Int J Pharm. 2006;324:128–135. doi: 10.1016/j.ijpharm.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 20.Mahmoud EA, Bendas ER, Mohamed MI. Preparation and evaluation of self-nanoemulsifying tablets of carvedilol. AAPS Pharm Sci Tech. 2009;10:183–192. doi: 10.1208/s12249-009-9192-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dixit RP, Nagarsenker MS. Formulation and in vivo evaluation of self-nanoemulsifying granules for oral delivery of a combination of ezetimibe and simvastatin. Drug Dev Ind Pharm. 2008;34:1285–1296. doi: 10.1080/03639040802071570. [DOI] [PubMed] [Google Scholar]
- 22.Balakrishnan P, Lee BJ, Oh DH, Kim JO, Hong MJ, Jee JP, Kim JA, Yoo BK, Woo JS, Yong CS, Choi HG. Enhanced oral bioavailability of dexibuprofen by a novel solid self-emulsifying drug delivery system (SEDDS) Eur J Pharm Biopharm. 2009;72:539–545. doi: 10.1016/j.ejpb.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 23.Tuleu C, Newton JM, Rose J, Euler D, Saklatvala R, Clarke A, Booth S. Comparative bioavailability study in dogs of a self emulsifying formulation of progesterone presented in a pellet and liquid form compared with an aqueous suspension of progesterone. J Pharm Sci. 2004;93:1495–1502. doi: 10.1002/jps.20068. [DOI] [PubMed] [Google Scholar]
- 24.Newton JM, Pinto MR, Podczeck F. The preparation of pellets containing a surfactant or a mixture of mono- and di-glycerides by extrusion/spheronization. Eur J Pharm Sci. 2007;30:333–342. doi: 10.1016/j.ejps.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 25.Xiongwei H, Chen L, Dingxiong C, Jing Z, Zhihong L, Wei W, Hongtao S. Sirolimus solid self-microemulsifying pellets: formulation development, characterization and bioavailability evaluation. Int J Pharm. 2012;438:123–133. doi: 10.1016/j.ijpharm.2012.07.055. [DOI] [PubMed] [Google Scholar]
- 26.Abdalla A, Mäder K. Preparation and characterization of a self emulsifying pellet formulation. Eur J Pharm Biopharm. 2007;66:220–226. doi: 10.1016/j.ejpb.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 27.Date AA, Nagarsenker MS. Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for cefpodoxime proxetil. Int J Pharm. 2007;329:166–172. doi: 10.1016/j.ijpharm.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 28.Pouton CW, Porter CJH. Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev. 2008;60:625–637. doi: 10.1016/j.addr.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 29.Porter CJH, Pouton CW, Cuine JF, Charman WN. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv Drug Deliv Rev. 2008;60:673–691. doi: 10.1016/j.addr.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 30.Gershanik T, Benita S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur J Pharm Biopharm. 2000;50(1):179–188. doi: 10.1016/S0939-6411(00)00089-8. [DOI] [PubMed] [Google Scholar]
- 31.Newton M, Petersson J, Podczeck F, Clarke A, Booth S. The influence of formulation variables on the properties of pellets containing a self-emulsifying mixture. J Pharm Sci. 2001;90:987–995. doi: 10.1002/jps.1051. [DOI] [PubMed] [Google Scholar]
- 32.Shah NH, Phuapradit W, Zhang Y, Ahmed H, Malick AW. Lipid-Based Isotropic Solutions: Design Considerations. In: Hauss DJ, editor. Oral lipid-based formulations enhancing the bioavailability of poorly water-soluble drugs. Informa Healthcare USA, Inc; 2007. p. 129–148.