

Abstract
Tablet compression of softwood cellulose and lignin prepared by a new catalytic oxidation and acid precipitation method were investigated and compared with the established pharmaceutical direct compression excipients. Catalytic pretreated softwood cellulose (CPSC) and lignin (CPSL) were isolated from pine wood (Pinus sylvestris). The compaction studies were carried out with an instrumented eccentric tablet machine. The plasticity and elasticity of the materials under compression were evaluated using force-displacement treatment and by determining characteristic plasticity (PF) and elasticity (EF) factors. With all biomaterials studied, the PF under compression decreased exponentially as the compression force increased. The compression force applied in tablet compression did not significantly affect the elasticity of CPSC and microcrystalline cellulose (MCC) while the EF values for softwood lignins increased as compression force increased. CPSL was clearly a less plastically deforming and less compactable material than the two celluloses (CPSC and MCC) and hardwood lignin. CPSL presented deformation and compaction behaviour almost identical to that of lactose monohydrate. In conclusion, the direct tablet compression behaviour of native lignins and celluloses can greatly differ from each other depending on the source and isolation method used.
KEY WORDS: elasticity, lignin, plasticity, pretreated cellulose, tablet compression
INTRODUCTION
Tablets are the most preferred solid oral unit dosage form for many reasons. Tablets are widely used in human medication but different forms of tablets also play an important role in veterinary medication applications (1,2). Today direct compression is considered as the most convenient technique for manufacturing tablets because of a number of advantages compared with wet granulation: fewer unit operations, shorter processing time, a need for fewer excipients in a tablet formula, reduced stability risks for drugs that are sensitive to heat or moisture and faster dissolution of the final product (3).
In recent years, a lot of attention has been focused on the development of new excipients and/or co-processed excipients with improved bulk powder and compression properties for tablet direct compression (4–6). The reasons for this interest includes the growing popularity of the direct compression process, increasing speed capabilities of modern tableting machines, technological and end-user limitations of existing excipients and increased performance expectations of excipients (6,7). In addition, recent attempts have also been made to develop new methods to classify the powders (excipients) with respect to their direct compression mechanism and behaviour, i.e. particle rearrangement, densification, fragmentation and particle plastic deformation (8,9). The fundamental knowledge of the compression of pharmaceutical powders is essential for the improvement of the quality of the final tablets and for the development of the compaction process.
Both cellulose and lignin are by-products of the pulping or bio-ethanol industries, and they are readily available, cheap and biocompatible. Natural cellulose is a linear polymer of glucose (with a β-1,4 orientation of the glucosidic bonds) in plants or woody materials (10). Various grades of microcrystalline cellulose (MCC) have been successfully used for years as a filler-binder in tablet compression but MCC has also some technological shortcomings such as loss of compacting upon wet granulation, high moisture sensitivity and poor die filling due to agglomeration (6,11). Recently, Kolakovic et al. (12) evaluated the potential of nanofibrillar cellulose (cellulose nanofibers) as novel tabletting material. Lignin is one of the three major (along with cellulose and hemicellulose) polymeric components found in the cell walls of higher-order plants (13). It is a polydisperse three-dimensional polymer in which the molecules are slightly cross-linked with each other (14,15) which makes it an interesting material for various applications. To our knowledge, this is the first work that reports the tablet compression properties of lignin materials.
Recently, Hakola et al. (16) introduced a new isolation method for cellulose and lignin from lignocellulosic biomass for enzymatic hydrolysis. The present technology has significant advantages compared to the other state-of-the-art pretreatment/isolation methods (1) making cellulose that can easily undergo enzymatic hydrolysis, (2) avoiding the loss of hydrolysable carbohydrates, (3) avoiding the formation of toxic compounds and (4) offering an environmentally benign concept for cellulosic ethanol production. In addition, chemical purity, physical material and bulk powder properties as well as solubility of cellulose and lignin can be modified with this new pretreatment/isolation technique (16,17). Therefore, these pretreated biomaterials would be interesting and potentially useful excipients to be applied as novel diluents (or co-diluents) for tablet direct compression.
The purpose of the present study was to investigate the direct compression behaviour and deformation mechanisms (elasticity, plasticity or fragmentation) of catalytic pretreated wood cellulose (CPSC) and lignin (CPSL) when compressed into tablets. The biomaterials studied were isolated from softwood (Pinus sylvestris) with a catalytic pretreatment procedure described by Hakola et al. (16) with some modification. The compression behaviour of the biomaterials was classified by using compression parameters derived from the force–displacement curves, and the method introduced by Antikainen and Yliruusi (18,19). Industrial softwood kraft lignin, hardwood lignin and microcrystalline cellulose (MCC) were used as reference materials in direct compression.
MATERIALS AND METHODS
Materials
Catalytic pre-treated softwood cellulose (CPSC) and lignin (CPSL) were prepared from pine soft wood (Pinus sylvestris) by using a catalytic oxidation and subsequent acid precipitation treatments. Industrial softwood kraft lignin (Indulin AT), hardwood lignin (PC 1369) and three commercial pharmaceutical direct compression excipients, microcrystalline cellulose, MCC (Avicel® PH 101, FMC Biopolymer, USA), lactose monohydrate (Pharmatose® 80 M; DFE Pharma, Germany) and dibasic calcium phosphate, DBCP (Emcompress®, E. Mendell, NY, USA) were used as reference tableting materials. Magnesium stearate (Ph.Eur.) and acetone (E. Merck, Germany) were used for preparing a lubricant suspension for tablet compression.
Preparation of CPSC and CPSL
Isolation of cellulose and lignin from pine soft wood (Pinus sylvestris) was carried out according to the method described by Hakola et al. (16) with some modification. The pine chips were first extracted with hexane for 2 days and subsequently with acetone for 1 day to remove the extractives. The aqueous alkaline solution was prepared by mixing 5.2 g (49.0 mmol) Na2CO3 to 200 ml of water, and it was combined with 10 g of extractive free pine chips (dry weight) to a preheated autoclave equipped with magnetic stirrer and oil bath heating. The reaction was continued for 20 h at elevated temperature (120°C) and with oxygen pressure (10 bar). After pretreatment, the solution was filtered. The solid cellulose fraction (CPSC) was dried in room temperature and subsequently ball milled with a Planetary Mono Mill pulverisette 6 (Fritsch GmbH, Germany) at 400 rpm for 3 min. The filtrate, which contained the solubilised lignin was acidified with HCl and the precipitated lignin (CPSL) was collected with additional filtration. The obtained solid lignin was vacuum dried and used in tablet compression studies without further size reduction.
Particle Size, Shape and Surface Morphology
The particle size and size distribution of the direct compression materials were determined by using a 3D-surface image analysis method (Flashsizer FS3D, Intelligent Pharmaceutics Ltd, Helsinki, Finland) described by Soppela et al. (20). The FS3D image analysis setup consists of a camera connected to a computer and glass cuvette towards the camera. The size of the measurement field was 1,280 × 960 pixels, i.e. 15.1 × 11.3 mm. The particle size was recorded in triplicate and eight images were taken per batch. The number of particles per image measured varied from 200 to 5,600. For a more detailed description of the principle of this 3D surface imaging and particle size measurement technique, readers are referred to references Soppela et al. (20) and Sandler (21).
Particle shape, surface morphology and microstructure were studied by using a high-resolution scanning electron microscope, SEM (Quanta FEG 250, FEI Company, USA). The samples were mounted on aluminium stubs with a double-sided tape and then coated with 3 nm platinum layer (Quorum Q150TS, turbomolecular-pumped high-resolution coater, Quorum Technologies, UK) in an argon atmosphere prior to microscopy.
Moisture Content
Pre-weighed powder samples of biopolymers and direct compression materials were kept (as thin layers) under controlled temperature and humidity conditions at 21°C and 50% RH for 48 h. The powder samples were then transferred into a silica gel containing desiccators (21°C and 0% RH) and stored for a subsequent 48 h before re-weighing. The moisture content (free water) of the powders was obtained by calculating the weight difference between the initial and end weight of the sample (n = 6). The following moisture contents (% w/w) for the materials were obtained: 8.1% (CPSC), 6.6% (CPSL), 3.0% (Indulin AT), 4.7% (PC 1369), 4.5% (MCC), 0% (lactose monohydrate) and 0% (DPCP).
Tablet Compression
The materials were stored and dried under controlled temperature and low-humidity conditions (21°C/35% RH) for at least 1 week before testing. Another additional set of compaction tests was performed with non-dried CPSC and milled fractions of it. Prior to tablet compression, the materials were kept (as thin layers) under controlled room temperature and humidity conditions at 21°C and 50% RH for at least 48 h.
Tablets were compressed with an instrumented Korsch EK-0 eccentric tableting machine (Erweka Apparatebau, Germany) equipped with 9-mm flat-faced punches. The upper punch was first placed in its lower position and the position of the lower punch was adjusted by using a 3-mm calibration plate between the punches. For compression studies, excipients were individually weighed out and poured into a pre-lubricated die (acetone solution of magnesium stearate 5% w/w). The operating speed of the tablet machine (36 rpm) and the height of tablets (3.0 mm) were kept constant in all compactions.
Immediately after compression, the height of each tablet was measured with a digital micrometer (Sony DZ 521, Tokyo, Japan). The mass of each tablet was measured with an analytical balance (Sartorious CP 2245, Raute, Goettingen, Germany). The crushing strength of tablets was determined using a tablet hardness tester (Schleuniger 2E, Dr. Schleuniger Pharmatorn AG, Solothurn, Switzerland).
Determination of Plasticity and Elasticity
The plasticity and elasticity of the materials under compression were determined by the method described by Antikainen and Yliruusi (19). In this method, the ratio of two areas of work, W1 and W2, obtained from the force–distance curve near the maximum force, is calculated. The plasticity factor (PF) determines the extent of plastic flow at a certain compression force level and gives a comparable numerical value (Eq. 1):
| 1 |
The elasticity factor (EF) was calculated with Eq. (2):
| 2 |
where H is the height of the tablet after removing it from the die, Hmin is the height of the tablet during maximum compression force. For a more detailed description of the method for the evaluation of plasticity and elasticity of the powders, readers are referred to references Antikainen (18), Antikainen and Yliruusi (19) and Mir et al. (22).
RESULTS AND DISCUSSION
Particle and Powder Properties
Particle shape, size and size distribution are important factors affecting powder flow, packing, densification and deformation under compression of tablets. For example, particle size and size distribution could greatly influence the tableting key attributes and thus the overall success of the direct tablet compression process (23). Scanning electron micrographs and FS3D particle size and size distributions of the biomaterials and the reference materials are shown in Figs. 1 and 2. Both the powdered CPSC and MCC presented particles with an elongated or fibre-like shape (Figs. 1a and 2a), and the FS3D particle size ranging from 125 to 1,400 μm (d50 685 μm) and below 250 μm (d50 105 μm), respectively (Fig. 1). The pre-milled CPSL powder particles were irregular and sharp-edged in shape (Fig. 1b) while reference softwood and hardwood industrial kraft lignins exhibited almost round granular particles (Fig. 1c, d). The FS3D maximum particle size of a pre-milled CPSL powder was 355 μm (d50 141 μm), and this was clearly smaller than the particle size of industrial reference lignin powders (softwood Indulin AT and hardwood PC 1369) with a maximum particle size of 710 μm (d50 363 μm) and 1,000 μm (d50 471 μm), respectively (Fig. 1). The histogram of both industrial reference lignins exhibited a clear negative skewness thus indicating a smaller proportion of fine particles than the powder. Lactose 80M and DBCP (Emcompress®) exhibited almost round and porous granular particles with a FS3D particle size below 355 μm (d50 212 μm) and 125–850 μm (d50 431 μm), respectively (Fig. 2).
Fig. 1.
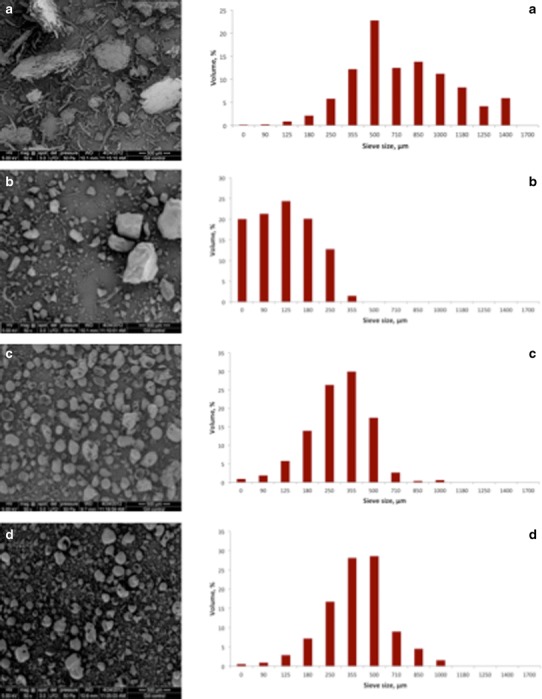
Scanning electron micrographs and FS3D particle size distributions of a catalytic pretreated powdered softwood cellulose, CPSC; b catalytic pretreated softwood lignin, CPSL; c industrial softwood kraft lignin, Indulin AT and d industrial hardwood kraft lignin, PC 1369. Magnification of SEMs ×50
Fig. 2.

Scanning electron micrographs and FS3D particle size distributions of a microcrystalline cellulose, MCC; b lactose monohydrate (Pharmatose® 80M) and c dibasic calcium phosphate, DBCP (Emcompress®). Magnification of SEMs ×50
The results on particle size and size distribution obtained with a novel 3D-surface image analysis method (Flashsizer FS3D) were found to be in good accordance with the respective particle size findings based on inspection of SEMs.
Densification and Deformation Under Compression
Initial particle rearrangement, densification and deformation under compression can have a significant effect on the compaction of tablets (23). To date, a number of methods have been described in the literature to analyse and understand these phenomena under compression. Most of these methods and mathematical models applied for tablet compression originate from metallurgic research and industry (18). We studied the deformation of biomaterials under compression by using a force–distance curve treatment method developed especially for pharmaceutical materials (19). The method allowed us to classify the deformation behaviour of different biomaterials under direct (tablet) compression.
In the modified force–distance curve treatment method, the ratio of the two compression work values (W1, W2) obtained from the compression force–distance curve near the maximum force is used to calculate a material-related PF value and to determine the extent of plastic flow. The more plastic the material, the higher PF value is obtained for the material, since plastic materials need a longer time to deform before the maximum punch displacement is achieved under compression (18,19). Elastic behaviour of the materials under compression was determined by calculating a ratio (EF) between the height of the tablet immediately after removing it from the die and the tablet height during maximum compression force. High values for PF and EF indicate low tendency or absence of fragmentation behaviour of the biomaterials.
The dependence of PF and EF of the biomaterials on the compression force is shown in Figs. 3 and 4, respectively. With increasing upper punch compression force, the PF values for all materials studied decreased exponentially (Fig. 3). It is known that MCC undergoes mainly time-dependent plastic deformation during tablet compression (24,25), and also in this study MCC was the most plastic material studied. MCC exhibited the highest plastic properties even at higher upper punch compression forces. CPSC presented significantly lower PF values under compression compared to those obtained with MCC at the respective compression forces. This may be explained by the great differences in particle size and size distribution of the materials (as shown in Figs. 1 and 2).
Fig. 3.

Effect of compression pressure on the plasticity factor (PF) of catalytic pretreated powdered softwood cellulose (CPSC) and lignin (CPSL), industrial softwood kraft lignin (Indulin AT), industrial hardwood kraft lignin (PC 1369) and direct-compression reference excipients (n = 10)
Fig. 4.

Effect of compression pressure on the elasticity factor (EF) of catalytic pretreated powdered softwood cellulose (CPSC) and lignin (CPSL), industrial softwood kraft lignin (Indulin AT), industrial hardwood kraft lignin (PC 1369) and direct-compression reference excipients (n = 10)
Interestingly, lactose monohydrate and the two softwood lignins (CPSL and industrial kraft lignin, Indulin AT) presented very similar compression behaviour (plasticity) in a range of upper punch compression forces 7–10 kN. At lower compression forces, however, CPSL exhibited clearly more plastic deformation than lactose monohydrate and industrial softwood kraft lignin (Indulin AT). Hardwood lignin (PC 1369) was found to be more plastic material than the two softwood lignins under compression with the PF values closed to MCC. This behaviour may be explained by the differences in the degree of substitution (and degree of crosslinking) as well as differences in the rigidity of the structure between hardwood and softwood lignin (15). As expected, DBCP which is reported to deform under compression mainly by fragmentation (25,26), was shown to be a less plastic material as the PF values were clearly the smallest with different upper punch compression forces applied (Fig. 3).
Due to the largest particle size, CPSC was found to be the most elastic material with the highest EF values ranging from 16 to 20% (Table I and Fig. 4). The EF values for CPSC were higher than the respective values for MCC (12–14%) within the same range of compression forces applied. This is in accordance with the findings of Kolakovic et al. (12) who reported that spray-dried nanofibrillar cellulose was more brittle (less elastic) in tablet compression than MCC. Interestingly, the compression force applied did not significantly affect the elasticity of both CPSC and MCC. In contrast, the EF value for softwood lignins (CPSL and industrial kraft lignin, Indulin AT), hardwood lignin (PC 1369), lactose monohydrate (Pharmatose® 80M) and DBCP (Emcompress®) increased as the compression force increased. For softer materials, such as pharmaceuticals and other organic materials, that have a low Young’s moduli, elasticity under compression is increased at higher pressures (23).
Table I.
Values for the Elasticity Factors (EF) of Catalytic Pretreated Powdered Soft-Wood Cellulose (CPSC), Catalytic Pretreated Softwood Lignin (CPSL), Industrial Softwood Kraft Lignin (Indulin AT), Industrial Hardwood Kraft Lignin (PC 1369) and the Three Direct Compression Reference Excipients under Different Compression Pressures (CP)
| Material | CP (MPa) | EF (%) | Material | CP (MPa) | EF (%) |
|---|---|---|---|---|---|
| CPSC | 45.4 | 16.9 | MCC PH 101 | 25.6 | 11.0 |
| 48.3 | 18.4 | 30.5 | 13.1 | ||
| 57.7 | 19.1 | 36.0 | 13.9 | ||
| 67.3 | 16.6 | 44.1 | 14.1 | ||
| 74.4 | 19.6 | 53.0 | 11.2 | ||
| 83.5 | 16.8 | 81.1 | 11.6 | ||
| CPSL | 84.8 | 12.0 | Lactose 80M | 106.5 | 13.7 |
| 102.0 | 12.7 | 108.5 | 10.5 | ||
| 101.2 | 14.4 | 135.2 | 14.5 | ||
| 111.4 | 12.7 | 150.8 | 14.2 | ||
| 131.8 | 14.3 | 168.5 | 14.4 | ||
| 156.0 | 15.0 | 189.4 | 14.5 | ||
| Indulin AT | 81.7 | 10.1 | Dibasic caphosphate | 23.5 | 6.2 |
| 106.2 | 10.7 | 34.7 | 5.8 | ||
| 114.8 | 11.8 | 50.6 | 7.2 | ||
| 118.2 | 11.5 | 89.2 | 10.7 | ||
| 135.7 | 10.5 | 136.7 | 11.3 | ||
| 156.8 | 12.0 | ||||
| PC 1369 | 66.0 | 9.4 | |||
| 80.9 | 10.1 | ||||
| 90.5 | 11.2 | ||||
| 107.5 | 9.2 | ||||
| 135.9 | 9.9 | ||||
| 148.5 | 11.6 |
The EF values for CPSL ranged from 12 to 15% which were close to the EF values obtained with lactose monohydrate (11–14%) and slightly higher than those obtained with industrial softwood kraft lignin (Indulin AT) and hardwood lignin (PC 1369). DBCP was found to be a less elastic material at a range of lower upper punch compression forces from 25 to 100 mPa.
Compaction Properties
The compactibility of biomaterials was evaluated by determining the relationship between the upper punch compression force and tablet crushing strength (Fig. 5). Direct compaction of the two celluloses (MCC and CPSC) resulted in the highest mechanical strength values for tablets (i.e. the highest slopes of the curve). Both celluloses were capable of forming tablets with a high mechanical strength even in the range of relatively low upper punch compression forces (from 2 to 5 kN) or compression pressures (from 25 to 75 MPa). Industrial softwood kraft lignin (Indulin AT) and hardwood lignin (PC 1369) exhibited almost identical compression force–crushing strength profiles, being more compactable than CPSL. Industrial hardwood kraft lignin presented surprisingly good compaction properties without any signs of tableting defects. The compaction behaviour of CPSL was close to that obtained with lactose monohydrate (with the low slope of a compression force–crushing strength curve). In addition, some capping was observed for tablets prepared from CPSL at higher compression forces. DBCP tablets showed capping even at the lowest compression forces used, and consequently, the poorest compaction behaviour among the materials studied.
Fig. 5.

Effect of compression pressure on the crushing strength of catalytic pretreated powdered softwood cellulose (CPSC) and lignin (CPSL), industrial softwood kraft lignin (Indulin AT), industrial hardwood kraft lignin (PC 1369) and direct-compressed reference tablets (n = 10)
In order to investigate the effects of particle size of CPSC on the mechanical strength of tablets, the compactibility of two additional (smaller) particle size fractions of CPSC were also studied (Fig. 6). No differences in compactibility between the three particle size fractions of CPSC were found, thus confirming particle-size-independent plastic behaviour of CPSC under compression. The compression force vs tablet crushing strength profile, however, was found to be different with the pre-dried and non-dried samples of CPSC. Tablets compressed from the pre-dried CPSC had lower mechanical strength compared to those compressed from non-dried CPSC. This suggests clear moisture sensitivity (dependency) of tablet compression when CPSC is used as a filler-binder. The influence of moisture on the tablet compression characteristics of MCC has been reported in the literature (11,27). In addition, the higher fine part and specific surface area of MCC have shown to result in tablets with higher crushing strengths (27).
Fig. 6.
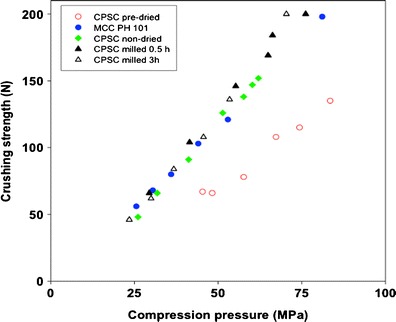
Effects of drying and milling (particle size) of catalytic pretreated powdered softwood cellulose (CPSC) on the crushing strength of tablets (n = 10). Microcrystalline cellulose (MCC) is used as a commercial reference direct-compression excipient
CONCLUSIONS
The particle properties (size and shape), consolidation and deformation under compression can greatly differ depending on the source and isolation method of wood celluloses and lignins. Powdered softwood cellulose isolated by a catalytic oxidation method (CPSC) undergoes mainly plastic deformation under compression, and the tablet compactibility is independent of the particle size. Plastic deformation is also the predominant consolidation mechanism for hardwood kraft lignin which exhibits surprisingly good compaction properties without any signs of tableting defects. Compression properties of catalytic pretreated softwood lignin are not as good as those of celluloses but the properties could be modified by using, e.g. a co-processing approach.
ACKNOWLEDGEMENTS
This research was supported by the European Social Fund’s Doctoral Studies and Internationalisation Programme DoRa and by NordForsk. This work is part of the targeted financing project no. SF0180042s09 and ETF grant project no. ETF7980. The Estonian Ministry of Education and Research is acknowledged for their financial support. We gratefully acknowledge H. Räikkönen, M.Sc. (Physics) for his assistance in the physical characterisation of powders.
Contributor Information
Anna Penkina, Phone: +372-737-5292, FAX: +372-737-528, Email: anna.penkina@hotmail.com.
Osmo Antikainen, Email: osmo.antikainen@helsinki.fi.
Maija Hakola, Email: maija.hakola@helsinki.fi.
Sirpa Vuorinen, Email: sirpa.vuorinen@helsinki.fi.
Timo Repo, Email: timo.repo@helsinki.fi.
Jouko Yliruusi, Email: jouko.yliruusi@helsinki.fi.
Peep Veski, Email: peep.veski@ut.ee.
Karin Kogermann, Email: karin.kogermann@ut.ee.
Jyrki Heinämäki, Email: jyrki.heinamaki@ut.ee.
REFERENCES
- 1.Rothen-Weinhold A, Dahn M, Gurny R. Formulation and technology aspects of controlled drug delivery in animals. PSTT. 2000;3(7):222–231. doi: 10.1016/s1461-5347(00)00276-5. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed I, Kasraian K. Pharmaceutical challenges in veterinary product development. Adv Drug Deliv Rev. 2002;54:871–882. doi: 10.1016/S0169-409X(02)00074-1. [DOI] [PubMed] [Google Scholar]
- 3.Jivraj M, Martini LG, Thomson CM. An overview of the different excipients useful for the direct compression of tablets. PSTT. 2000;3(2):58–63. doi: 10.1016/s1461-5347(99)00237-0. [DOI] [PubMed] [Google Scholar]
- 4.Gonnissen Y, Remon JP, Vervaet C. Development of directly compressible powders via co-spray drying. Eur J Pharm Biopharm. 2007;67:220–226. doi: 10.1016/j.ejpb.2006.12.021. [DOI] [PubMed] [Google Scholar]
- 5.Gohel MC, Parikh RK, Brahmbhatt BK, Shah AR. Preparation and assessment of novel coprocessed superdisintegrant consisting of crospovidone and sodium starch glycolate: a technical note. AAPS PharmSciTech. 2007;8(1), Article 9:E1–7. [DOI] [PubMed]
- 6.Marwaha M, Sandhu D, Marwaha RK. Coprocessing of excipients: a review on excipient development for improved tabletting performance. Int J Appl Pharm. 2010;2(3):41–47. [Google Scholar]
- 7.Patel RP, Bhavsar M. Directly compressible materials via co-processing. Int J PharmTech Res. 2009;1(3):745–753. [Google Scholar]
- 8.Klevan I, Nordström J, Tho I, Alderborn G. A statistical approach to evaluate the potential use of compression parameters for classification of pharmaceutical powder materials. Eur J Pharm Biopharm. 2010;75:425–435. doi: 10.1016/j.ejpb.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 9.Nordström J, Klevan I, Alderborn G. A protocol for the classification of powder compression characteristics. Eur J Pharm Biopharm. 2012;80:209–216. doi: 10.1016/j.ejpb.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 10.Weil J, Westgate P, Kohlmann K, Ladisch MR. Cellulose pretreatments of lignocellulosic substrates. Enzyme Microb Technol. 1994;16(11):1002–1004. doi: 10.1016/0141-0229(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 11.Tobyn MJ, McCarthy GP, Staniforth JN, Edge S. Physicochemical comparison between microcrystalline cellulose and solidified microcrystalline cellulose. Int J Pharm. 1998;169:183–194. doi: 10.1016/S0378-5173(98)00127-6. [DOI] [Google Scholar]
- 12.Kolakovic R, Peltonen L, Laaksonen T, Putkisto K, Laukkanen A, Hirvonen J. Spray-dried cellulose nanofibers as novel tablet excipient. AAPS PharmSciTech. 2011;12(4):1366–1373. doi: 10.1208/s12249-011-9705-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hatakeyama H, Hatakeyama T. Lignin structure, properties, and applications. Adv Polym Sci. 2010;232:1–63. doi: 10.1007/12_2009_12. [DOI] [Google Scholar]
- 14.Li Y, Sarkanen S. Alkylated kraft lignin-based thermoplastic blends with aliphatic polyesters. Macromolecules. 2002;35:9707. doi: 10.1021/ma021124u. [DOI] [Google Scholar]
- 15.Doherty WOS, Mousavioun P, Fellows CM. Value-adding to cellulosic ethanol: lignin polymers. Ind Crop Prod. 2011;33:259–276. doi: 10.1016/j.indcrop.2010.10.022. [DOI] [Google Scholar]
- 16.Hakola M, Kallioinen A, Kemell M, Lahtinen P, Lankinen E, Leskelä M, et al. Liberation of cellulose from the lignin cage: a catalytic pretreatment method for the production of cellulosic ethanol. ChemSusChem. 2010;3(10):1142–1145. doi: 10.1002/cssc.201000217. [DOI] [PubMed] [Google Scholar]
- 17.Penkina A, Hakola M, Paaver U, Vuorinen S, Kirsimäe K, Kogermann K, et al. Solid-state properties of softwood lignin and cellulose isolated by a new acid precipitation method. Int J Biol Macromol. 2012;51(5):939–945. doi: 10.1016/j.ijbiomac.2012.07.024. [DOI] [PubMed] [Google Scholar]
- 18.Antikainen O. New methods to evaluate applicability of powders and granules for tablet compression. PhD Theses, University of Helsinki, Dissertationes Biocentri Viikki Universitatis Helsingiensis; 2003.
- 19.Antikainen O, Yliruusi J. Determining the compression behaviour of pharmaceutical powders from the force-distance compression profile. Int J Pharm. 2003;252:253–261. doi: 10.1016/S0378-5173(02)00665-8. [DOI] [PubMed] [Google Scholar]
- 20.Soppela I, Airaksinen S, Hatara J, Räikkönen H, Antikainen O, Yliruusi J, et al. Rapid particle size measurement using 3D surface imaging. AAPS PharmSciTech. 2011;12(2):476–484. doi: 10.1208/s12249-011-9607-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sandler N. Photometric imaging in particle size measurement and surface visualization. Int J Pharm. 2011;417:227–234. doi: 10.1016/j.ijpharm.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 22.Mir VG, Heinämäki J, Antikainen O, Revoredo OB, Colarte AI, Nieto OM, et al. Direct compression properties of chitin and chitosan. Eur J Pharm Biopharm. 2008;69:964–968. doi: 10.1016/j.ejpb.2008.01.029. [DOI] [PubMed] [Google Scholar]
- 23.Patel S, Kaushal AM, Bansal AK. Effect of particle size and compression force on compaction behavior and derived mathematical parameters of compressibility. Pharm Res. 2007;24(1):111–124. doi: 10.1007/s11095-006-9129-8. [DOI] [PubMed] [Google Scholar]
- 24.David ST, Augsburger LL. Plastic flow during compression of directly compressible fillers and its effect on tablet strength. J Pharm Sci. 1977;66:155–159. doi: 10.1002/jps.2600660205. [DOI] [PubMed] [Google Scholar]
- 25.Roberts RJ, Rowe RC. The effect of the relationship between punch velocity and particle size on the compaction behaviour of materials with varying deformation mechanisms. J Pharm Pharmacol. 1986;38:567–571. doi: 10.1111/j.2042-7158.1986.tb03082.x. [DOI] [PubMed] [Google Scholar]
- 26.McKenna A, McCafferty DF. Effect of particle size on the compaction mechanism and tensile strength of tablets. J Pharm Pharmacol. 1982;34:347–351. doi: 10.1111/j.2042-7158.1982.tb04727.x. [DOI] [PubMed] [Google Scholar]
- 27.Shlieout G, Arnold K, Müller G. Powder and mechanical properties of microcrystalline cellulose with different degrees of polymerization. AAPS PharmSciTech. 2002;3(2), Article 11:1–10. [DOI] [PMC free article] [PubMed]