

Abstract
The purpose of this study is to enhance the dissolution rate of prednisone by co-grinding with Neusilin to form a complex that can be incorporated into a mini-tablet formulation for pediatrics. Prednisone–Neusilin complex was co-grinded at various ratios (1:1, 1:3, 1:5, and 1:7). The physicochemical properties of the complex were characterized by various analytical techniques including: differential scanning calorimetry (DSC), X-ray powder diffraction (XRPD), scanning electron microscope (SEM), particle size, surface area, solubility, and dissolution rate. The co-grinded prednisone–Neusilin complex (1:7) was blended with other excipients and was formulated into a 2-mm diameter mini-tablet. The mini-tablets were further evaluated for thickness, weight, content uniformity, and dissolution rate. To improve taste masking and stability, mini-tablets were coated by dip coating with Eudragit® EPO solution. DSC and XRPD results showed that prednisone was transformed from crystalline state into amorphous state after co-grinding with Neusilin. Particle size, surface area, and SEM results confirmed that prednisone was adsorbed to Neusilin’s surface. Co-grinded prednisone–Neusilin complex (1:7) had a solubility of 0.24 mg/mL and 90% dissolved within 20 min as compared to crystalline prednisone which had a solubility of 0.117 mg/mL and 30% dissolved within 20 min. The mini-tablets containing co-grinded prednisone–Neusilin complex (1:7) exhibited acceptable physicochemical and mechanical properties including dissolution rate enhancement. These mini-tablets were successfully dip coated in Eudragit® EPO solution to mask the taste of the drug during swallowing. This work illustrates the potential use of co-grinded prednisone–Neusilin to enhance solubility and dissolution rate as well as incorporation into a mini-tablet formulation for pediatric use.
Key words: mini-tablet, Neusilin, pediatric, prednisone, solubility
INTRODUCTION
In the past several years, the regulatory agencies in the USA and Europe have initiated continual emphasis on the necessity of developing novel pediatric formulations. In response, pharmaceutical companies have developed various novel pediatric formulations; some of which have already been launched into the market (1).
In general, oral delivery of pediatric drugs is preferred over other delivery routes since it is convenient, economical, and user-friendly (1). Prednisone is a glucocorticoid prodrug which converts to its active metabolite prednisolone in the liver (2). For decades, this drug has been involved widely in pediatric therapies. At low doses, prednisone is involved in the treatment of emergency rescues such as acute asthma (3). At relatively high doses, prednisone is prescribed for the treatment of pediatric cancer (4). Although prednisone has therapeutic uses for many pediatric diseases, there are several challenges that should be addressed with the oral delivery of this drug. These challenges include: (1) it is sparingly soluble in water and has low bioavailability, therefore, any liquid formulation must include organic solvents; (2) it has low tolerance depending on the dose strength and can cause fluctuation in overall absorption; (3) it should be in a dosage form which could be swallowed readily; and (4) it has extremely bitter taste which requires better palatability through taste masking.
In order to improve the dissolution rate of poorly soluble drugs, a number of approaches have been reported including amorphilization (5), nanocrystal (6,7), complexation (8), salt formation (9), and polymorph transformation (10). Although an amorphous form dissolves faster than a crystalline form, amorphous solids retain higher free energy levels which cause the solid to become unstable and inherit the tendency to convert to the crystalline form. In order to stabilize amorphous molecules during the storage and dissolution process, preparing solid dispersion is one approach and various polymers such as polyvinylpyrrolidone, polyethylene glycol, or cellulose derivatives have been widely used as dispersants (11,12). In contrast to organic polymers, Neusilin can also function as a carrier to keep drug in amorphous state (13,14). Neusilin® US2 is a porous granule of amorphous magnesium aluminosilicate which retains a tremendous surface area (around 300 m2/g). The surface of Neusilin contains a large amount of silanol functional groups which facilitate the formation of hydrogen bonds (15–17). After cogrinding a drug with Neusilin® US2, an amorphous drug–Neusilin complex is formed (13,14,18). One possible mechanism for the formation of amorphous complex is linked to the crystalline structure disruption which is caused by the force of mechanical shearing during milling then followed by the adherence of amorphous pieces into Nuesilin® US2 surfaces by hydrogen bonds or salt bridge formation (15). It is well-known that Neusilin® US2 is involved in solid dosage form not only as an adsorbent but also as a low-toxicity excipient which can increase tablet hardness (19).
Swallowing ability is critical for pediatric formulations. Regular solid dosage forms such as capsules or tablets are unfortunately not suitable for pediatric population since children less than 10 years old are not able to grasp the skills of swallowing regular size tablets or capsules. Although granules and powders are suitable replacements for tablets or capsules, particle size must be characterized to prevent any dose fluctuations. To achieve swallowing appropriateness and uniform unit dose strength, mini-tablet is a suitable candidate. Mini-tablet is defined as a tablet with a diameter less than 3 mm (20). A recent study conducted by Thomson et al. illustrated that out of 100 preschoolers, no one suffered the potential of choke caused by swallowing placebo mini-tablets and up to 86% of 5-year-old children swallowed mini-tablets successfully (21). Additionally, the manufacturing procedures of mini-tablets can be scaled-up since mini-tablets can be compressed by regular tablet press with reproducible sizes, weights, and dose strengths (22).
To enhance pediatric compliance due to unpleasant taste of a drug, film coating with a polymer can be a realistic solution. For instance, Eudragit® EPO is a taste-masking and moisture-protective material which has been extensively applied in film coating (23). Since it is a pH-dependent copolymer, it is only slightly swellable when the pH is higher than 5 and is completely soluble when the pH is lower than 5. Therefore, Eudragit® EPO film coating can be used to achieve taste masking in the oral cavity (pH = 6.8) while keeping the immediate release characteristics for the dosage form in stomach (pH = 1.2).
The objective of this study was to develop a novel pediatric formulation of prednisone by enhancing dissolution rate of this compound after cogrinding with Neusilin. The prednisone–Neusilin complex was compressed with other excipients into 2 mm diameter mini-tablets and Eudragit® EPO multiple layers were coated on the mini-tablet for the purpose of taste masking.
MATERIALS AND METHODS
Materials
Prednisone was purchased from Sigma Aldrich (Missouri, USA). Neusilin® US2 was kindly donated by Fuji Chemicals (New Jersey, USA). Eudragit® EPO is poly (butyl methacylate-co-(2-dimethylaminoethyl) methacrylate-co-methyl methacrylate) and was kindly donated by Evonik (New Jersey, USA). Croscarmellose sodium was kindly donated by FMC Biopolymer (Pennsylvania, USA). Magnesium stearate was kindly donated by Mallinckrodt (Missouri, USA). Silicified microcrystalline cellulose was kindly donated by JRS Pharma (New York, USA).
Milling
A Retsch Ball mill (Model MM200; Pennsylvania, USA), attached with a 25-mL stainless steel jar and 5-mm diameter stainless steel ball, was used to grind prednisone with and without Neusilin® US2. The frequency of vibration was kept constant at 4 Hz and the milling operation time was maintained for 90 min. One gram of prednisone with variable amounts of Neusilin® US2 at various ratios (1:1, 1:3, 1:5, and 1:7) were physically mixed and then milled at room temperature. After ball milling, samples were transferred to a loosely capped glass vial and stored in vacuum at room temperature.
Differential Scanning Calorimetry Studies
The thermobehavior of samples were explored using a differential scanning calorimetry (DSC) Q2000 (TA Instruments, New Castle, Delaware, USA) which is equipped with a nitrogen cooler at 50 mL/min nitrogen purge. The instrument was calibrated using Indium standard. Seven samples (about 5 mg each) were tested including prednisone, Neusilin® US2, 1:1 prednisone–Neusilin® US2 physical mixture, and co-ground 1:1, 1:3, 1:5, and 1:7 prednisone–Neusilin® US2 complex. Samples were first sealed in an aluminum pan and held at 40°C for 2 min. Thereafter, samples underwent modulated heating rate at 5°C/min associated with 0.5°C modulation amplitude every 40 s. The overall temperature scan ranged from 40°C to 300°C.
X-RAY POWDER DIFFRACTION AND PERCENT CRYSTALLINITY
The X-ray powder diffraction of prednisone, Neusilin, and co-ground prednisone–Neusilin complexes at different ratios were obtained using Bruker AXS D8 advance diffractometer (Bruker, Germany) which is equipped with a copper anode (target, Cu Kα radiation; voltage, 40 kV; current, 30 mA; divergence slit, 3.722 mm; antiscatter slit, 3.722 mm; detector slit, 5 mm; and Goniometer radius, 300 mm) at ambient temperature. A thin layer of powder sample was first laid on a sample holder and then measured at 2θ in a continuous range from 3° to 40° with a step size 0.048. The percent crystallinity of samples was determined using Eva Software (Bruker). A two-phase model was employed in the determination of the degree of crystallinity; that is the sample is composed of crystals and amorphous and no regions of semicrystalline organization. Thus, the diffraction profile was divided in two parts: peaks related to diffraction of crystallites and broad region related to the scattering from amorphous phase. The assumption is that the areas are proportional to the scattering intensities of crystalline and amorphous phases. The percent crystallinity was calculated from the integrated intensities of all crystalline peaks and the amorphous region under the diffraction curve. Percent crystallinity of sample is defined as X% = Icrystalline/(Icrystalline + Iamorphous). The contribution to the intensity from the amorphous region in the sample was identified by properly adjusting the curvature of the background curve. The contribution to the intensity from the crystalline region was obtained after subtracting the above background curve from the XRD pattern.
Solubility Measurements
Excess amounts of prednisone or co-ground prednisone–Neusilin® US2 complex (1:7) were dispersed into 30 mL of simulated gastric fluid. The suspensions were placed in a mechanical shaker at a constant shaking rate of 50 rpm and a constant temperature of 25°C. Two milliliters of liquid were withdrawn at each selected time point, filtered through a 0.22 μm membrane filter, and diluted to an appropriate ratio using simulated gastric fluid. The concentration of prednisone was determined at 242 nm using UV spectrometer (PerkinElmer, Massachusetts, USA). Three replicates were carried out for each sample.
Particle Size Measurement
Laser diffractometry (Micromeritics, Georgia, USA) was employed to analyze particle size distribution. Saturated solution of prednisone was chosen as the background solution. Particle size measurement of suspension containing excess powder blend was performed. Three replicates were performed for each sample. Particle size was expressed as volume distribution according to the Mie theory. Relative refractive index of water (1.33) was inserted in this experiment.
Surface Area Measurement
The specific surface area of each sample was determined by the gas adsorption method according to the BET theory using “Gemini” surface area analyzer (Micromeritics). Nitrogen gas was adsorbed to powder surface, and the amount adsorbed gas was converted into the surface area of powder. Analysis was performed at five relative pressures ranging from 0.05 to 0.2.
Morphology
Morphologies of prednisone, Neusilin, and co-ground 1:7 prednisone–Neusilin, were characterized using the Environmental Scanning Electron Microscope (Philips, Netherland) equipped with a Peltier stage. Prior to microscope observation, all samples underwent platinum sputter-coating in argon.
Core Mini-Tablet Preparation
Three core mini-tablet formulations were prepared. Formulation 1 was directly compressed from a powder blend of 75.2% co-ground 1:7 prednisone–Neusilin complex containing 9.4% prednisone, 18.8% silicified microcrystalline cellulose, 5% croscarmellose sodium, and 1% magnesium stearate. Formulations 2 and 3 were prepared as control groups. In formulation 2, drug and excipients were identical to formulation 1, except for the use of 1:7 prednisone–Neusilin physical mixtures. In formulation 3, no Neusilin granules were added; thus, 9.4% prednisone was blended and compressed with 84.6% silicified microcrystalline cellulose, 5% croscarmellose sodium, and 1% magnesium stearate. All mini-tablets were compressed into a biconvex tablet with a diameter of 2 mm using a Carver hydraulic press (Carver INC, Indiana, USA). The compression force was kept constant at 125 lb.
Characterization of Core Mini-Tablets
Thickness, Weight Variation, and Unit Dose Strength
Thickness of ten core mini-tablets for each formulation (formulations 1, 2, and 3) was determined using a vernier caliper. Weight variation was evaluated by weighing each mini-tablet (n = 10).
To evaluate the average strength of unit dose, each core mini-tablets (n = 10) of each formulation (1, 2, and3) were individually dissolved in 25 mL solution containing water (68.8%), tetrahydrofuran (25.0%), and methanol (6.2%), followed by mechanical shaking overnight to dissolve all prednisone. The suspension was then filtered through a 0.45 μm filter membrane, and the concentration of prednisone was determined at 242 nm using UV spectrophotometer.
Tensile Strength and Friability
Tensile strength and friability of mini-tablets (formulation 1) were conducted. Tensile strength was calculated using the following equation:
| 1 |
Where Fd is tablet hardness, D is tablet diameter, and H is tablet central cylinder thickness. Tablet hardness was determined using a manual tablet testing instrument (Vortex Sales Group, North Carolina, USA).
Friability tests were conducted using a friabilator (Sotax, Massachusetts, USA). Twenty mini-tablets were first de-dusted, weighed, and laid into the friabilator drum with around 6 g of 3-mm glass beads. One hundred revolutions were set. Friability was calculated according to Eq. (2):
| 2 |
Core Mini-Tablet Coating
Due to the small mini-tablet size, mini-tablets could not be coated by fluid bed or pan coating. Alternatively, dip-coating process was utilized to coat mini-tablets. A coating solution consisted of Eudragit® EPO dissolved in acetone (20%, w/w) was utilized for coating. The mini-tablets (formulation 1) were dipped in Eudragit® EPO solution for approximately 40 s and then air dried. Thereafter, a 15-s dip-coating process followed by air-drying was repeated eight times. This group of coated mini-tablets was defined as formulation 4. All coated mini-tablets were dried and stored in a vacuum oven at room temperature.
Physical Stability Tests
The purpose of this study is to examine the influence of moisture on the physical stability of prednisone in the amorphous state over a 4-week period. In this experiment, core mini-tablets (formulation 1) and coated mini-tablets (formulation 4) were stored in isolated desiccators at 75% RH (supersaturated sodium chloride solution) in a 25°C oven.
Powder and Mini-Tablet Dissolution Tests
In vitro powder dissolution tests were performed using the USP paddle apparatus (Distek INC, New Jersey, USA). Five hundred milliliters of simulated gastric fluid (2 g/L sodium chloride; pH adjusted to 1.2 with hydrochloric acid) was maintained at 37°C. Paddle rotation speed was kept constant at 50 rpm. Briefly, 10 mg of prednisone powder or drug content equivalent to prednisone–Neusilin complex was uniformly sprinkled to the surface of the dissolution medium. Co-ground prednisone–Neusilin complexes at different ratios were evaluated and conventional prednisone was utilized as the control group. The concentration of prednisone was monitored at 242 nm using an in-line fiber optic cables connected to ultraviolet detector (pION, Massachusetts, USA). Three replicates were carried out for each formulation.
Dissolution test parameters of core mini-tablets were kept constant as previous powder dissolutions. Eighteen mini-tablets (equivalent to 10 mg of prednisone) were directly dropped into the dissolution vessel. Three formulations (formulations 1, 2, and 3) were evaluated and three replicates were carried out for each formulation.
Dissolution behavior of coated mini-tablets (formulation 4) was evaluated in simulated gastric fluid (pH = 1.2) as well as in simulated saliva (pH = 6.8). In addition, dissolution rates of mini-tablets (formulations 1 and 4) after stability test were evaluated in gastric fluid (pH = 1.2). Dissolution parameters (temperature, paddle rotation speed, medium volume, and number of tablets) were kept identical to those of core mini-tablets. Three replicates were carried out for each formulation. Statistical analysis (t test) was applied at 15-min time points for all dissolution profiles of prednisone–Neusilin complexes.
RESULTS AND DISCUSSION
Differential Scanning Calorimetry Studies
DSC is utilized to evaluate thermal behavior and phase transitions of solids. It provides useful information about the solid-state properties (crystalline or amorphous) of solid materials. All DSC thermograms are shown in Fig. 1. The conventional prednisone thermogram shows an endothermic sharp peak at 235°C. This refers to prednisone melting point and also suggests that prednisone is in crystalline state. On the other hand, Neusilin has no melting peak which is indicative of its amorphous state. However, the 1:1 physical mixture of prednisone and Neusilin did not show considerable phase transformation. Actually, the 1:1 prednisone–Neusilin physical mixture showed an endothermic peak at around 225°C which indicates that prednisone maintained some crystallinity. We also noticed a shift in melting peaks for 1:1 physical mixture and co-ground. This melting point reduction is linked to the interaction between prednisone and Neusilin. In a physical mixture, the addition of Neusilin attracts prednisone crystal aggregates and slightly lowers the melting point of prednisone. In a co-ground mixture, Neusilin and prednisone have closer interaction since some of the aggregates may have smaller size due to grinding and therefore prednisone melting peak shifts to a lower melting point. Hence, the heat of melting of 1:1 physical mixture of prednisone and Neusilin was 31.13 J/g and decreased to 25.13 J/g for 1:1 co-ground mixture which suggests a small reduction in crystallinity of prednisone. It is likely that only a certain amount of prednisone could exist in amorphous state at 1:1 ratio, and the rest exists as small crystals after milling due to insufficient Neusilin adsorption surface. As the amount of Neusilin increases, the DSC curves of 1:3, 1:5, and 1:7 (prednisone–Neusilin) show a complete disappearance of prednisone endothermic peak. Thus, according to the DSC thermograms, further addition of Neusilin caused major reduction in drug crystallinity. It was observed that when the content of Neusilin became three times in magnitude the drug content or even higher (co-ground 1:3 prednisone–Neusilin complex), the majority of prednisone shifted to the amorphous state. Therefore, based on the DSC data, we conclude that amorphization of prednisone was related to the amount of Neusilin added. A possible explanation for this phenomenon suggests that as the total surface area of Neusilin® US2 increased, prednisone slices adhered and settled onto the surface of Neusilin® US2 granules, thus inhibiting the reversion back to the ordered shape in the crystalline structure (15). It is worth noting that previous reports in the literature suggested that mixing Neusilin with volatile crystalline organic compounds may transform such compounds from crystalline state into amorphous state (24).
Fig. 1.
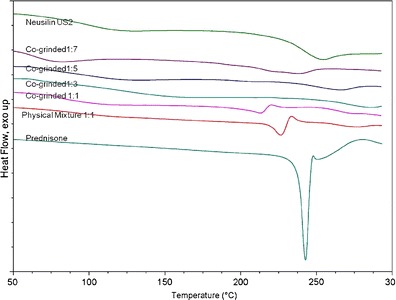
DSC thermograms of various prednisone–Neusilin powder samples from top to bottom: Neusilin® US 2 prednisone–Neusilin, Co-grinded 1:7, Co-grinded 1:5, Co-grinded 1:3, Co-grinded 1:1, physical mixture 1:1, and crystalline prednisone
X-ray Powder Diffraction
Solids in crystalline state have intensive constructive reflections; however, those in amorphous state do not reflect X-ray beam since they do not have long-range molecular arrangements. The X-ray powder diffraction of conventional prednisone and prednisone–Neusilin complexes at different ratios are shown in Fig. 2. The X-ray diffraction of Neusilin shows the absence of any peaks which exhibits the amorphous nature of Neusilin. Prednisone reveals multiple diffraction peaks including two predominantly split peaks at 14°, and one peak at 18° as well as other minor peaks. Both the 1:1 physical mixture and co-ground complexes indicate similar diffraction characteristics similar to conventional prednisone. Moreover, and as expected, the intensity of X-ray diffraction of 1:1 co-ground complex was slightly smaller than that of 1:1 physical mixture. As the amount of Neusilin increased, all intensities of characteristic peaks decreased remarkably. When the ratio of prednisone to Neusilin increased to 1:7, the two predominant split peaks at 14° and the one at 18° were extremely low in intensity and all other minor peaks completely disappeared. Therefore, the X-ray diffraction suggested that most prednisone in co-ground 1:7 complex was in amorphous state. In order to determine if the reduction in peak intensity at the co-ground 1:7 was due to reduced crystallinity or just Neusilin addition effect, we prepared a 1:7 physical mixture and noticed that the larger peak intensities were also observed as compared to 1:7 co-ground. This suggests a dilution effect (i.e., addition of Neusilin) and reduction in crystallinity. We have calculated the percent crystallinity for all the samples shown in the X-ray diffraction patterns (see Table I).
Fig. 2.
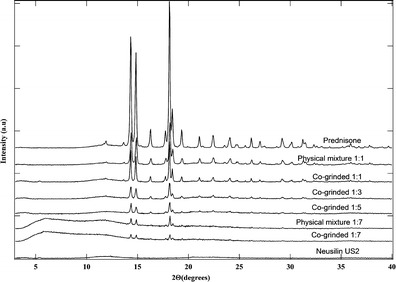
X-ray powder diffraction spectra of different powder samples from top to bottom: crystalline prednisone, drug–neusilin 1:1 physical mixture, Co-grinded 1:1 complex, Co-grinded 1:3 complex, Co-grinded 1:5 complex, 1:7 physical mixture, Co-grinded 1:7 complex, and Neusilin® US 2
Table I.
Crystallinity Obtained from X-ray Data for Prednisolone–Neusilin Mixture
| Sample | Percent crystallinity |
|---|---|
| Prednisone | 86.87 |
| 1:1 Physical mixture | 76.14 |
| 1:1 Co-ground | 62.90 |
| 1:3 Co-ground | 40.54 |
| 1:5 Co-ground | 32.68 |
| 1:7 Co-ground | 25.10 |
| Neusilin | 0 |
The percent crystallinity varies from 86.87% for prednisone to 25.10% for 1:7 co-ground. Neusilin has zero crystallinity. This finding suggests that co-grinding with Neusilin decreases the crystallinity of prednisone.
Solubility Measurements
It is reported in the literature that the solubility and dissolution rate enhancement of a drug in amorphous state is caused by the formation of disordered structure of molecules which acquire higher free energy as compared to the molecules in ordered crystalline structure (25). The solubility of conventional prednisone and co-ground 1:7 prednisone–Neusilin complex were evaluated as a function of time in Fig. 3. Prednisone exists in the crystalline state with highly hydrophobic properties. Figure 3 shows that crystalline prednisone reaches an equilibrium solubility of 0.117 mg/mL within 3 h then attains a plateau up to 72 h. As for co-ground 1:7 prednisone–Neusilin complex, prednisone reaches a maximal solubility of 0.28 mg/mL within 10 h then plateaus to 72 h. These results indicate that addition of Neusilin in the prednisone–Neusilin complex displayed about twofold increase in solubility. Therefore, a prednisone-saturated solution in the presence of Neuslin, can maintain solubility for at least 72 h. Additionally, precipitation or solubility reduction were not observed during this experimental design, which suggests that Neusilin granules are acting as crystallization inhibitors and would prevent the recrystallization process of prednisone in the saturated solution.
Fig. 3.
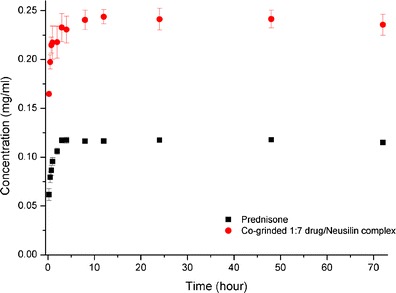
Black square solubility of crystalline prednisone, red circle solubility of co-grinded 1:7 drug–Neusilin complex
Surface Area and Particle Size Measurements
It has been reported that the silanol groups which are situated on the surface of Neusilin granules act as potential hydrogen bond acceptors (15,16). Therefore, it is likely to form hydrogen bonds between prednisone and these groups. This assumption would suggest that amorphous prednisone is expected to adsorb onto the surface of Neusilin® US2. It was suggested in the literature that by combining the results of surface area and particle size analysis, the modality of the drug either as adsorbed to Neusilin surface or kept free in the crystal form could be evaluated (14). Results of particle size and surface area measurements were listed in Table II. The manufacturer of Neusllin ® US2 reported that the mean particle size for Neusilin ranged from 60 to 120 μm and the specific surface area was 300 m2/g. This was consistent with the experimental values that we obtained for milled Neusilin® US2 (mean particle size, 88.62 μm; surface area, 329.60 m2/g). Hence, Neusilin® US2 granules were kept intact or with minimum fracture during the milling process under this shaking frequency. The high value for the surface area of Neusilin is related to its porous nature and its free-exposed surface. Prednisone crystals had smaller mean particle size of 6.36 μm and a surface area of 3.31 m2/g. We need to pinpoint that prednisone crystals were disrupted during ball milling and slices of prednisone in the amorphous form were adsorbed onto Neusilin surfaces. Thus, the particle size distribution of co-ground prednisone–Neusilin complex was expected to become close to that of free Neusilin granules alone. We also noticed that the surface area of the complex was lower than that of Neusilin granules because prednisone slices covered and/or occupied the surfaces of Neuslin. The mean values of particle size of co-ground 1:7 prednisone–Neusilin complex was 87.74 μm and total surface area was 271.79 m2/g which explains the particle size distribution and the lower surface area of prednisone as compared to those of Neusilin. It was noted that as the content of Neusilin in the complex decreased, particle size distribution D10, D50, D90, and Mv shifted to smaller values. This in part is due to the existence of drug crystals in 1:1 complex (mean particle size, 66.67 μm) and 1:3 complex (mean particle size, 75.95 μm). Moreover, the decrease of Neusilin content in the complex resulted in a decrease in surface area; e.g., 1:1 complex (surface area, 157.15 m2/g); 1:3 complex (surface area, 229.04 m2/g). These surface area values could be reasoned by (a) drug crystals that remained in the complex after co-grinding had relatively smaller surface area and (b) porous surface on the Neusilin were covered by amorphous drug so that surface area was further decreased.
Table II.
Surface Area and Particle Size Analysis of Various Prednisone–Neusilin Powder Samples
| Sample | D10 (μm) | D50 (μm) | D90 (μm) | Mv (μm) | Surface area (m2/g) |
|---|---|---|---|---|---|
| Prednisone | 1.44 | 4.83 | 12.12 | 6.36 | 3.31 |
| Co-ground 1:1 D/N | 5.71 | 54.14 | 144.05 | 66.67 | 157.15 |
| Co-ground 1:3 D/N | 11.44 | 78.48 | 144.06 | 75.95 | 229.04 |
| Co-ground 1:5 D/N | 16.16 | 88.81 | 158.59 | 83.29 | 252.07 |
| Co-ground 1:7 D/N | 33.24 | 90.85 | 160.58 | 87.74 | 271.79 |
| Milled Neusilin® | 34.16 | 90.89 | 161.63 | 88.62 | 329.60 |
D/N prednisone–Neusilin complex
Morphology
The morphologies of prednisone and Neusilin were directly visualized by scanning electron microscope (SEM). This experiment enabled us to observe the adsorption of prednisone to Neusilin surfaces. As seen in Fig. 4a and after cogrinding (ball milling), Neusilin US2 granules were kept intact. It could be seen that Neusilin granules had a spherical shape and wide particle size distribution as was also verified by particle size measurement using laser diffraction. In addition, the surfaces of granules were not smooth, rather porous. The morphology of prednisone crystals is shown in Fig. 4b. It was noted the crystals had a much smaller size as compared to Neusilin granules. The particle size of prednisone as obtained by SEM was less than 10 μm. This is consistent with the previous particle size measurement. The morphology of co-ground 1:7 complex is shown in Fig. 4c. We noticed that most prednisone particles were adsorbed onto the surface of Neusilin and only some particles were left free besides Neusilin. This observation verified our assumption that Neusilin functioned as an adsorbent that enabled the amorphization of prednisone. It was also observed that the porous surface of Neusilin was partially covered by prednisone particles which were responsible for surface area reduction of Neusilin granule. Finally, we need to mention that Neusilin maintained its original spherical shape and size after grinding.
Fig. 4.

SEM micrographs of a Milled Neusilin® US2, b crystalline prednisone, c co-grinded 1:7 drug–Neusilin complex
Characterization of Core Mini-Tablets
The three mini-tablet formulations which were prepared are shown in Table III. The unit dose of prednisone in each tablet was about 0.5 mg which was therapeutically appropriate for pediatric indications. Tablet thickness, total weight, and unit dose strength were consistent among the three formulations which suggested that single-compression process maintained consistent mini-tablet characteristics during the compression process. In addition, results of tensile strength (4.01 ± 0.31 MPa) and friability tests (0.21%) indicated that mini-tablets of formulation 1 which contained co-ground 1:7 prednisone–Neusilin complex had acceptable mechanical properties. The hardness of the co-ground prednisone–Neusilin tablet has increased and could be detected by the hardness machine. We were not able to use the same method to determine the tensile strength and friability for formulations 2 and 3 because of the low hardness values which were not sensed by the hardness machine.
Table III.
Properties of the Prednisone Mini-Tablets
| Formulation | Thickness (mm) | Weight (mg) | Unit dose strength (mg)n | Tensile strength (MPa) | Friability (%) |
|---|---|---|---|---|---|
| 1 | 1.93 ± 0.03 | 5.23 ± 0.14 | 0.55 ± 0.01 | 4.01 ± 0.31 | 0.21 |
| 2 | 1.92 ± 0.01 | 5.23 ± 0.19 | 0.54 ± 0.01 | NA | NA |
| 3 | 1.84 ± 0.01 | 5.40 ± 0.25 | 0.56 ± 0.01 | NA | NA |
n = 10 tablets
Powder and Core Mini-Tablet Dissolution Profiles
Dissolution profiles of co-ground prednisone–Neusilin complexes at different ratios of 1:1, 1:3, 1:5, and 1:7; conventional (crystalline) prednisone are shown in Fig. 5. The dissolution profile of crystalline prednisone (control) indicated that up to 30% of the drug had dissolved within 20 min and around 70% dissolved within 90 min. While the dissolution profiles for 1:1, 1:3, 1:5, and 1:7 showed significant increase in the dissolution rate as compared to the control. Student’s t test was applied to 15-min data points and showed statistical significance for all the cumulative release data points as compared to conventional prednisone (p < 0.05).
Fig. 5.
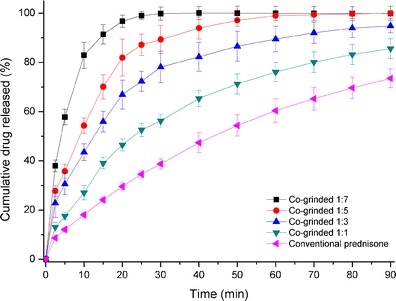
Dissolution profiles of black square co-grinded 1:7 drug–Neusilin complex, red circle co-grinded 1:5 drug–Neusilin complex, blue triangle co-grinded 1:3 drug–Neusilin complex, green down-pointing triangle co-grinded 1:1 drug–Neusilin complex, pink left-pointing triangle crystalline prednisone in simulated gastric fluid (pH = 1.2)
The observed differences in dissolution rates were related to the co-existence of amorphous and crystalline forms of prednisone. At the low ratio of prednisone in the complexes (i.e., a ratio of 1:7), more amorphous form resulted in faster dissolution rate. This was consistent with the observations of other researchers who investigated dissolution rate enhancement caused by different ratios of drug to excipient (26,27). As the Neusilin ratio decreased, more prednisone is present in the crystalline state and less amorphous form is adsorbed onto the surface of Neusilin granules which leads to slower dissolution rate. Moreover, it is important to mention that incomplete release of amorphous form may also be related to the formation of gel-like mass, which was discussed by Law et al. (28).
In order to prepare mini-tablets, and since powder complex ratio at 1:7 exhibited the fastest dissolution profile as shown in Fig. 5, a co-ground prednisone to Neusilin complex at 1:7 was selected for mini-tablet (formulation 1). In addition, physical mixture at the same ratio and crystalline prednisone mini-tablets were also utilized as controls (formulations 2 and 3).
The dissolution profiles of the three mini-tablets (formulations 1, 2, and 3) which were described are shown in Fig. 6. The mini-tablets dissolution profiles in Fig. 6 show that 1:7 co-ground prednisone–Neusilin tablets exhibited the fastest dissolution profile (87% in 20 min) followed by 1:7 physical mixture (68% in 20 min), then the mini tablet with crystalline prednisone (60% in 20 min). Previous reports indicated that the disintegration time can positively influence the dissolution rate of the drug from tablets (29). Therefore, mini-tablets are expected to disintegrate within a short period of time (1 min) in order to release prednisone rapidly in gastric fluids. The presence of Neusilin in the formulation improves the hardness and may cause a delay to the disintegration process. In order to obtain a reasonable disintegration time, croscarmellose sodium was added as a super-disintegrant. The dissolution profiles presented in Fig. 6 indicate that disintegration of mini-tablets was acceptable and did not inhibit the dissolution rate of prednisone. It is important to mention that the crystalline form of prednisone in formulations 2 and 3 dissolved much faster as compared to prednisone powders particularly during the initial stage. A possible explanation is linked to the hydrophilic properties of the co-blended excipients which include silicified microcrystalline cellulose and/or Neusilin. Both can enhance the wettability of prednisone powder. We can conclude from Fig. 6 that formulation 1 showed an enhanced dissolution rate of prednisone; therefore, it is reasonable to say that prednisone remained in amorphous state during mini-tablet preparation and dissolution processes.
Fig. 6.
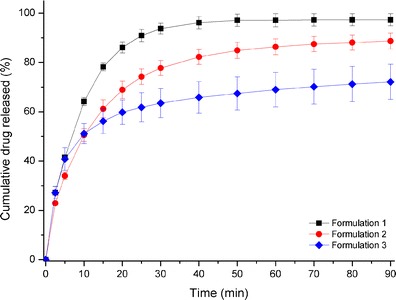
Dissolution profiles of fresh core mini-tablets: black square formulation 1 (1:7 co-grinded Drug–Neusilin) in simulated gastric fluid (pH = 1.2); red circle formulation 2 (1:7 physical mixture of Drug–Neusilin) in simulated gastric fluid (pH = 1.2), blue diamond formulation 3 (crystalline prednisone) in simulated gastric fluid (pH = 1.2)
Coated Mini-Tablets Dissolution Profiles
Film coating of tablets is a widespread approach to inhibit contact between a drug with unpleasant taste and sensors of taste buds. It was reported that suppression of drug release for only few minutes at the initial stage could reduce unpleasant sensations associated with bitter taste of drugs (30). Eudragit® EPO, a pH-dependent copolymer, is insoluble at pH above 5 but becomes soluble at pH below 5. Therefore, this polymer has been selected as an appropriate material for taste masking purpose (23,31). Figure 7 shows release behavior of prednisone within coated mini-tablet in both simulated saliva solution (pH = 6.8) and in simulated gastric fluid solution (pH = 1.2). It was observed that when coated mini-tablets were placed in simulated saliva, the shape of the tablets was unchanged for approximately 70–90 s then afterwards gel-like flocculants were formed. Hypothetically, this may be due to the incomplete disintegration of coated mini-tablets after water penetration across Eudragit® EPO coating layer. The dissolution profile of coated mini-tablet in simulated saliva confirmed the assumption that Eudragit® EPO coating layer suppressed prednisone release significantly (2% within 2.5 min). However, in simulated gastric fluid, Eudragit® EPO dissolved rapidly and prednisone has dissolved with no barrier.
Fig. 7.
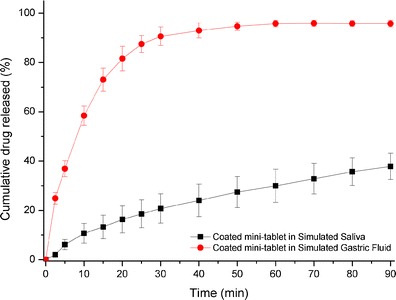
Dissolution profiles of coated mini-tablets: red circle in simulated gastric fluid (pH = 1.2), black square in simulated saliva (pH = 6.8)
It is well known that exposure to moisture accelerates the recrystallization process of amorphous drugs and reduces the drug dissolution rate (32). Eudragit® EPO coating has been noted to protect against moisture penetration (23). It is expected in this study that it would decrease the moisture uptake of amorphous prednisone, thereby decreasing the recrystallization process. Drug release behavior of formulations 1 and 4 (after 1 month of physical stability evaluation) in simulated gastric fluid were plotted and shown in Fig. 8. The similarity factor value (f2) was calculated and a comparison was made between the release profile of fresh coated mini-tablets (formulation 4), and coated mini-tablets (formulation 4) after 1 month on physical stability (the f2 value was 66.5). Also, a comparison was made between release profiles of fresh core mini-tablets (formulation 1) and core mini-tablets (formulation 1) after 1 month on physical stability (the f2 value was 46.6). In addition, a comparison was made between release profiles of coated mini-tablets (formulation 4) after 1 month on physical stability and core mini-tablets (formulation 1) after 1 month of physical stability (the f2 value was 70.7), respectively.
Fig. 8.
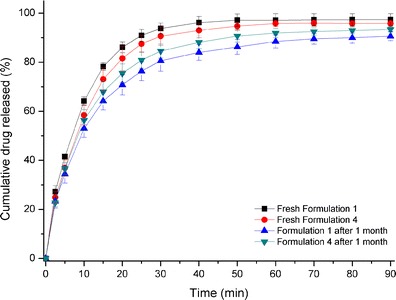
Dissolution profiles of: black square Fresh core mini-tablet (formulation 1) in simulated gastric fluid (pH = 1.2), red circle Fresh coated mini-tablet (formulation 4) in simulated gastric fluid (pH = 1.2), blue triangle core mini-tablet (formulation 1) after 1-month physical stability evaluation in simulated gastric fluid (pH = 1.2), green down-pointing triangle coated mini-tablet (formulation 4) after 1-month physical stability evaluation in simulated gastric fluid (pH = 1.2)
These values indicate that although there is no significant difference between drug release profiles for coated and uncoated mini-tablets after 1 month of stability testing, Eudragit® EPO coating had some influence on maintaining (moisture decreases dissolution rate and therefore coating helps to maintain dissolution rate) drug dissolution rate. A possible reason is that Eudragit® EPO coating somewhat decreased the exposure of drug to moisture, which can minimize the drug recrystallization; however, it was concluded that recrystallization of amorphous drug could not totally be inhibited by the Eudragit® EPO coating during this limited time frame of 4 weeks. We like to show a visual comparison in size between formulations 1 and 4 (see Fig. 9).
Fig. 9.
Size comparison between core mini-tablet (formulation 1) and coated mini-tablet with Eudragit EPO (formulation 4)
CONCLUSION
This study demonstrated that co-ground prednisone–Neusilin complex enhanced drug dissolution rate due to a decrease in prednisone crystallinity. Co-ground prednisone–Neusilin (1:7) complex yielded reliable and reproducible preparation processes for mini-tablets. Eudragit® EPO coating (taste masking) suppressed the drug release from mini-tablets in simulated saliva (pH + 6.8) while maintained immediate release properties in simulated gastric media (pH = 1.2). Thus, due to its small size, coated and uncoated mini-tablets of co-ground prednisone–Neusilin can potentially be appropriate for pediatric use.
References
- 1.Strickley RG, Iwata Q, Wu S, Dahl TC. Pediatric drugs—a review of commercially available oral formulations. J Pharm Sci. 2008;97:1731–1774. doi: 10.1002/jps.21101. [DOI] [PubMed] [Google Scholar]
- 2.Frey FJ, Escher G, Frey BM. Pharmacology of 11β-hydroxysteroid dehydrogenase. Steroids. 1994;59:74–79. doi: 10.1016/0039-128X(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 3.Schuh S, Reisman J, Alshehri M, Dupuis A, Corey M, Arseneault R, et al. A comparison of inhaled fluticasone and oral prednisone for children with severe acute asthma. New Engl J Med. 2000;343:689–694. doi: 10.1056/NEJM200009073431003. [DOI] [PubMed] [Google Scholar]
- 4.Kung FH, Nyhan WL, Cuttner J, Falkson G, Lanzkowsky P, Duca VD, et al. Vincristine, prednisone and l-asparaginase in the induction of remission in children with acute lymphoblastic leukemia following relapse. Cancer. 1978;41:428–434. doi: 10.1002/1097-0142(197802)41:2<428::AID-CNCR2820410208>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 5.Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50:47–60. doi: 10.1016/S0939-6411(00)00076-X. [DOI] [PubMed] [Google Scholar]
- 6.Hecq J, Deleers M, Fanara D, Vranckx H, Amighi K. Preparation and characterization of nanocrystals for solubility and dissolution rate enhancement of nifedipine. Int J Pharm. 2005;299:167–177. doi: 10.1016/j.ijpharm.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 7.Mauludin R, Müller RH, Keck CM. Development of an oral rutin nanocrystal formulation. Int J Pharm. 2009;370:202–209. doi: 10.1016/j.ijpharm.2008.11.029. [DOI] [PubMed] [Google Scholar]
- 8.Challa R, Ahuja A, Ali J, Khar R. Cyclodextrins in drug delivery: an updated review. AAPS PharmSciTech. 2005;6:E329–E357. doi: 10.1208/pt060243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berge SM, Bighley LD, Monkhouse DC. Pharmaceutical salts. J Pharm Sci. 1977;66:1–19. doi: 10.1002/jps.2600660104. [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi Y, Ito S, Itai S, Yamamoto K. Physicochemical properties and bioavailability of carbamazepine polymorphs and dihydrate. Int J Pharm. 2000;193:137–146. doi: 10.1016/S0378-5173(99)00315-4. [DOI] [PubMed] [Google Scholar]
- 11.Chiou WL, Riegelman S. Pharmaceutical applications of solid dispersion systems. J Pharm Sci. 1971;60:1281–1302. doi: 10.1002/jps.2600600902. [DOI] [PubMed] [Google Scholar]
- 12.Serajuddin ATM. Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs. J Pharm Sci. 1999;88:1058–1066. doi: 10.1021/js980403l. [DOI] [PubMed] [Google Scholar]
- 13.Vadher A, Parikh J, Parikh R, Solanki A. Preparation and characterization of co-ground mixtures of aceclofenac and Neusilin US2 for dissolution enhancement of aceclofenac. AAPS PharmSciTech. 2009;10:606–614. doi: 10.1208/s12249-009-9221-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maclean J, Medina C, Daurio D, Alvarez-Nunez F, Jona J, Munson E, et al. Manufacture and performance evaluation of a stable amorphous complex of an acidic drug molecule and neusilin. J Pharm Sci. 2011;100:3332–3344. doi: 10.1002/jps.22583. [DOI] [PubMed] [Google Scholar]
- 15.Qian KK, Bogner RH. Application of mesoporous silicon dioxide and silicate in oral amorphous drug delivery systems. J Pharm Sci. 2012;101:444–463. doi: 10.1002/jps.22779. [DOI] [PubMed] [Google Scholar]
- 16.Maciel GE. Probing hydrogen bonding and the local environment of silanols on silica surfaces via nuclear spin cross polarization dynamics. J Am Chem Soc. 1996;118:401–406. doi: 10.1021/ja951550d. [DOI] [Google Scholar]
- 17.Chuang IS, Maciel GE. A detailed model of local structure and silanol hydrogen bonding of silica gel surfaces. J Phys Chem B. 1997;101:3052–3064. doi: 10.1021/jp9629046. [DOI] [Google Scholar]
- 18.Gupta MK, Vanwert A, Bogner RH. Formation of physically stable amorphous drugs by milling with neusilin. J Pharm Sci. 2003;92:536–551. doi: 10.1002/jps.10308. [DOI] [PubMed] [Google Scholar]
- 19.http://www.neusilin.com/product/pharmaceutical_application.php
- 20.Lennartz P, Mielck JB. Minitabletting: improving the compactability of paracetamol powder mixtures. Int J Pharm. 1998;173:75–85. doi: 10.1016/S0378-5173(98)00206-3. [DOI] [Google Scholar]
- 21.Thomson SA, Tuleu C, Wong ICK, Keady S, Pitt KG, Sutcliffe AG. Minitablets: new modality to deliver medicines to preschool-aged children. Pediatrics. 2009;123:E235–E238. doi: 10.1542/peds.2008-2059. [DOI] [PubMed] [Google Scholar]
- 22.Tissen C, Woertz K, Breitkreutz J, Kleinebudde P. Development of mini-tablets with 1 mm and 2 mm diameter. Int J Pharm. 2011;416:164–170. doi: 10.1016/j.ijpharm.2011.06.027. [DOI] [PubMed] [Google Scholar]
- 23.Cerea M, Zheng W, Young CR, McGinity JW. A novel powder coating process for attaining taste masking and moisture protective films applied to tablets. Int J Pharm. 2004;279:127–139. doi: 10.1016/j.ijpharm.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 24.Konno T, Kinuno K, Kataoka K. Physical and chemical changes of medicinals in mixtures with adsorbents in the solid state. I.: Effect of vapor pressure of the medicinals on changes in crystalline properties. Chem Pharm Bull. 1986;34:301–307. doi: 10.1248/cpb.34.301. [DOI] [PubMed] [Google Scholar]
- 25.Gupta P, Kakumanu VK, Bansal AK. Stability and solubility of celecoxib-PVP amorphous dispersions: a molecular perspective. Pharm Res. 2004;21:1762–1769. doi: 10.1023/B:PHAM.0000045226.42859.b8. [DOI] [PubMed] [Google Scholar]
- 26.Johansen H, Møller N. Solvent deposition method for enhancement of dissolution rate: importance of drug-to-excipient ratio. J Pharm Sci. 1978;67:134–136. doi: 10.1002/jps.2600670140. [DOI] [PubMed] [Google Scholar]
- 27.Law D, Schmitt EA, Marsh KC, Everitt EA, Wang W, Fort JJ, et al. Ritonavir–PEG 8000 amorphous solid dispersions: in vitro and in vivo evaluations. J Pharm Sci. 2004;93:563–570. doi: 10.1002/jps.10566. [DOI] [PubMed] [Google Scholar]
- 28.Law D, Krill SL, Schmitt EA, Fort JJ, Qiu Y, Wang W, et al. Physicochemical considerations in the preparation of amorphous ritonavir–poly(ethylene glycol) 8000 solid dispersions. J Pharm Sci. 2011;90:1015–1025. doi: 10.1002/jps.1054. [DOI] [PubMed] [Google Scholar]
- 29.Middleton EJ, Davies JM, Morrison AB. Relationship between rate of dissolution, disintegration time, and physiological availability of riboflavin in sugar-coated tablets. J Pharm Sci. 1964;53:1378–1380. doi: 10.1002/jps.2600531122. [DOI] [PubMed] [Google Scholar]
- 30.Albertini B, Cavallari C, Passerini N, Voinovich D, González-Rodríguez ML, Magarotto L, et al. Characterization and taste-masking evaluation of acetaminophen granules: comparison between different preparation methods in a high-shear mixer. Eur J Pharm Sci. 2004;21:295–303. doi: 10.1016/j.ejps.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 31.Kayumba PC, Huyghebaert N, Cordella C, Ntawukuliryayo JD, Vervaet C, Remon JP. Quinine sulphate pellets for flexible pediatric drug dosing: formulation development and evaluation of taste-masking efficiency using the electronic tongue. Eur J Pharm Biopharm. 2007;66:460–465. doi: 10.1016/j.ejpb.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 32.Andronis V, Yoshioka M, Zografi G. Effects of sorbed water on the crystallization of indomethacin from the amorphous state. J Pharm Sci. 1997;86:346–351. doi: 10.1021/js9602711. [DOI] [PubMed] [Google Scholar]