

Abstract
Physiologically based pharmacokinetic (PBPK) modeling has become a useful tool to estimate the performance of orally administrated drugs. Here, we described multiple in silico/in vitro/in vivo tools to support formulation development toward mitigating the positive food effect of NVS123, a weak base with a pH-dependent and limited solubility. Administered orally with high-fat meal, NVS123 formulated as dry filled capsules displayed a positive food effects in humans. Three alternative formulations were developed and assessed in in vitro and in vivo preclinical and/or clinical studies. By integrating preclinical in vitro and in vivo data, the PBPK model successfully estimated the magnitude of food effects and the predicted values were within ±30% of the observed results. A model-guided parameter sensitivity analysis illustrated that enhanced solubility and longer precipitation times under fed condition were the main reason for enhanced NVS123's exposure in presence of food. Eventually, exposure after an amorphous formulation was found to be not significantly altered because of remarkably enhanced intestinal solubility and reduced precipitation. Gastroplus population simulations also suggested that the amorphous formulation is promising in mitigating a clinically significant food effect. Overall, these efforts supported the rationale of clinical investigation of the new formulation, and more importantly, highlighted a practical application of PBPK modeling solving issues of undesirable food effects in weakly basic compounds based on preclinical in vitro/in vivo data.
Electronic supplementary material
The online version of this article (doi:10.1208/s12249-013-0018-2) contains supplementary material, which is available to authorized users.
KEY WORDS: amorphous formulation, food effects, physiologically based pharmacokinetic (PBPK) modeling, population simulation, precipitation
INTRODUCTION
Food can induce various changes in physiological conditions, such as delayed gastric emptying, change of gastrointestinal (GI) pH, stimulation of bile flow, and interaction of intestinal influx or efflux transporters (1–4). Briefly, a food effect is manifested if the 90% confidence interval (CI) for the ratio of population means between fed and fasted treatments fails to meet the limits of 80–125% for either area under the curve (AUC) or maximum concentration (Cmax) (4). Positive food effects are commonly seen for Biopharmaceutics Classification System (BCS) Class 2 drugs which have low solubility and high permeability (2,5) and positive food effects occur when a higher systemic exposure is observed under fed conditions compared with the fasted state. When a Class 2 drug is administered shortly after a meal is ingested, food may enhance the solubilization of drug in the intestinal lumen (e.g., via the formation of micelle) and inhibit the efflux transporters in the intestine, and thus improve the absorption. Conversely, food can delay gastric emptying and prolong intestinal transit time, resulting in a delayed tmax of the drug product. Weak bases can be Class 2 compounds, and they can be highly soluble in the low-pH environment of the fasted stomach but poorly soluble at higher pH in the intestine. Therefore, in the presence of meal, compounds of weak bases are susceptible to variable dissolution and intestinal precipitation due to changes in GI pH and stimulated secretion of bile salts.
Compound NVS123 (Table I) is a weakly basic, BCS Classes 2 to 4 molecule with a steep pH-dependent solubility profile where solubility limits for drug dissolution above pH 4 and the solubility can be as low as ∼1 μg/mL at pH 7. Moreover, while NVS123 dissolution in 0.1 N HCl or simulated gastric fluids (SGF) is nearly complete within 30 min, most dissolved drug can be susceptible for rapid precipitation in intestinal fluid. Hence, such a drug may be prone to incomplete oral absorption, reduced systemic exposure, and lack of dose proportionality in clinical trials. NVS123 is mainly eliminated by hepatic metabolism mediated by CYP3A4 and is not a substrate for intestinal efflux transporters (e.g., P-gp). A clinical food effect study using dry filled capsules showed a significant increase in Cmax and AUC by 1.8- and 1.6-fold after a high fat meal, respectively. The compound has a somewhat narrow therapeutic index, and thus high Cmax is undesirable. In this paper, we describe the application of biopharmaceutical tools such as in vitro two-step dissolution, canine food effect model, and physiologically based absorption simulation to support development of a solid dosage form to overcome the positive food effect. Prior publications have also reported the utility of physiologically based pharmacokinetic models (PBPK) to quantitatively predict the magnitude of food effects and estimate the concentration vs. time profiles for compounds in development project (6–8). However, few attempts have been considered to predict the effect of meal for formulations which contain various excipients and complex matrices. We hereby describe a novel PBPK model approach to guide the formulation design for mitigation of positive food effects by combining the in vitro/in vivo data. In addition, this mechanism-based PBPK model simulated the human concentration vs. time profiles of a promising dosage form of NVS123 in the presence or absence of meal, which laid the theoretical foundation for a future pivotal clinical study to evaluate the performance of the formulation on suppressing the food effect.
Table I.
Physicochemical and Biopharmaceutical Properties of NVS123
| Input parameter | Values |
|---|---|
| Caco-2 permeability | 1.6 × 10−4 cm/min |
| Human permeability | 10 × 10−3 cm/min |
| Dog effective permeability | 30 × 10−3 cm/min |
| Molecular weight | >450 |
| LogP | >4.5 |
| pK a | 3–3.5 |
| Mean particle size | 20 μm |
| pH-solubility profile | |
| pH 1.0 | 2 mg/mL |
| pH 6.8 (crystalline) | 1 μg/mL |
| pH 6.8 (amorphous) | 0.01 mg/mL |
| Blood/plasma ratio(R b/p) | 0.7 |
| Plasma unbound (%) | 2 |
| First-pass hepatic extraction (%): | 7.0 |
| Human plasma clearance (mL/min/kg): | 1.0 |
| Human volume of distribution (L/kg) | 1.5 |
MATERIALS AND METHODS
Chemicals and Reagents
NVS123 was provided by Novartis Pharmaceuticals Corporation (East Hanover, NJ). All other reagents and chemicals were at least ACS reagent grade. Pentagastrin was purchased from Sigma (St. Louis, MO). The human FDA diet (two eggs, two bacon strips, two pieces of toast with butter/jelly, two slices of white bread hash brown potatoes, and 8 oz whole milk) was prepared on site, homogenized, and weighed about 580 g with ∼1,100 calories. A custom high-fat dog food (∼50 g) was scaled down from human FDA high diet (Sodexho Services, Wallingford, CT).
Computer Hardware and Software
GastroPlus (version 7.0, Simulations Plus, Inc, CA, USA) was run on a Lenovo computer with Intel® Core™ i5 processor. This program enables the prediction of rate and extent of oral drug absorption from the GI tract using the Advanced Compartmental Absorption Transit (ACAT) model based on an earlier absorption model originally established by Yu and Amidon (9). With the input of physicochemical properties (e.g., solubility, permeability, LogP, pKa, and particle sizes), dissolution rates for solid formulations, and systemic PK parameters human concentration time profiles can be generated.
Formulations
Several NVS123 formulations were developed. A dry filled capsule (F1) was initially used for clinical evaluation, where the active drug substance was mixed with regular fillers and lubricants (e.g., lactose and magnesium stearate). In our early clinical studies, significant food effect is still the major obstacle for the improvement of patient compliances (the capsule must be taken without food). To address the positive food effect in the clinic, three new capsule formulations for NVS123, including solid suspended formulation (F2), roller compaction (F3), and amorphous formulation (F4), were also sequentially developed. The crystalline form was used as the active pharmaceutical ingredient (API) for F1, F2, and F3. Clinical food effect data were available for F1, F2, and F3; F4 was only evaluated preclinically.
Two-Step In Vitro Dissolution and Precipitation Studies
The dissolution and precipitation behaviors of each formulation were evaluated using a two-step dissolution method. The USP type II dissolution apparatus was used for this study. Initially, the drug dissolution profiles under fasted or fed states were investigated with one unit of drug product (200 mg) in 250 ml of SGF media at a rotating speed of 100 rpm at 37°C. After 60 min, the pre-warmed (37°C) fasted or fed simulated small intestinal fluid (FaSSIF or FeSSIF) medium (2×, 250 mL) was added into the beaker under quick stirring for about 1 min. The pH in the mixture was adjusted to 6.8 or 5.8 by addition of 10 M NaOH for FaSSIF or FeSSIF medium, respectively. A 0.5-mL sample of the solution was taken out periodically from 0 to 180 min, and the same amount of the medium at the same temperature was replaced. The samples (∼100 μL) were immediately filtered through a 0.45-μm PVDF filtration membrane. The filtrates were diluted with suitable volume of acetonitrile and the concentration of NVS123 was determined using HPLC with a UV detector.
Bridging In Vitro–In Vivo Dissolution
When evaluating the in vivo performance of formulations with complex excipients, understanding the relationship between in vitro and in vivo dissolution is a prerequisite for estimation of the drug input rate into the systemic circulation. It is well known that dissolution testing conditions (e.g., medium composition, paddle speed, apparatus, etc.) affects the in vitro dissolution rate which can be significantly different from apparent in vivo profiles. Such disconnection may be more pronounced under fed condition than under fasted condition. Immediately after food is ingested, the gastric pH increases from the basal values (∼1 to 2 in the fasted stomach) because gastric acid is instantly buffered by meal constituents and then gradually decreases and returns to basal level because of increased gastric acid secretion stimulated by meal; the time required for latter process may be highly dependent on the type of meals (10). Therefore, considering that the complex GI physiology changes under fed conditions, it is difficult to anticipate the in vivo dissolution kinetics of a weakly basic compound based on the results of in vitro assays where dissolution media with fixed pH values are typically used. We optimized the parameter of the Weibull equation (11) using the Gastroplus optimization module to identify the best in vivo relevant dissolution profile (as *.crd files) that can capture the absorption phase of PK data. Briefly, the GastroPlus ACAT model which incorporated in vitro dissolution profiles described by the Weilbull equations was used to predict the systemic availability vs. time curves. Conversely, the observed concentrations vs. time curves were deconvoluted to generate the systemic availability vs. time profiles. When the systemic availability as predicted by Gastroplus is in agreement with the deconvoluted systemic availability, the in vitro and in vivo dissolution can be well correlated. Otherwise, the Weibull parameters of the dissolution profiles need to be adjusted to describe the deconvoluted in vivo dissolution profiles. For BCS Class 2 compounds, such as weak bases, the most common adjustment of dissolution curve for fed PK prediction is to decrease the release rate of the compound by increasing the time scaling and shape factors in the Weibull equations.
Dog Studies and Pharmacokinetic Analysis
All animal studies were conducted with the approval of Animal Care and Use Committee of Novartis or the contract research organization where we outsourced one arm of the dog study. The animal experiments are conducted in full compliance with local, national, ethical, and regulatory principles and local licensing regulations, per the spirit of Association for Assessment and Accreditation of Laboratory Animal Care. Beagle dogs (n = 4 or 5) were dosed in a nonrandomized, single-sequence design, with at least a 7-day washout between treatments. Each dogs received an intramuscular dose of pentagastrin (6 μg/kg) 30 min prior to doing or feeding. For the fasted arm, dog was restrained from food overnight before the dosing and until 4 h after the dose. For the fed arm, the human FDA high-fat meal was homogenized (∼580 g of paste and ∼ 1,000 cal/serving), and an aliquot of 50 g of paste (∼ 95 cal/serving) was given to dogs immediately after the administration of pentagastrin. All dogs completed the meal in 10 min. After dosage forms were orally administrated, a 50-mL of sterile water was administrated by oral syringe. Approximately 1.5–3 mL of venous whole blood was collected at 0 (pre-dose), 0.083, 0.25, 0.5, 1, 2, 4, 6, 8, 12, 24, and 48 h post-dose. Water was provided ad libitum. No sedatives or anesthetics were used for dosing purposes. The dog PK profiles after a single intravenous bolus dose of 1 mg/kg has also been studied previously. The serum concentrations of NVS123 were measured using a validated liquid chromatography/electrospray ionization tandem mass spectrometry (supplemental materials). The dog food effect studies for F1, F2, and F3 formulation were conducted internally while the dog study for F4 was outsourced and conducted in a contract research organization under good laboratory practice.
Food Effect Studies in Healthy Subjects
A randomized, open-label, two-arm, four-period crossover study was conducted to determine the effect of food on the pharmacokinetics after a single 200-mg oral dose of F1, F2, and F3 in healthy subjects. This study was conducted in accordance with the ethical principles of the Declaration of Helsinki. The study protocol and informed consent forms were approved by the local institutional review board.
The participants were healthy male or female subjects between 18 and 65 years of age without significant deviation from normal in medical history, physical examination, vital signs, cardiograms, and clinical laboratory determination. Subjects were not allowed to take any CYP3A4 enzyme inducing or inhibiting drugs within 4 weeks prior to study. In addition, no prescription drugs were allowed within 2 weeks prior to dosing, and no over-the-counter medication and herbal supplements were permitted within 72 h prior to dosing.
The healthy volunteers received the F1 capsule or two new formulations (e.g., F2 and F3) during four study periods and two treatment arms per the following regimens: (A1) a single oral dose of 200 mg F1 capsule was administered under fasted conditions (after an overnight fast of ∼ 10 h), (B1) a single oral dose of 200 mg F1 capsule was taken under fed conditions (30 min after a FDA human high-fat meal with about 1,000 calories and 50% fat), (C1) a single oral dose of 200 mg F2 capsule under fasted conditions, and (D1) a single oral dose 200 mg F2 capsule under fed conditions; (A2) same dosing regimen as A1, (B2) same dosing regimen as B1, (C2) a single oral dose of 200 mg of F3 capsule under fasted conditions, and (D2) a single oral dose of 200 mg F3 capsule under fed conditions. A total of 40 healthy adult subjects were randomly assigned in equal numbers to four sequences of regimens A1, B1, C1, and D1 for treatment arm 1 and regimens A2, B2, C2, and D2 for treatment arm 2 of the study. Each dose of NVS123 was taken orally with 240 mL of water (see Electronic Supplementary Materials for sample size selection). All subjects were allowed to consume a standard lunch 4 h after the dose. The blood samples were scheduled to be collected pre-dose and at 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 24, 36, and 48 h post-dose. A 12-day washout period was applied to separate each dosing regimen assigned for each subject. The plasma concentrations of NVS123 were measured using a validated liquid chromatography/electrospray ionization tandem mass spectrometry (see Electronic Supplementary Materials).
Data Analysis of Observed and Simulated PK Data
The plasma concentration vs. time data were analyzed with noncompartmental analysis methods to calculate the PK parameters (e.g., AUC, Cmax, tmax, etc.) using WinNonlin Phoenix software v6.1 (Pharsight Corporation, Mountain View, CA). To assess the food-related effects on NVS123 PK, a linear mixed effects model was fitted to the log-transformed PK parameters of AUC0–inf and Cmax of NVS123 using SAS 9.0 (SAS Institute Inc., Cary, NC, USA). In this model, sequence, period, and treatment were included as fixed effects and subject was considered as a random effect. A food effect was identified if the fed state/fasting state arithmetic mean ratios of AUC0–inf and Cmax and their 90% confidence intervals were below 0.80 or above 1.25.
PK and Absorption Simulations
Biopharmaceutical Characterization of NVS123 Formulations
The performance of the formulations was simulated or modeled using ACAT model in GastroPlus v8.0. The model consisted of the numerical integration of differential equations to quantitatively describe multiple drug absorption processes, including release, dissolution, precipitation, re-dissolution, luminal degradation, and passage across GI membranes. The default GI physiology parameters under fasted and fed conditions within the ACAT model were used based on the average values obtained from published population data, including pH, volume, length, radius, transit time, and bile salt concentration. The physiology parameters used in the GastroPlus ACAT model have been listed in Table S1.
The experimental in vitro dissolution profiles for all formulations (Fig. 1) in SGF were incorporated directly in GastroPlus program under both fasted and fed condition. The in vitro dissolution profiles of each formulation in simulated gastric fluid were loaded as *.dsd files for both dog and human models. The Weibull release functions were then used to fit the in vitro dissolution data and the resulting Weibull release profiles (regarded as a dissolution profile) were further incorporated in the model as *.crd file. When needed, the Weibull release parameters for each dosing cohort (e.g., formulation and fasted/fed) were optimized to provide reasonably closest simulation to the observed mean PK profile after judged by visual inspection, correlation coefficient (R2) between predicted and observed profiles, root of mean squared prediction error (RMSE), and the prediction deviation of the key PK parameters (Cmax, tmax, and AUC). According to measured FaSSIF and FeSSIF solubilities, the solubility at a particular pH and bile salt concentrations at each GI segment was approximated using the following equation:
| 1 |
where [NaTC] is the milimolar concentration of sodium taurocholate used in the dissolution medium or defined in the GI compartment in the GastroPlus ACAT model, Csx and Cso are the predicted drug biorelevant solubility and the intrinsic aqueous solubility in the unit of mg/ml, respectively, Mw is the molecular weight, SR is the bile salt solubility ratio, which can be estimated based using the equation developed by Mithani et al. (12).
Fig. 1.
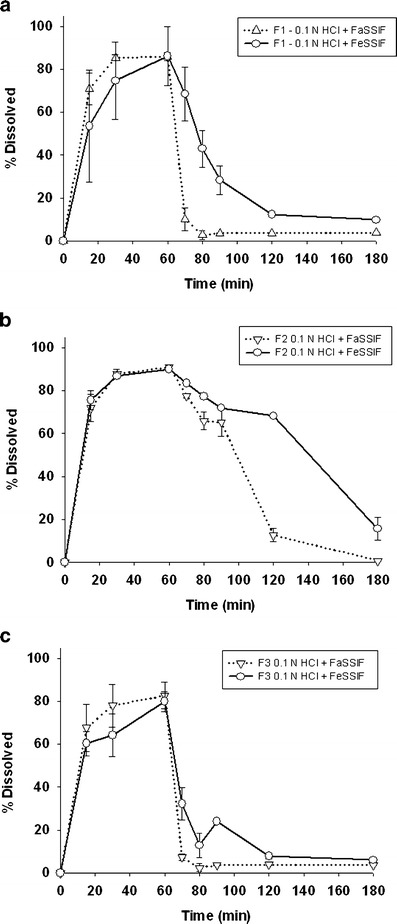
In vitro two-step dissolution/precipitation profiles of three formulations of NVS123, including F1 (a), F2 (b), and F3 (c) formulations in SGF/FaSSIF or SGF/FeSSIF media
Drug precipitation was described by the mean precipitation time (Tp) which represents mean time for particles to precipitate from solution when the local concentration exceeds the drug solubility. For BCS Class 2 weak basic compounds with pH-dependent solubility, like NVS123, compounds are susceptible for rapid precipitation in intestinal fluid. Under the fasted state, a relatively short precipitation time in intestinal is expected, whereas, under the fed state, bile acids stimulated by food will sometimes slow the precipitation or stop it completely, resulting a relatively longer precipitation time. The values of Tp are difficult to measure quantitatively in vitro and were fitted against the observed data. Additionally, it was more important to know how to estimate the Tp values in human model based on the preclinical data. Dogs have been used as surrogates to predict human food effects for multiple compounds (13,14), with adjustments of meal amount (e.g., 50 g) and doses along with a reduction of gastric pH using pentagastrin. With efforts to optimize a predictive canine food effect model, we may reasonably assume that Tp is likely similar between dogs and humans when preclinical studies are well designed. The values of mean Tp were optimized to fit to the dog concentration-time profile following each dosage regimen as dosing formulation under fasted or fed conditions using ACAT dog models. Then, the obtained values of Tp in dog models were incorporated in human GastroPlus model to simulate the absorption under corresponding dosing scenarios. By comparing the simulated results with observed human data, we could establish that Tp between dogs and humans may be similar. The precipitated drug particles were then assumed to be re-dissolved with the dissolution rate described by Wang-Flanagan model as follows (Eq. 2)
| 2 |
where MD is the dissolved amount, Mu, t is the undissolved amount at time t, Cs is the solubility at a particular pH, Cl is the concentration of dissolved drug in gut lumen, ρ is the drug density, Dw is the diffusion coefficient, rt is the current particle radius, and T is the diffusion layer thickness which is equal to the particle radius (15).
Disposition Parameters for NVS123 in Humans
PK data after oral administration of the F1 in each study arm under fasted state were fitted by compartmental PK model using PKPlus™ (Simulation Plus, CA, USA). It was found that one-compartmental disposition model with first-order absorption can well describe to the plasma concentration vs. time data. The oral human PK parameters were then calculated (arm 1: V/F = 5.07 L/kg, CLpo = CLIV/F = 0.219 L h−1 kg−1; arm 2: V/F = 5.01 L/kg, CLpo = CLIV/F = 0.197 L h−1 kg−1). The human PK profiles of NVS123 after i.v. doses have not been determined and thus absolute bioavailability data are not available. According to unpublished data from a human absorption, metabolism, and excretion study under fasted conditions, the excretion of intact NVS123 in feces is ∼70% of the administered dose. As only ∼1% of NVS123 was found in the bile of bile-duct cannulated rats after an intravenous dose (in-house data), hepatobiliary elimination of the parent compounds was assumed negligible in human. Therefore, the fraction absorption (Fa) of NVS123 under fasted state was about 30% based on a human ADME study. Assuming that drug degradation in the GI tract is minimal, the bioavailability (F) of NVS123 can be calculated based on the following equations (Eqs. 3 and 4).
| 3 |
After rearrangement of Eq. 3, we obtained Eq. 4.
| 4 |
where Rb/p is the blood to plasma distribution ratio (∼0.7 based on an internal in vitro assay), QH is the hepatic blood flow (1.24 L h−1 kg−1), and CLIV and CLPO are the plasma CL after a single intravenous and oral dose, respectively. Based on the calculated F value, the systemic clearance (CLiv) and apparent volume of distribution (Vz) were derived from clinically observed CLpo and Vz/F. The calculated pharmacokinetic disposition parameters of NVS123 were summarized in Table I.
Parameter Sensitivity Analysis
As a rule of thumb, we can assume that fed bioequivalence can be achieved for a BCS Class 2 weak basic drug if the Fa of the drug product is greater than 70% under fasted condition. To design a formulation that can mitigate the positive food effect, it was important to explore the interaction between the in vivo solubility and precipitation and their impacts on the extent of absorption. As described previously in Eq. 1, the in vivo solubility is assumed to correlate with the intrinsic solubility of the active pharmaceutical ingredient (API) at a particular pH and bile salt concentrations at each GI segment. Therefore, simulations were run using parameters sensitivity analysis (PSA) to compare the sensitivity of Fa to the bile salt SR and mean Tp after a single 200 mg oral dose of IR dosage form under fasted condition. The duodenum solubility, represented as the solubility in the GI tract, can be further calculated using Eq. 1 and simulated SR values. The PSA of Fa was shown in contour plots as a function of duodenal solubility and mean Tp.
Virtual Simulation for Pharmacokinetic Profiles and Fed Bioequivalence Studies
The model was employed to predict exposure in humans for the new formulations, F4. While F4 has been tested in canine food effect model, the human model was established using measured or adjusted in vitro dissolution profile and the Tp values fitted from the dog PK data. The population simulator was applied to run a virtual food effect bioequivalent trial simulation with 25 subjects in a crossover design. All selected physiological and PK parameters are randomly sampled from log-normal distribution with defined coefficient of variance of 20–30%. The population simulation can estimate 90% CI of Cmax, AUC0–inf, and the mean plasma concentration.
RESULTS
In Vitro Two-step Dissolution Tests
The two-step dissolution tests were studied in F1, F2, and F3 formulation to mimic the drug dissolution in stomach as well as the precipitation in small intestine under fasted and fed conditions. Figure 1 showed the dissolved amount (as percentage of dose) at each time point from these formulations. In all of the tested formulations, NVS123 was nearly completed dissolved (>80%) within 1 h in SGF while precipitated after FaSSIF or FeSSIF were added into the dissolution beaker. It appeared that precipitation rate of NVS123 is slower in FeSSIF medium than that in FaSSIF medium, suggesting that the mean Tp is higher under fed condition than fasted condition in F1, F2, and F3 formulation. These results supported our modeling assumption that the difference of precipitation rate is a key-driven fact for the observed food effect.
Evaluation of the Oral PK and Precipitation Kinetics in Dogs
A two compartmental model best described the concentration-time profiles after an intravenous dose of 1 mg/kg in dogs, and the model was thereby used to calculate the PK parameters, such as CL, Vss, and micro-constants (Table S2). From the observed PK data in dog food effect studies, a high-fat meal increased Cmax and AUC of F1, F2, and F3 oral formulations, but yielded no significant changes for AUC with the F4 formulation (Table II). Interestingly, for each formulation tested in the dog studies, no significant difference was observed in drug onset (represented by tmax) between fed and fasted condition. The calculated PK disposition parameters were then incorporated in the Gastroplus ACAT model. Using the Gastroplus optimization module, the value of Tp was fitted against the mean observed dog oral PK profiles under fasted or fed conditions (Fig. 2). For all simulations for dog PK data, the R2 was greater than >0.90 and RMSE were in reasonable range (Table II). For all formulations except for the F4 formulation, the Tp (fitted against dog PK profile data) under the fed state was consistently higher than that under the fasted condition, suggesting that solubilized NVS123 remained in supersaturated state in the intestine for a longer time after high fat meals. Conversely, for formulation F4, Tp was significantly longer under fasted condition (∼4,000 s) compared with other formulations and was not influenced by food (Table II).
Table II.
Comparison of Simulated (SIM) vs. Observed (OBS) Mean Plasma PK Parameters of NVS123 in Dogs
| Parameters | F1 | F2 | F3 | F4 | ||||
|---|---|---|---|---|---|---|---|---|
| OBS | SIM | OBS | SIM | OBS | SIM | OBS | SIM | |
| Dose (mg) | ||||||||
| Fasted | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
| Fed | ||||||||
| C max (ng/mL) | ||||||||
| Fasted | 510 | 610 | 670 | 735 | 595 | 737 | 560 | 801 |
| Fed | 880 | 920 | 929 | 935 | 934 | 932 | 695 | 720 |
| AUC0–inf (μg × h/mL) | ||||||||
| Fasted | 3.22 | 3.20 | 4.57 | 4.71 | 3.31 | 3.81 | 3.15 | 2.80 |
| Fed | 6.60 | 6.10 | 7.02 | 6.12 | 7.75 | 6.16 | 2.89 | 3.13 |
| t max (h) | ||||||||
| Fasted | 1 | 1.4 | 2 | 1.7 | 1.5 | 1.4 | 1 | 1.6 |
| Fed | 2 | 2.1 | 2 | 2 | 2 | 2.2 | 1.5 | 2.3 |
| Precipitation time (s)a | ||||||||
| Fasted | N/A | 1,000 | N/A | 1,800 | N/A | 1,500 | N/A | 4,000 |
| Fed | N/A | 3,500 | N/A | 3,000 | N/A | 3,000 | N/A | 4,000 |
| Correlation coefficient (R 2)b | ||||||||
| Fasted | N/A | 0.98 | N/A | 0.94 | N/A | 0.9 | N/A | 0.91 |
| Fed | N/A | 0.9 | N/A | 0.99 | N/A | 0.98 | N/A | 0.92 |
| RMSEc | ||||||||
| Fasted | N/A | 32.1 | N/A | 77.7 | N/A | 99.5 | N/A | 95.0 |
| Fed | N/A | 104 | N/A | 37.3 | N/A | 47.3 | N/A | 77.0 |
aPrecipitation time (T p) was fitted against in vivo dog PK profiles in the model
bCorrelation coefficient between the observed concentration and simulated values
cRoot mean square prediction error (RMSE) of plasma concentration. RMSE = where N is the number of observed data points
Fig. 2.
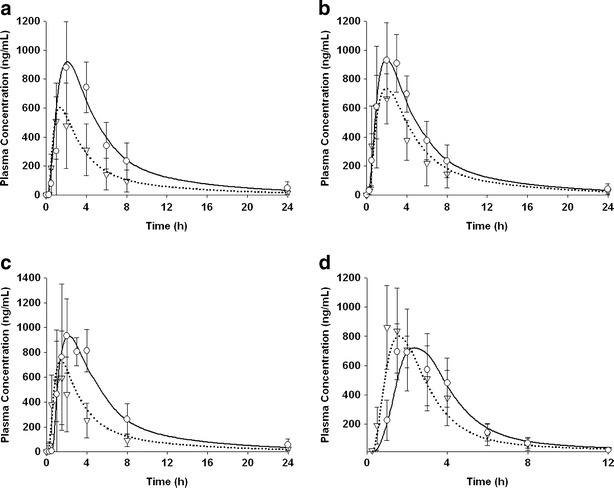
Observed and simulated mean plasma concentrations after a single administration of 50 mg of NVS123 given as F1 (a), F2 (b), F3 (c), or F4 (d) formulation in dogs under fasted and fed state. Symbol annotation: open triangles observed fasted concentration with standard deviation; open circles observed fed concentration with standard deviation; dotted curve simulated mean fasted concentration; and solid curve simulated fed concentration
Development of a Human Physiologically based Absorption Model
Correlations Between In Vitro Dissolution Profiles and In Vivo Dissolution Rate
The in vitro dissolution profiles successfully simulated the drug release and dissolution in all dog studies with and without the food intake. Therefore, we assumed that the changes of systemic exposure caused by meals were mainly associated with precipitation and intestinal solubility. In the clinical study with the F1 formulation, the in vitro dissolution appeared to correlate directly with in vivo dissolution if the drug product was taken without the food. However, the absorption rate was slower when meal was given prior to the dose, as evidenced by delayed tmax. Apparently, the in vitro profile tested in SGF (pH 1.6) did not describe the in vivo dissolution under fed condition, and hypothetical in vivo dissolution profiles (as a *.crd file in Gastroplus) were further optimized to fit the PK data and to match the deconvoluted systemic availability profiles. The adjusted profile reflected at least two dissolution phases in the presence of a high fat meal. Within 1 h after the dose, only a minor portion (<20%) of the API in the F1 dosage form was dissolved due to increased stomach pH after the meal. After 1 h to 2 h post-dose, the API gradually dissolved, but the rate was ∼50% less than that under fasted state (Fig. 3). The simulated in vivo systemic availability profile was approximately in agreement with the deconvoluted profile. A similar approach was also successfully used to obtain the optimal dissolution profile for F2 and F3 formulation. Therefore, there was sufficient confidence to apply this approach to simulate the PK of F4 formulation with adjusted fed dissolution profile. The detailed Weibull parameters for different formulations have been listed in Table S3.
Fig. 3.
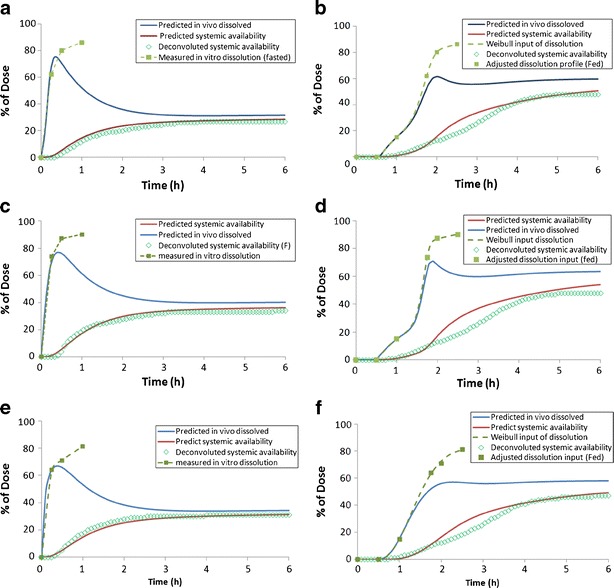
Comparison of in vitro dissolution, adjusted Weibull input dissolution, and predicted in vivo dissolution as well as deconvoluted and predicted systemic availability for F1 (a, b), F2 (c, d), and F3 (e, f) formulation under fasted (a, c, d) and fed (b, d, f) state
In Silico PBPK Absorption Model of Food Effect in F1, F2, and F3 Formulations
The mean PK profiles after oral administration of F1, F2, and F3 were simulated using the PK and physicochemical parameters listed in Table I and physiology parameters in Table S1, under both fasted and fed condition. The model captured the dog and human PK profiles under fasted and fed conditions for all tested formulations (Figs. 2 and 4; Tables II and III). For all simulations for human PK data, the R2 was greater than >0.91 and RMSE were in reasonable range (Table III). A high-fat meal increased the oral absorption and systemic exposure following the administration of F1, F2, and F3 in dogs and humans. The simulated magnitude of food effects in human in terms of changes on AUC, tmax, and Cmax was less than 1.5-fold of the observed effects (Table III). The simulated extent of absorption (Fa) were 34.7%, 43.9%, and 38.5% under fasted state while 64.0%, 65.7%, and 59.9% under fed state for F1, F2, and F3 formulation, respectively. Overall, the physiologically based absorption model predicted the in vivo performance of new pharmaceutical formulations (e.g., F4) with confidence.
Fig. 4.
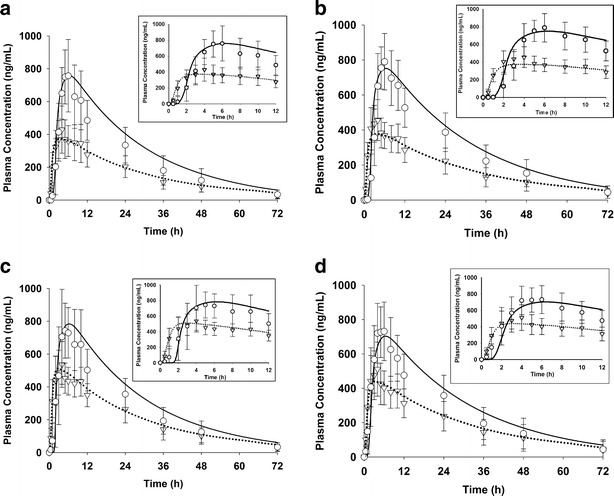
Observed and simulated mean plasma concentrations after a single administration of 200 mg of NVS123 given as F1-arm 1 (a), F1-arm 2 (b), F2 (c), or F3 (d) formulation in humans under fasted and fed state. Symbol annotation: open triangles observed fasted concentration with standard deviation; open circles observed fed concentration with standard deviation; dotted curve simulated mean fasted concentration, and solid curve simulated fed concentration. Insert panel: observed and simulated mean plasma concentrations of each formulation from 0 to 12 h
Table III.
Comparison of Simulated (SIM) vs. Observed (OBS) Mean Plasma PK Parameters of NVS123 in Humans
| Parameters | F1 (Arm 1) | F1 (Arm 2) | F2 | F3 | ||||
|---|---|---|---|---|---|---|---|---|
| OBS | SIM | OBS | SIM | OBS | SIM | OBS | SIM | |
| Dose (mg) | ||||||||
| Fasted | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 |
| Fed | ||||||||
| C max (ratio) | ||||||||
| Fed vs. fasted | 1.76 | 2.03 | 1.72 | 2.05 | 1.38 | 1.53 | 1.38 | 1.66 |
| FE of predictiona | 1.16 | 1.19 | 1.11 | 1.21 | ||||
| AUC0–inf (ratio) | ||||||||
| Fed vs. Fasted | 1.57 | 1.84 | 1.58 | 1.84 | 1.33 | 1.51 | 1.37 | 1.57 |
| FE of predictiona | 1.17 | 1.17 | 1.13 | 1.15 | ||||
| t max (ratio) | ||||||||
| Fed vs. fasted | 1.50 | 1.91 | 1.50 | 1.80 | 1.25 | 2.25 | 1.50 | 2.06 |
| FE of predictiona | 1.27 | 1.20 | 1.80 | 1.38 | ||||
| F a (%) | ||||||||
| Fasted | N/A | 34.7 | N/A | 34.7 | N/A | 43.9 | N/A | 38.5 |
| Fed | N/A | 64.0 | N/A | 64.0 | N/A | 65.7 | N/A | 59.9 |
| F (%) | ||||||||
| Fasted | N/A | 32.1 | N/A | 32.3 | N/A | 40.5 | N/A | 35.9 |
| Fed | N/A | 59.2 | N/A | 59.7 | N/A | 60.7 | N/A | 55.8 |
| Precipitation time (s)b | ||||||||
| Fasted | N/A | 1,000 | N/A | 1,000 | N/A | 1,800 | N/A | 1,500 |
| Fed | N/A | 3,500 | N/A | 3,500 | N/A | 3,000 | N/A | 3,000 |
| Correlation coefficient (R 2)c | ||||||||
| Fasted | N/A | 0.95 | N/A | 0.93 | N/A | 0.97 | N/A | 0.96 |
| Fed | N/A | 0.93 | N/A | 0.91 | N/A | 0.93 | N/A | 0.92 |
| Root mean squared prediction error (RMSE)d | ||||||||
| Fasted | N/A | 28.3 | N/A | 32.8 | N/A | 30.4 | N/A | 28.2 |
| Fed | N/A | 81.3 | N/A | 71.8 | N/A | 81.4 | N/A | 84.5 |
aFold error (FE) of the prediction.
bPrecipitation time (T p) in the human model was assumed to be similar to that in the dog model because of relatively similar intestinal pH and bile salt concentration. This assumption was verified by comparison of simulated and observed PK profiles (Fig. 3) and parameters (Table III)
cCorrelation coefficient between the observed concentration and simulated values
dRoot mean squared prediction error (RMSE) of plasma concentration. RMSE = where N is the number of observed data points
Sensitivity of the Extent of Drug Absorption to Tpand Intestinal Solubility
Although extensive formulation efforts have been made to increase the solubility/dissolution of NVS123 API which partly mitigated food effects, fed bioequivalence has not been achieved. PSA have been proven a potential powerful tool to implement quality by design by validating the key factors related to the formulation performance (16). Here, the PSA was represented by the contour plot under fasted condition with the interaction between duodenum solubility and Tp and their impacts on NVS123's extent of absorption. As expected, the contour plot clearly indicated that the effects of intestinal solubility and Tp were both significant to Fa (Fig. 5). These findings suggested that the ideal dosage form required providing sufficient intestinal solubility and/or mitigating drug precipitation. In general, it is very challenging to design a formulation which can maximize only one factor by either completely suppressing the precipitation (Tp > 10,000 s) or dramatically enhance the intestinal solubility. A more practical approach is to balance the enhancement of solubility and the inhibition of precipitation. For the formulations with short Tp (e.g., F1, 1,000 s; F2, 1,800 s; and F3, 1,500 s) under fasted state, the targeted duodenum solubility for a favorable Fa under fasted condition (Fa > 70%) is at least larger than 0.15 mg/mL, which is not very practical to achieve. A significantly longer Tp (∼4,000 s) under fasted state was estimated in the dog study for formulation F4, and thereby a lower targeted solubility (>0.07 mg/mL) will be sufficient to reach desirable Fa. Based on the Eq. 1, theoretically calculated duodenum solubility of NVS123 in F4 is about 0.072 mg/mL. Therefore, F4 is likely a promising formulation with high in vivo absorption (>70% Fa) in the fasted state thus potentially eliminating the positive food effects. Overall, this knowledge provided important insights into formulation design and further allowed the formulation scientists to justify the acceptance criteria for the key factors contributing to the desired in vivo performance of drug products.
Fig. 5.
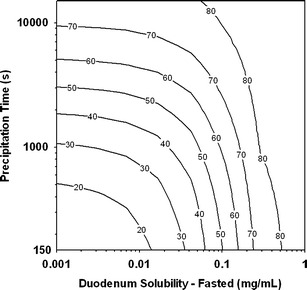
Sensitivity analysis displayed as contour plot of fraction absorbed (F a) as a function of precipitation time and duodenum solubility under fasted state (200 mg)
Virtual Food Effect Studies for the F4 Formulation
As observed in the preclinical food effect study, no significant PK shift was observed with F4 in presence and absence of a meal. A theoretical understanding of the experimental results in the dog studies has also been manifested by the PSA. To validate further our predictions that could be influenced by other counteracting effects, virtual population food-effect bioequivalence trials were conducted. These virtual population studies randomly incorporate related parameters with predefined distribution to perform a stochastic simulation in which counteracting effects of many sensitivity parameters can be examined (16). The virtual tests were conducted after a single dose of 200 mg F4 formulation under fasted and fed state. The in vitro dissolution profile was directly used to simulate the PK profile under fasted condition while the dissolution profile was adjusted similarly as described previously in the simulation under fed condition (Fig. 6a). The F4 population food effect trials showed that food did neither significantly alter Cmax nor AUC of NVS123 using the 80–125% criteria whereas the upper 90% CI of Cmax and AUC ratio (fed vs. fasted) was slightly above the upper BE limits (Table IV; Fig. 6b). The mean values of Fa for F4 formulation were predicted to be 79.5% and 86.3% under fasted and fed state, respectively. This virtual simulation suggested that F4 formulation may be promising to eliminate clinically significant food effects. As a result of the modeling, a clinical study was proposed with the F4 formulation.
Fig. 6.
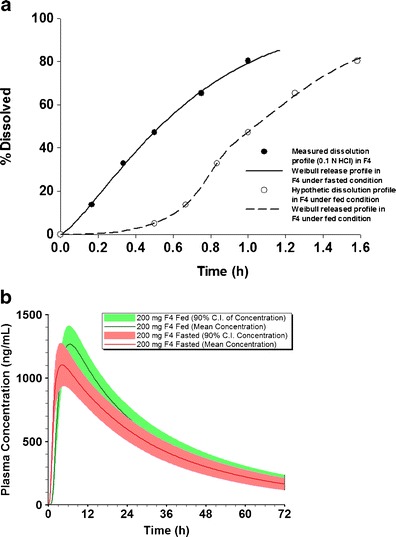
a Comparison of in vitro dissolution and adjusted Weibull input dissolution for F4. b Simulated plasma concentration with 90% confidence interval (CI) after a single dose of F4 formulation (200 mg) under fasted and fed state
Table IV.
Virtual Food Effect Study for F4 Formulation
| Parameters | Fasted (values) | Fed (values) | Fed vs. fasted (ratio) | |||
|---|---|---|---|---|---|---|
| Mean | 90% CI | Mean | 90% CI | P.E. | 90% CI | |
| C max (ng/mL) | 1,141 | [962, 1,322] | 1,311 | [1,163, 1,459] | 1.14 | [1.00, 1.28] |
| AUC0–inf (μg × h/mL) | 43.8 | [36.4,51.1] | 49.0 | [41.5, 56.5] | 1.12 | [0.95, 1.28] |
| t max (h) | 5.0 | [4.56, 6.21] | 6.86 | [6.48, 7.61] | ||
DISCUSSION
Presence of a food effect can affect drug labeling (4) and convenience to the patient. A positive food effect is manifested by a higher systemic exposure when the drug is given with food compared with the fasted state and is mainly seen for BCS/BDDCS 2 drugs, such as NVS123. This work reported the utility of PBPK modeling for the formulation development of solid dosage formulations of a weakly basic compound. The in silico absorption model was developed based on the preclinical in vitro and in vivo findings. Based on the in vitro two-step dissolution/precipitation and in silico predicted biorelevant solubility, we have demonstrated that solubility and precipitation can significantly influence absorption and exposure in the fasted state. Under fed state conditions, NVS123 is anticipated to have higher solubility in the GI tract and may be supersaturated in the intestine for a longer time after high fat meals. According to our mechanistic understanding of the positive food effect of NVS123, we started the modeling with the market formulation of dry filled capsule (F1) in the canine food effect data, to generate Tp, a key parameter, for the human model. In addition, NVS123 drug dissolution behavior tends to be influenced by the food and formulations. The Gastroplus ACAT model was used to establish correlation between in vitro and in vivo dissolution. The food-related delayed drug dissolution was captured by adjusting dissolved % vs. time profile with multiple phases of dissolution rates. Using these approaches, the current model successfully simulated the absorption and concentration vs. time curves for F1, F2, and F3 formulation under both fasted and fed condition.
One of the major challenges in developing a PBPK model is to estimate key parameters that are not easily measured directly, such as precipitation time and solubility in the GI tract. Preclinical animal models have been widely used to evaluate the performance of new dosage forms to support the decision-making for subsequent clinical study. However, most of the studies use the animal model as surrogates to estimate qualitatively the clinical outcomes in human. Here, we propose a method to estimate human precipitation time under fasted and fed condition by fitting PK data from dog food effect studies. Dog is the most studied species for predicting human food effects (17–19) because clinical service forms (e.g., capsule or tablet) can be directly administered to dogs. The Tp obtained from the optimized dog model was further incorporated into the human model and the resulted human model captured the mean observed data reasonably well under fasted and fed conditions for all of the three formulations. A high-fat meal increased the extent of absorption following the administration of F1, F2, and F3 formulation in dogs and humans. For all of the formulations tested in the clinic, the predicted Tp under fed state were consistently higher than the values under fasted conditions. The simulated results support the fact that meals can stimulate bile flow and enhance solubility of poorly water-soluble compound in small intestine. The predicted magnitude of these food effects on AUC and Cmax fell in less than 1.3-fold of the observed effects. This success case study has proved that the key parameters (e.g., Tp) in human PBPK model can be successfully estimated using preclinical PBPK model and results. On the other hand, the prediction accuracy using such an approach may depend on the compound properties and formulations. It is also critical that human relevant disposition parameters and physicochemical properties are used.
PSA plays an important role in the modeling and simulation process. It can provide the fundamental understanding of mechanism of absorption for particular drug products, which further guide the formulation strategy. The current PSA was depicted using a contour plot to illustrate the potential region with optimal intestinal solubility and precipitation time which is targeted by the further formulation design. Based on this analysis, if only focusing on the solubility enhancement, it may nearly impossible to design a formulation that can dramatically increase the drug intestinal solubility over 0.25 mg/mL which is about >250-fold higher than the buffer solubility (1 μg/mL at pH 7). A promising formulation design will need to balance the precipitation suppression and solubility enhancement. The positive food effects was partially reduced in the F2 and F3 formulation but not fully eliminated. These two formulations improved API GI solubility to some extent under fasted condition, whereas the API GI solubility under fed condition was also improved significantly. As the higher solubility under fasted conditions was not sufficient to enhance the Fa over 70%, the positive food effect remained. Another promising method of enhancing the solubility is to use different polymorphs for API, such as amorphous forms. It was found that AUC and Cmax of F4 formulation were not significantly changed under fasted and fed condition in dogs, indicating that the new amorphous form dramatically improved the solubility/dissolution and reduced prolonged precipitation (evidenced by fitted Tp > 4,000 s in dog model) of NVS123 in the intestine. As a result, the absorption is nearly complete (>87%) in the dog model under either fasted or fed state conditions. Prior to the clinical study, population simulations were conducted to estimate the clinical PK outcomes and potential clinical food effects. However, a debatable point of using preclinical canine models for evaluation of the formulation performance is whether the absorption and metabolism are different between different species. To this end, the preclinical results need to be further confirmed using human physiological absorption models prior to the decision of clinical evaluation. In all of the formulation tested in dogs and humans, the absorption of NVS123 is generally higher in dogs than that in humans under fasted and fed condition while the drug clearance and first pass elimination in dogs are higher than those in humans (e.g., NVS123 showed moderate blood CL in dogs but was predicted to show low blood CL in humans based on the hepatic extraction ratio). We showed a crossover virtual fed bioequivalence study using the population simulation module in the ACAT model which enabled introduction of population variability to the model. The virtual clinical trial simulation was capable of estimating important bioequivalent parameters including the mean values and 90% confidential intervals, which may further help to design clinical food effect studies. According to the simulated results, a clinical study of F4 is to be conducted.
CONCLUSIONS
The successful integration of multiple biopharmaceutical tools, such as two-step dissolution test, canine model and physiologically based in silico simulation allowed the mitigation of positive food effects in drug development. We simulated four formulations of NVS123 in a PBPK model and the simulations were in agreement with the currently available clinical PK data. Furthermore, the PSA and a virtual bioequivalence simulation allowed establishing model-guided formulation strategy and assisting the decision-making of clinical investigation. The work also highlighted that dog in vivo data, if appropriately translated and analyzed, can greatly support the implementation of human absorption model by providing key input parameters. Overall, the mechanism-based modeling with solid assumptions based on preclinical in vitro and in vivo results is a key bridging step for current formulation design prior to the clinical study, and it will greatly facilitate the implementation of Quality by design in drug development.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
(DOCX 32 kb)
Acknowledgments
The authors would like to thank Dr. Akash Jain and members of the Novartis Food Effect Quality Plus, preclinical PK/PD, clinical pharmacology, and oral formulation development team for conducting preclinical and clinical studies as well as general scientific discussions and inputs.
Conflict of Interests
None.
References
- 1.Fleisher D, Li C, Zhou Y, Pao LH, Karim A. Drug, meal and formulation interactions influencing drug absorption after oral administration. Clinical implications. Clin Pharmacokinet. 1999;36(3):233–254. doi: 10.2165/00003088-199936030-00004. [DOI] [PubMed] [Google Scholar]
- 2.Benet LZ. WCY. Using a biopharmaceutics drug disposition classification system to predict bioavailability and elimination characteristics of new molecular entities. Somerset, NJ: NJDMDG; 2006.
- 3.Custodio JM, Wu C-Y, Benet Leslie Z. Predicting drug disposition, absorption/elimination/transporter interplay and the role of food on drug absorption. Adv Drug Deliv Rev. 2008;60(6):717–733. doi: 10.1016/j.addr.2007.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.FDA. Food-Effect Bioavailability and Fed Bioequivalence Studies. Guidance for Industry 2002. Available from: http://www.fda.gov/downloads/regulatoryinformation/guidances/ucm126833.pdf. Accessed 02 Jun 2012
- 5.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22(1):11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 6.Parrott N, Lukacova V, Fraczkiewicz G, Bolger MB. Predicting pharmacokinetics of drugs using physiologically based modeling-application to food effects. AAPS J. 2009;11(1):45–53. doi: 10.1208/s12248-008-9079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones HM, Parrott N, Ohlenbusch G, Lave T. Predicting pharmacokinetic food effects using biorelevant solubility media and physiologically based modelling. Clin Pharmacokinet. 2006;45(12):1213–1226. doi: 10.2165/00003088-200645120-00006. [DOI] [PubMed] [Google Scholar]
- 8.Heimbach T, Xia B, Lin TH, He H. Case studies for practical food effect assessments across BCS/BDDCS class compounds using in silico, in vitro, and preclinical in vivo data. AAPS J. 2013;15(1):143–158. doi: 10.1208/s12248-012-9419-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu LX, Amidon GL. Characterization of small intestinal transit time distribution in humans. Int J Pharm. 1998;171(2):157–163. doi: 10.1016/S0378-5173(98)00174-4. [DOI] [Google Scholar]
- 10.Gardner JD, Ciociola AA, Robinson M. Measurement of meal-stimulated gastric acid secretion by in vivo gastric autotitration. J Appl Physiol. 2002;92(2):427–434. doi: 10.1152/japplphysiol.00956.2001. [DOI] [PubMed] [Google Scholar]
- 11.Langenbucher F. Linearization of dissolution rate curves by the Weibull distribution. J Pharm Pharmacol. 1972;24(12):979–981. doi: 10.1111/j.2042-7158.1972.tb08930.x. [DOI] [PubMed] [Google Scholar]
- 12.Mithani SD, Bakatselou V, TenHoor CN, Dressman JB. Estimation of the increase in solubility of drugs as a function of bile salt concentration. Pharmaceut Res. 1996;13(1):163–167. doi: 10.1023/A:1016062224568. [DOI] [PubMed] [Google Scholar]
- 13.Lentz KA, Quitko M, Morgan DG, Grace JE, Jr, Gleason C, Marathe PH. Development and validation of a preclinical food effect model. J Pharm Sci. 2007;96(2):459–472. doi: 10.1002/jps.20767. [DOI] [PubMed] [Google Scholar]
- 14.Lentz KA. Current methods for predicting human food effect. AAPS J. 2008;10(2):282–288. doi: 10.1208/s12248-008-9025-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Flanagan DR. General solution for diffusion-controlled dissolution of spherical particles. 1. Theory. J Pharm Sci. 1999;88(7):731–738. doi: 10.1021/js980236p. [DOI] [PubMed] [Google Scholar]
- 16.Zhang X, Lionberger RA, Davit BM, Yu LX. Utility of physiologically based absorption modeling in implementing quality by design in drug development. AAPS J. 2011;13(1):59–71. doi: 10.1208/s12248-010-9250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akimoto M, Nagahata N, Furuya A, Fukushima K, Higuchi S, Suwa T. Gastric pH profiles of beagle dogs and their use as an alternative to human testing. Eur J Pharm Biopharm. 2000;49(2):99–102. doi: 10.1016/S0939-6411(99)00070-3. [DOI] [PubMed] [Google Scholar]
- 18.Lui CY, Amidon GL, Berardi RR, Fleisher D, Youngberg C, Dressman JB. Comparison of gastrointestinal Ph in dogs and humans—implications on the use of the beagle dog as a model for oral absorption in humans. J Pharm Sci. 1986;75(3):271–274. doi: 10.1002/jps.2600750313. [DOI] [PubMed] [Google Scholar]
- 19.Meyer JH, Dressman J, Fink A, Amidon G. Effect of size and density on canine gastric-emptying of nondigestible solids. Gastroenterology. 1985;89(4):805–813. doi: 10.1016/0016-5085(85)90576-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 32 kb)