

Abstract
The aim of the present work was to use GastroPlus™ software for the prediction of pharmacokinetic profiles and in vitro–in vivo correlation (IVIVC) as tools to optimize the development of new generic medications. GastroPlus™ was used to simulate the gastrointestinal compartment and was based on the advanced compartmental absorption and transit model. Powder dissolution and efavirenz tablet dissolution studies were carried out to generate data from which correlation was established. The simulated plasma profile, based on the physicochemical properties of efavirenz, was almost identical to that observed in vivo for biobatches A and B. A level A IVIVC was established for the dissolution method obtained for the generic candidate using the Wagner–Nelson (r2 = 0.85) and for Loo–Riegelman models (r2 = 0.92). The percentage of fraction absorbed indicated that 0.5% sodium lauryl sulfate may be considered a biorelevant dissolution medium for efavirenz tablets. The simulation of gastrointestinal bioavailability and IVIVC obtained from immediate-release tablet formulations suggests that GastroPlus™ is a valuable in silico method for IVIVC and for studies directed at developing formulations of class II drugs.
KEY WORDS: bioavailability, computational simulation, efavirenz, GastroPlus™, in vivo–in vitro correlation
INTRODUCTION
Worldwide, the use of generic medicines represents a cost-effective and technologically viable way of providing broadened access of quality medications to a country's population. However, a significant barrier, to the rapid development and market availability of novel generic medicines, is the regulatory requirements behind the approval and registration of a drug product, particularly in regards to the requirement of performing the bioequivalence studies (1). Therefore, waiving the need for bioequivalence studies can fast track the process of developing generic medicines, where the bioavailability of drugs can instead be estimated by assessment of solubility, permeability, and dissolution parameters. Based on the Biopharmaceutics Classification System (BCS), as proposed by Amidon et al. (2), the Food and Drug Administration published a guide for waiving bioavailability and bioequivalence studies for the immediate-release solid oral dosage forms (3). According to this guide, waiving these studies is recommended for highly soluble and permeable drugs (class I), extending to class II and III drugs under certain situations (3–6). In the case of class II drugs, which exhibit properties of low solubility and high permeability, dissolution is the rate-limiting step for drug absorption, whereas class III drugs rapidly dissolve and permeability is the rate-limiting step in their absorption. Accordingly, their distinct pharmacokinetic properties can be simulated by in vitro–in vivo correlation (IVIVC) which can be used in combination with computational simulation tools, such as the software GastroPlus™, to safely reduce the cost and time to develop new generic drugs. A level A IVIVC demonstrates that the conditions of the dissolution test are able to simulate in vivo performance (1,5,7). The most effective approaches for biowaiving class III drugs are performing in vitro permeation tests, as well as the use of Caco-2 cells or intestinal segments isolated from animals. These tests must be validated and the results correlated with the in vivo data (8).
The in silico method estimates parameters with the aid of computational technology and has proven useful in many studies in the field of pharmaceutical sciences for predicting bioavailability during the process of developing formulations (4,5,7,9). Computer simulations have been employed in the assessment of waiving bioequivalence studies and the determination of IVIVC for class II and III drugs, reducing both research time and the cost of developing new generic medicines (4,5,7,9). An example of an in silico method is GastroPlus™, a software which has applications based on BCS theory and the advanced compartmental transit and absorption model, which simulates gastrointestinal absorption and the different pharmacokinetic parameters of medications (10,11). GastroPlus™ can also aid in the development of new formulations and the selection of biorelevant dissolution conditions with IVIVC (12,13). This software has been used for predicting pharmacokinetic behavior and establishing the IVIVC for various class II drugs (10,12–15). For instance, in a recent study involving the drug carbamazepine, IVIVC was determined through computational simulations that used GastroPlus™ to justify waiving the bioequivalence studies for this drug (14).
Efavirenz is an antiviral drug from the class of nonnucleoside reverse-transcriptase inhibitors used for the treatment of acquired immunodeficiency syndrome and has known action against the human immunodeficiency virus type 1 (16). It has poor water solubility and is categorized as a class II drug under the BCS (17,18), with a solubility of approximately 10 μg/mL (17). Efavirenz has similar behavior to the so-called brick dust drugs, drugs with a high melting point and stable crystalline structure in which strong intramolecular bonds significantly limit dissolution and bioavailability of their pharmaceutical forms (19) hampering the development of generic medicines for this class. Furthermore, the bioequivalence and bioavailability studies require an increase in expended effort and greatly limit the development of these new formulations. Thus, the registration process has become quite costly and cumbersome which compromises rapid access of this formulation to the patient population. The use of in silico bioavailability prediction based upon the drug's physicochemical properties and pharmacokinetic parameters provides a promising alternative. At the same time, the predictive capacity of the (active pharmaceutical ingredients (API)) physicochemical properties on the safety and efficacy of medicines produced after the sanitary registration depend on the development of a dissolution method that accurately reflects what is observed in vivo. These aspects of drug behavior can also be anticipated by in silico methods.
Thus, the aim of this study was to assess the viability of using GastroPlus™ software as an in silico tool for studying its effectiveness in formulation development studies of brick dust drugs. The software was tested and compared the bioequivalence results obtained from oral solid formulations that were produced using API efavirenz with differing physicochemical characteristics and the simulated results produced by this software. This in silico method was also used to calculate the level of IVIVC suited to the dissolution method used for the immediate-release efavirenz tablets while considering data obtained with different dissolution methods and absorption models.
MATERIALS AND METHODS
Materials and Reagents
All solvents used in this study were of chromatographic grade, and all reagents used were of analytic grade and purchased from Tedia (Rio de Janeiro, Brazil). The API, efavirenz tablets, as well as the bioequivalence results were provided by Farmanguinhos Laboratory (Rio de Janeiro, Brazil). The API batches, denominated batches A (712847) and B (0640/07), were also acquired from the same company. Two batches of efavirenz tablets, named biobatch A (0708EX216) and biobatch B (0806EX180), containing 600 mg of efavirenz were donated by the Farmanguinhos Laboratory. The formulation biobatch A contained cellulose, lactose, croscarmellose sodium, sodium lauryl sulfate, and hydroxypropylcellulose. However, biobatch B contained the same excipients as biobatch A with the exception of hydroxypropylcellulose. Biobatch B has 50% less cellulose, 360% more sodium lauryl sulfate, and 50% more croscarmellose sodium compared to biobatch A. Biobatches A and B were produced using the API batches A and B, respectively. The efavirenz standard contained 100.0% efavirenz and was prepared according to the ISO Guide 34 (20) and supplied by Globe Química (São Paulo, Brazil). All solutions were prepared using MilliQ water (Millipore, Bedford, MA, USA).
Drug Characterization
The solubility of each batch of API was assessed by adding an excess of drug into separate beakers which contained 10 mL of three different media (at a temperature of 37 ± 0.5°C): simulated gastric fluid (SGF) pH 1.2, water pH 5.5, and simulated enteric fluid (SEF) pH 6.8 (21). Each beaker was stirred at 50 rpm using a magnetic stirrer for 24 h. After this period, the resultant solution was centrifuged at 3,500 rpm for 30 min. The supernatant was immediately filtered through a 0.45-μm filter, and the sample concentration was determined by UV–Vis spectrophotometry (Vankel 50, Varian Inc., Palo Alto, CA, USA) at a detection wavelength of 248 nm. A five-level calibration curve was prepared for each medium at the following concentrations: 4.0, 6.0, 8.0, 10.0, and 12.0 μg/mL (22). All experiments were performed in triplicate.
The particle size distribution in the efavirenz batches was determined by laser diffraction using the wet mode (Malvern Mastersizer 2000, Hydro 2000 SM, Worcestershire, UK). The samples were prepared by dispersing approximately 10 mg of drug in a beaker containing 10 mL 0.02% (w/v) polysorbate 80 solution. A volume of 100-mL water was used as the dispersion medium and the mixture was stirred at 2,000 rpm.
The specific surface area was determined by gas adsorption (Micromeritics Gemini VI 2385C). Also, the analytic gas was nitrogen while the reference gas was helium which was used to measure free space. The samples were degassed under nitrogen for 24 h at 25°C prior to analyses. The vacuum pressure that was used was 500 mmHg/min during a 1-min period, and the equilibrium time for adsorption was 10 s. The amount of nitrogen gas that was adsorbed in the relative pressure range of 0.05 < P/Po < 0.35 was determined. The specific surface area was calculated using Brunauer, Emmett, and Teller methods.
The efavirenz content was measured by HPLC according to the method published in the US Pharmacopeia (21). The quantification was performed using the LaChrom Elite chromatograph system from Merck-Hitachi (Darmstadt, Germany) coupled to a diode array detector (DAD L-2130). The stationary phase used was the PerkinElmer Ciano column (4.6 mm × 15 cm; 5 μm) which was kept at 35°C with a flow rate of 1.5 mL/min, an injection volume of 35 μL, and detection wavelength of 250 nm. The mobile phase that was utilized was a methanol, trifluoroacetic acid, and water mixture (solution A 1:0.005:9 and solution B 9:0.005:1) using a gradient mode from 60:40 to 20:80 (solution A/solution B).
Drug Characterization Studies
The influence of (batches A and B) API physicochemical properties on the preparation of biobatches A and B was assessed using the powder dissolution method which implemented a dissolutor, Hanson Research Model SR6 (Chatsworth, USA), with a paddle apparatus (described as apparatus 2) in the US Pharmacopeia. The dissolution procedure consisted of placing a known amount of the drug (~600 mg), dispersed in 5 mL of medium, directly into the dissolutor vessel (23). The dissolution conditions were 900 mL of aqueous sodium lauryl sulfate (SLS) solution with concentrations of 0.5%, 1.0%, and 2.0% w/v at 37°C with a rotation speed of 50 rpm. The sampling was performed at 5, 10, 15, 30, 45, and 60 min using a 10-μm cannula filter and subsequently filtered through a 0.45-μm filter. Samples of 500 μL were diluted in a 25-mL volumetric flask using the dissolution medium. The quantification was performed on a UV–Vis spectrophotometer (Vankel 50, Varian Inc., Palo Alto, CA, USA) at a wavelength of 248 nm. The efavirenz standard solution was prepared by diluting 50.0 mg of standard into a 50-mL volumetric flask containing 0.5 mL of methanol, and the final volume was adjusted with the dissolution medium after 15 min of ultrasound treatment. A five-level curve was prepared from this standard solution for each SLS concentration (22). The percentage of the cumulative amount of dissolved drug was determined.
The efavirenz tablets in biobatches A and B were analyzed according to the dissolution methods described in the US Pharmacopeia (USP), the Brazilian Pharmacopoeia (BrP), and the method developed internally by Farmanguinhos Laboratories (Far) (21,24). The three methods were compared in order to verify the most discriminative conditions for the different tablets and determine the IVIVC (Table I). The tablets were dissolved in 900 mL medium at 37°C in a paddle apparatus. Measurements were performed using a UV–Vis spectrophotometer and a similar procedure to that described for the powder dissolution test utilized for the APIs. Statistical analysis of the dissolution data was performed using one-way ANOVA and Tukey's multiple comparisons test, with the aid of GraphPad Prism® software (version 5.0, GraphPad Software® Inc., 2007).
Table I.
Dissolution Conditions of Efavirenz Tablets
| Parameters | Far | BrP | USP |
|---|---|---|---|
| Medium (900 mL) | SLS 0.5% | SLS 1.0% | SLS 2.0% |
| Rotation speed (rpm) | 50 | 100 | 50 |
| Sampling time (min) | 45 | 45 | 30 |
| Q (%) ≥80 for the three methods | |||
Far Farmanguinhos methodology, BrP Brazilian Pharmacopoeia, USP US Pharmacopeia, SLS sodium lauryl sulfate
Bioequivalence Study
Two different batches of the reference drug (Stocrin®) were used in the bioequivalence studies of biobatches A and B, since biobatch A was produced upon verification of non-bioequivalence of biobatch B. The bioequivalence studies were carried out independently and at different times. The batches of the reference drug used were those that were commercially available to the market at the time of each study. The bioequivalence study A (BE A) was a two-period BE study designed to compare the commercially available 600-mg tablets (Stocrin®, reference drug batch A) with a generic tablet formulation (biobatch A). The medicines were administered to healthy volunteers, which included men and women aged between 18 and 45 years with a mean body weight of 64.72 kg under fasting conditions. The volunteers ingested a standardized volume of 200-mL water, and blood samples were collected up to 192 h post-administration at the following intervals: 0–pre-dose, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 24, 36, 48, 60, 72, 84, 96, 108, 120, 132, 144, 156, 168, 180, and 192 h after oral administration of each treatment, and the washout period was 35 days.
In the second bioequivalence study (BE B), the reference formulation (Stocrin® 600 mg tablets, reference drug batch B) and the tablet formulation (biobatch B) were administered to healthy volunteers, men and women aged between 18 and 50 years with a mean body weight of 66.9 kg under fasting conditions. A standardized volume of water, 200 mL, was ingested and blood samples were collected up to 168 h post-administration, in the following intervals: 0–pre-dose, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 24, 36, 48, 60, 72, 84, 96, 108, 120, 132, 144, 156, and 168 h after oral administration of each treatment, and the washout period was 42 days. In both BE studies, an open, randomized, cross-over experimental protocol design was adopted. Before the volunteers were recruited, the ethics committee in research approved the protocols presented here. Tolerability was monitored by physical examination, including vital sign measurements, and ECG was performed at screening as well as laboratory analysis which included biochemistry tests, hematology tests, and urinalysis, which were performed at screening and during the study period. Blood samples were collected for plasma separation and immediately centrifuged for 10 min at 1,800×g, and the samples were promptly frozen at −70°C until further analysis. All volunteers underwent clinical assessments to confirm their health status during the study period. Additionally, the samples were analyzed using the LC-MS method. A high-performance liquid chromatographic tandem mass spectrometric assay, with turbo ion spray using the positive mode and multiple reaction monitoring scan type, was developed and validated in order to quantify the amount of efavirenz in plasma samples (22). The solid-phase extraction of the plasma samples was carried out on a C18 cartridge after being diluted 1:1 with phosphate buffer, pH 7. Chromatography analysis was performed using the C18 analytical column and 50:50 acetonitrile/phosphate buffer (pH 3.5) as the mobile phase. The response was linear over the concentration range of 0.1 μg–10 /mL in human plasma. A statistical analysis of the collected data was performed using the GraphPad Prism version 5.0 (GraphPad Software, Inc, 2007) for reporting the bioequivalence data (experimental results).
In Silico Studies
GastroPlus™ (version 7.0, Simulations Plus Inc., Lancaster, CA, USA) was used for computer simulations of the bioavailability of biobatches A and B as well as the references A and B. This program uses the ACAT model to calculate the fraction of the drug dose absorbed in each compartment of the intestine and comprised of three modules: compound, physiology, and pharmacokinetics. Input parameters for the compound module included pKa, log P, dose, dose volume, permeability, and particle density. In this study, solubility and particle size were determined experimentally for batches A and B and also obtained from the API Drug Master File database as a drug reference for the batches. These data were loaded using the option of solubility versus pH and particle size distribution, respectively, whereas the other parameters utilized were obtained from the literature to perform the software simulation.
The Johnson model was used as the dissolution model because it takes into account the changes in particle radius during dissolution as well as the properties regulating dissolution of cylindrical particles (25). The effect of bile salt concentration on solubility was also considered using this specific option in the software “adjust solubility for salt effect” in the “dissolution model” screen. In the absence of solubility data of the biorelevant medium (FaSSIF or FeSSIF), the theoretical solubilization ratio (SR = 3.34 · 105) for the bile salts was calculated based on the drug log P value using the Eq. 1, according to Mithani et al. (11,26). Then, the drug solubility was determined in each portion of the gastrointestinal tract based on its local bile salt concentration, e.g., duodenal solubility at the bile salt concentration 2.8 mM (0.148 mg/mL) (11).
| 1 |
where Solbile, pH is the in vivo solubility (milligrams per milliliter) in compartment with specific pH and bile salt concentration; Solaq, pH is the buffer solubility at given pH calculated from reference solubility, pKa, and solubility factor; Mwt and ρ are the molecular weight and the density of water, respectively; SR is bile salt solubilization ratio and represents drug's affinity to bile salt micelles; and Cbile is the in vivo concentration of bile salts in given compartment.
The drug absorption simulations in GastroPlus™ based on drug physicochemical and pharmacokinetic data were performed using the “IR tablet mode” that refers to immediate (IR)-release tablets. When the in vitro dissolution profile of the IR biobatch A tablet was used as a software input, the “CR-dispersed” dosage option that refers to the controlled release and the “tabulated in vitro dissolution data” function were selected (10).
In the physiology module, the ASF Opt logD Model SA/V 6.1 absorption model was selected using the setting human physiology in the fasting state. Therefore, the software adjusted the physiological variables such as pH, transit times, geometric parameters, lengths, radii, and volumes for the different compartments of the gastrointestinal tract according to the selected condition (11).
The data obtained from the biobatches A and B bioequivalence studies were loaded into the PKPlusTM module and evaluated by the software according to non-compartmental, 1-, 2-, and 3-compartment pharmacokinetic models. The best fit (the three-compartment model) was then imported into the pharmacokinetic module to enable software prediction of the curve at a given plasma concentration versus time (13). The summary of all input parameters for simulation is given in Table II.
Table II.
Input Data for Simulation of Bioavailability of Batches A and B
| Parameters | Batch A | Batch B |
|---|---|---|
| Molecular weight (g/mol) | 315.67a | 315.67a |
| Log P | 5.4a | 5.4a |
| pKa | 10.2a | 10.2a |
| Dose (mg) | 600b | 600b |
| Formulation | Tabletb | Tabletb |
| P app (cm/s × 10–5) | 8.92a | 8.92a |
| Blood/plasma ratio | 0.74a | 0.74a |
| Precipitation time (s) | 900d | 900d |
| Diffusion coefficient | 0.7473 ± 0.022c | 0.7473 ± 0.016c |
| Particle density (g/mL) | 1.2d | 1.2d |
| Physiology | Fastedb | Fastedb |
| AFS (model) | SA/V 6.1d | SA/V 6.1d |
| Body weight (kg) | 64.7b | 66.9b |
| Distribution volume (L/kg) | 1.56 ± 0.32c | 4.68 ± 0.11c |
| Depuration (L/h) | 2.65 ± 0.52c | 6.26 ± 0.91c |
| t 1/2 (h) | 266.3c | 163.72c |
| Dose volume (mL) | 200b | 200b |
| Simulation time (h) | 192b | 168b |
Simulations were also performed to assess how particle size affected the fractional absorption of the tablets produced from the API batches A and B in the model of absorption selected. The results simulated were compared statistically with the experimental data based upon the software manual and other literature references (10,11,15).
In Vitro–In Vivo Correlation
Numeric Deconvolution
The in vivo fraction of efavirenz that was absorbed was calculated by numeric deconvolution of the bioavailability data of biobatch A (generic approved on BE study) using GastroPlus™ by the Wagner–Nelson (one compartment) and the Loo–Riegelman (three compartment) models (10,11). Evaluation of IVIVC was made by comparing the fractions of absorbed drug with the percentage of drug dissolved in vitro at the same time points. Regression analysis was used to evaluate the obtained correlations.
Numeric Convolution
A numeric convolution was performed using the dissolution data obtained by applying the Farmanguinhos, Brazilian, and US Pharmacopeia methods as inputs for the GastroPlus™ so that not only the pharmacokinetics parameters of the dosage form were estimated, but also the best dissolution methods were identified for the in vivo conditions of the efavirenz tablets.
RESULTS AND DISCUSSION
Drug Characterization
The efavirenz APIs were assessed in this study using batches A and B that were previously characterized by the monograph from USP 34 (21). The API content was measured at 98.99 ± 0.5% for batch A and 99.00 ± 0.5% for batch B, with the percentage of moisture at 0.33% and 0.23%, respectively. The melting range was measured experimentally for both batches and was in the range of 139–141°C, and only isoform I of efavirenz was identified for the two batches by X-ray diffraction and differential scanning calorimetry analysis (27). Solubility in water for both batches was statistically similar (P > 0.05): 8.98 μg/mL for batch A and 8.23 μg/mL for batch B. The solubility values of efavirenz in SGF were 10.76 μg/mL for batch A and 8.40 μg/mL for batch B, whereas in SEF, they were 7.56 and 7.52 μg/mL for batches A and B, respectively. The solubility of both batches in SGF and SEF was statistically similar (P > 0.05). Samples from the solubility test were analyzed by HPLC coupled with DAD according to the USP 34 method (21) to confirm the stability of the APIs under those conditions. The particle size distribution analysis showed the D(0.5) of 3.288 and 17.077 μm for batches A and B, respectively, with polydispersion index of 2.752 and 2.2724. This indicated a smaller mean size for batch A and a highly homogenous population for both batches. Surface areas were determined to be 8.679 m2/g for batch A and 2.003 m2/g for batch B, i.e., the surface area of batch A was four times greater and consistent with the smaller mean size. Notably, batches A and B presented highly divergent particle sizes and surface areas. These physicochemical characteristics directly influence the pharmacokinetic properties of the pharmaceutical forms produced using these APIs (28).
Bioequivalence Studies
The simulated data obtained for batches A and B and those obtained for the reference drugs A and B are listed in Table III. The results of the bioequivalence studies showed that biobatch B was not bioequivalent to the reference medication Stocrin®. Moreover, biobatch A was bioequivalent and proved be an adequate candidate for generic formulation. The two batches of medications differ in terms of the physicochemical characteristics of the API used and according to the formulation. Biobatch B was prepared using an API with a mean surface area that is four times lower and a particle diameter that is five times higher than that of biobatch A. These differences in the physicochemical characteristics may promote alterations in the dissolution rate of the API as well as the tablets produced using these APIs. This would explain the differences observed in the experimental or simulated pharmacokinetic parameters for each biobatch, in accordance with that found by Wei et al. for glyburide (12). The composition of SLS in biobatch A was drastically reduced (73% less) compared to biobatch B and instead included the addition of hydroxypropylcellulose (Klucel® LF) at a concentration that stabilizes the supersaturated solutions that occur during the process of tablet dissolution (29). The use of Klucel® improved drug dissolution and, consequently, the bioavailability of the drug. Besides the changes cited, the biobatch A formulation had 35% less croscarmellose sodium compared to that in biobatch B (27). Despite being a superdisintegrant, an excess of sodium croscarmellose can retard API release from the formulation, as observed in the dissolution profile of biobatch B (30,31).
Table III.
Simulated and Experimental Results Obtained on Bioequivalence Studies of Efavirenz Biobatches A and B
| Pharmacokinetic parametera | Bioequivalence study A | Bioequivalence study B | ||||||
|---|---|---|---|---|---|---|---|---|
| Reference drug A | Biobatch A | Reference drug B | Biobatch B | |||||
| Simulated | Experimental results | Simulated | Experimental results | Simulated | Experimental results | Simulated | Experimental results | |
| C max (μg/mL) | 3.14 ± 0.23 | 3.74 ± 0.68 | 2.78 ± 0.43 | 2.93 ± 0.83 | 2.34 ± 0.87 | 2.92 ± 0.88 | 0.77 ± 0.62 | 1.09 ± 0.44 |
| T max (h) | 3.0 ± 1.2 | 2.7 ± 1.0 | 3.4 ± 0.76 | 4.5 ± 1.8 | 4.5 ± 0.98 | 3.0 ± 5.0 | 3.6 ± 0.91 | 4.5 ± 23.63 |
| AUC 0 – t (μg h/mL) | 139.42 ± 13.0 | 121.03 ± 42.64 | 131.27 ± 16.24 | 137.95 ± 42.85 | 99.80 ± 17.4 | 100.03 ± 24.55 | 50.64 ± 29.33 | 65.10 ± 16.45 |
| AUC 0 – ∞ (μg h/mL) | 163.28 ± 22.71 | 163.38 ± 74.37 | 187.6 ± 19.11 | 184.52 ± 52.63 | 135.39 ± 15.9 | 138.27 ± 49.23 | 69.75 ± 15.53 | 92.86 ± 34.68 |
Powder and Tablet Dissolution Studies
The dissolution profiles of batches A and B obtained using the powder dispersion method are presented in Fig. 1. Batch A yielded a statistically higher percentage of dissolved drug in comparison to batch B at the three different concentrations of the SLS tested (P < 0.01). An increased velocity of drug dissolution was observed upon increasing the concentration of SLS in the dissolution media, except in batch B, where increasing the SLS concentration from 1.0% to 2.0% did not yield a higher percentage of dissolved drug. In order to use discriminative conditions, capable of differentiating batches with different physicochemical characteristics, the aqueous dissolution medium 0.5% (w/v) SLS was selected for further dissolution tests (32). The API (batch A) used in the production of biobatch A was approved in the bioequivalence study and dissolved much faster than the API batch (batch B) which was used to produce biobatch B. The use of biobatch A led to a comparatively greater percentage of dissolved mass of the drug (Fig. 1). These results confirmed that the distinct physicochemical properties of batches A and B, as evidenced in the distribution of particle sizes and surface area, are reflected in the in vitro dissolution of powder in these batches. This finding corroborates the influence of API particle size distribution and surface area on the bioavailability of the prepared efavirenz tablets (27,33,34). Figure 2a, b shows the dissolution profile of the biobatch A and B tablets which were carried out using three different methods, presented at Table I (21,24). Comparison of the three methods revealed that the highest percentage of dissolved drug resulted from the BrP method, which recommends a faster rotation speed (100 rpm). A slight statistical difference was observed between batches A and B when using this method (P < 0.05). The USP dissolution method which used 2.0% SLS at 50 rpm was unable to produce a detectable difference between biobatches A and B. The Farmanguinhos method was more discriminating than the other methods applied, since the dissolution profiles obtained for biobatches A and B were statistically different (P < 0.01), than what was shown by the producer of the generic candidate (Fig. 2c). Besides having the lowest final percentage of drug dissolved of the three methods assessed, this method utilized less aggressive conditions (0.5% SLS at 50 rpm) and demonstrated it was possible to differentiate tablets produced using an API that has different physicochemical characteristics. This phenomenon was shown to be clearly differentiated for the powder dissolution method (P < 0.05). Some previous dissolution studies using SLS have shown that the use of lower percentages of surfactant in the media provides greater discriminatory power to the dissolution method (34–36). Based on the Farmanguinhos method, the percentage of efavirenz dissolved during a 60-min period was 87.51% for biobatch A versus 66.93% for biobatch B (Fig. 2c).
Fig. 1.
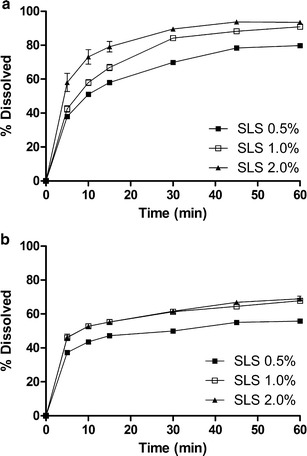
The intrinsic dissolution profile of efavirenz batch A (a) and batch B (b) by powder dissolution method using SLS at concentrations of 0.5%, 1.0%, and 2.0% (w/v)
Fig. 2.
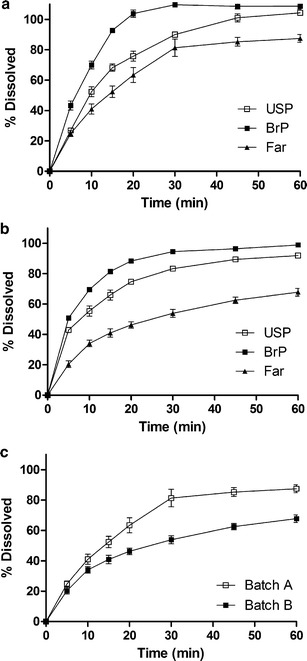
The comparative dissolution profile of efavirenz tablet biobatches A (a) and B (b) produced with IFA batches A and B using the US Pharmacopeia (USP), Brazilian Pharmacopoeia (BrP), and Farmanguinhos (Far) methods. Comparison between biobatches A and B using the Far dissolution method (c)
In Silico Studies
GastroPlus™ was used to simulate the absorption profile of biobatches A and B and the reference drugs A and B in order to predict the bioequivalence of the generic formulation. Figure 3 shows the profile curve of plasma concentration versus time from the bioavailability/bioequivalence studies and the predicted profile for batches A and B. The experimental/simulated parameters (Table III) were calculated based upon the physicochemical and physiological parameters shown in Table II. Furthermore, Table III indicates that the experimental results obtained for Cmax and AUC were close to the simulated values. This finding is in accordance with the results described in other studies that applied the GastroPlus™ (10,12,15,35,36). This provides evidence that the ASF Opt logD Model SA/V 6.1 was a good prediction model of the absorption properties of batch A based on in vivo studies. However, the simulated value for Tmax was lower than the corresponding observed value, indicating that the algorithm used by the software and/or inputs supplied do not allow accurate prediction of this parameter. For batch B and references A and B, it is evident that the simulations led to underestimation and overestimation of some values. These results may be attributed to the absence of some supplementary inputs related to the formulation aspects of the software, consequently hampering the prediction of pharmacokinetic parameters from different formulations.
Fig. 3.
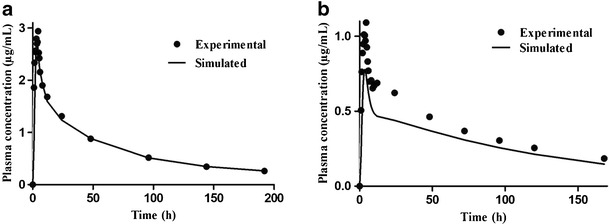
Profile of efavirenz plasma concentration versus time of experimental data (solid line) and predicted data (squares) of biobatches A (a) and B (b)
Differences in the pharmacokinetics parameters as determined by the PKPlusTM module (Table II) may affect fitting of PK for the clinical PK data. However, there is variability among individuals included in this study as mentioned by other groups in the literature (37–42). Thus, physiological differences including variable enzyme expression, blood perfusion into tissues, and tissue volume may explain the low CL value in our model, which is still within the range of values reported.
The prediction fraction of drug absorption was calculated after changing only the particle size related to batches A and B. Subsequently, a high absorption in the proximal compartments (duodenum and jejunum) and a low amount of absorption in the distal region of the gut were observed, as reported in the literature (43).
Since the fraction absorbed for batch A was higher (98.5%) than batch B (80.3%), we may conclude that the absorption model was sensitive to the particle size (Fig. 4). This difference may have collaborated directly upon the dissolution rate of each batch thus influencing the variability of the absorption profile and the simulated pharmacokinetic parameters (Fig. 3 and Table III). Other studies that reported optimization of class II drug formulations also demonstrated differences in bioavailability which depend on particle size (44). Therefore, this absorption model may be able to set the particle size of the drug and assay the acceptable or non-acceptable conditions according to the observed in vivo results, which may help in strategies for developing new formulations with efavirenz.
Fig. 4.
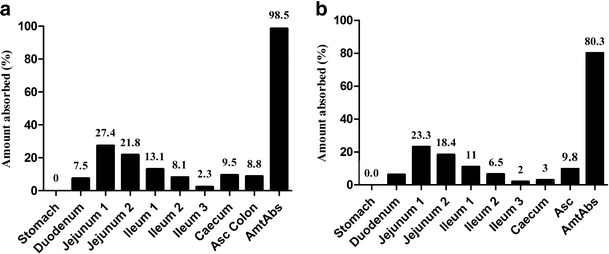
Compartmental absorption of the batches A (a) and B (b)
Although GastroPlus™ was efficient in predicting bioavailability based upon the different physicochemical properties of the API, the software was not able to anticipate effects on API release due to the presence of different excipients in the formulations of biobatches A and B.
In Vitro–In Vivo Correlation
The in vitro–in vivo correlation was established using the GastroPlus™ based upon evaluating different inputs of the in vitro dissolution and in vivo release data obtained from the biobatch A tablets, which was approved in the bioequivalence study, although not based on results of a legally protected product (Stocrin®). Table IV shows the IVIVC established using deconvolution approaches (the Wagner–Nelson and Loo–Riegelman models), whereas Fig. 5 shows the regression analysis data. The IVIVC did not show a characteristic linear distribution of its dosage form either in the modified or immediate release of class II drugs (reference), instead an inverted L-shaped distribution was obtained which has been observed by others (45). This indicated that the method utilized for the dissolution study of the efavirenz tablets resulted in a fast dissolution of the drug. Thus, in addition to the established IVIVC, these results suggest that variations in the dissolution profile can be obtained for each method, e.g., using the BrP method, in which a higher dissolution profile was obtained compared to the other methods which resulted in a lower correlation coefficient (R = 0.63 for the Wagner–Nelson and R = 0.50 for the Loo–Riegelman model). In this scenario, the in vitro dissolution data were not correlated with in vivo dissolution values of biobatch A. However, the dissolution method developed by Farmanguinhos produced in vitro dissolution data that better reflected the in vivo dissolution data obtained from bioavailability studies, which show a minimally adequate correlation coefficient (R = 0.85 for the Wagner–Nelson model and R = 0.93 for the Loo–Rielgeman model). Although apparently low, such correlation coefficient values were considered acceptable in other studies reported in the literature; however, this dissolution method was more discriminative and yielded a gradual release of efavirenz (15).
Table IV.
Correlation Coefficient and Pharmacokinetic Parameters Observed In Vivo (Biobatch A) and Simulated Using the Wagner–Nelson and Loo–Riegelman Models
| Model | Methods | r 2 | AUC (μg h/mL) | C max (μg/mL) |
|---|---|---|---|---|
| Biobatch A | ||||
| Wagner–Nelson | Far | 0.85 | 171.2 | 2.36 |
| BrP | 0.63 | 152.4 | 2.14 | |
| USP | 0.81 | 162.1 | 2.27 | |
| Loo–Riegelman | Far | 0.93 | 120.8 | 3.84 |
| BrP | 0.50 | 101.2 | 4.35 | |
| USP | 0.73 | 109.5 | 4.52 | |
| Stocrin® | ||||
| OBS | – | 137.95 ± 42.85 | 2.93 ± 0.83 | |
Stocrin® drug reference batch A
Far Farmanguinhos methodology, BrP Brazilian Pharmacopoeia, USP US Pharmacopeia, OBS observed pharmacokinetic parameters. Stocrin® drug reference batch A
Fig. 5.
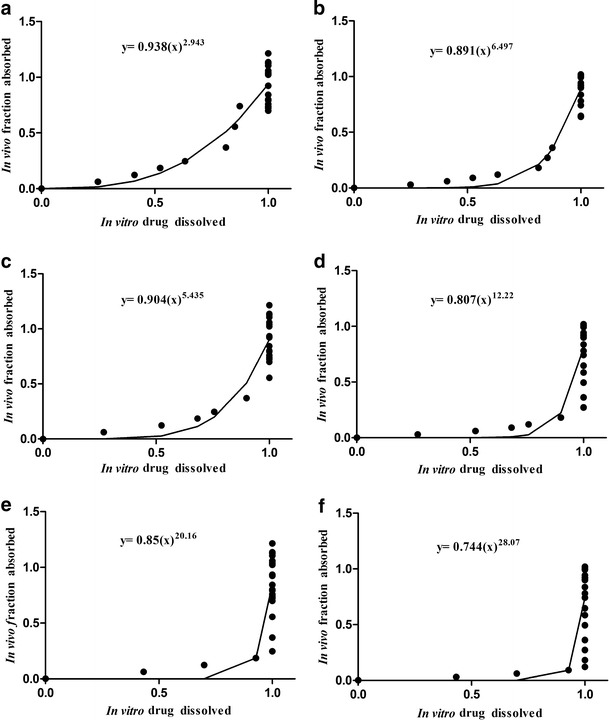
IVIVC plot for biobatch A IR tablets based on dissolution data using the USP, BrP, and Far methods and fraction absorption data according mono-compartment and tri-compartment models. a Mono-compartment Far. b Tri-compartment Far. c Mono-compartment USP. d Tri-compartment USP. e Mono-compartment BrP. f Tri-compartment BrP
After establishing the IVIVC, the capacity of the model to predict the in vivo behavior of the dosage forms from the in vitro dissolution studies (convolution) was verified. The Wagner–Nelson model was the most suitable model for use in the simulation of the convolution. Considering the three methods evaluated, the Farmanguinhos method presented the best simulated value for the maximum plasma concentration when compared with the experimental bioavailability/bioequivalence studies as shown in Table IV. The correlation between the simulated and experimental maximum plasma concentration for the Farmanguinhos method, as shown in Fig. 6 which confirmed that the Wagner–Nelson model, was better than the Loo–Riegelman model for this purpose. The best convolution between the three dissolution methods was obtained using the Farmanguinhos method presented in Fig. 7, which indicated that it was the preferable method for establishing in vitro–in vivo correlation for the efavirenz tablet.
Fig. 6.
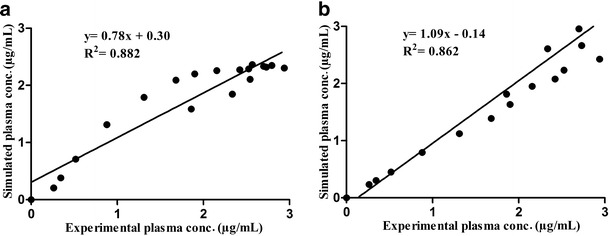
IVIVC plot experimental and simulated plasma concentration of the biobatch A IR tablet based on convolution of dissolution data using the Far methods using mono-compartment and tri-compartment models. a Mono-compartment Far. b Tri-compartment Far
Fig. 7.
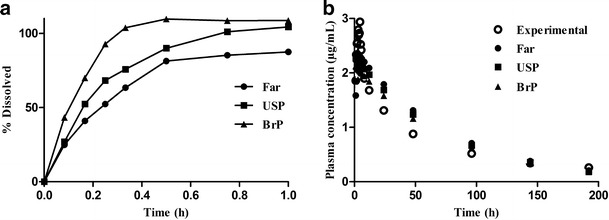
Biobatch A IR tablets dissolution profile and the experimental and simulated in vivo profile
CONCLUSION
This study showed that GastroPlus™ has the predictive capacity to calculate the bioavailability of oral solid formulations produced using API efavirenz which possessed different physicochemical characteristics. By using GastroPlus™, the IVIVC was established based upon the dissolution data obtained using different methods and different absorption models. The use of a dissolution method that has a level A IVIVC is desirable since it ensures the safety and effectiveness of the subsequent batches produced. In addition, the software was able to simulate pharmacokinetic parameters using the convolution method which provides in vitro dissolution data for the efavirenz tablets. However, the predictive capacity of the software can be improved by including options for varying input of the formulation parameters. This would help ensure a more accurate pharmacokinetic estimation for supporting the development of generic medicines. These results confirm that GastroPlus™ is a valuable in silico method for IVIVC and for studies directed at developing formulations of class II drugs.
Acknowledgments
This work was supported by FAPERJ, Edital CAPES Nanobiotecnologia 2008, and CNPq. We are grateful to Michelle Parvatiyar for her English review.
References
- 1.Food and Drug Administration. Guidance for industry: bioavailability and bioequivalence studies for orally administers drug products. General Considerations. US Department of Health and Human Services, CDER/FDA; 2003.
- 2.Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a Biopharmaceutic Drug Classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pham Res. 1995;12:413–20. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 3.Food and Drug Administration. Guidance for industry: waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a Biopharmaceutics Classification System. US Department of Health and Human Services, CDER/FDA; 2000.
- 4.Yu LX, Amidon GL, Polli JE, Zhao H, Mehta MU, Conner DP, et al. Biopharmaceutics Classification System: the scientific basis for biowaiver extensions. Pharm Res. 2002;19:921–5. doi: 10.1023/A:1016473601633. [DOI] [PubMed] [Google Scholar]
- 5.Tubic-Grozdanis M, Bolger MB, Langguth P. Application of gastrointestinal simulation for extensions for biowaivers of highly permeable compounds. AAPS J. 2008;10:213–26. doi: 10.1208/s12248-008-9023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsume Y, Amidon GL. The biowaiver extension for BCS class III drugs: the effect of dissolution rate on the bioequivalence of BCS class II immediate-release drugs predicted by computer simulation. Mol Pharm. 2010;7:1235–43. doi: 10.1021/mp100053q. [DOI] [PubMed] [Google Scholar]
- 7.Rinak E, Dokoumetzidis A, Valsami G, Macheras P. Identification of biowaivers among class II drugs: theoretical justification and practical examples. Pharm Res. 2004;21:1567–72. doi: 10.1023/B:PHAM.0000041450.25106.c8. [DOI] [PubMed] [Google Scholar]
- 8.Polli JE, Yu LX, Cook JA, Amidon GL, Borchardt RT, Burnside BA, et al. Summary workshop report: Biopharmaceutics Classification System—implementation challenges and extension opportunities. J Pharm Sci. 2004;93:1375–81. doi: 10.1002/jps.20064. [DOI] [PubMed] [Google Scholar]
- 9.Pelkonen O, Turpeinen M, Raunio H. In vivo-in vitro-in silico pharmacokinetic modelling in drug development. Clin Pharmacokinet. 2011;50:483–91. doi: 10.2165/11592400-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 10.Grbic S, Parojcic J, Ibric S, Djuric Z. In vitro-in vivo correlation for glicazide immediate-release tablets based on mechanistic absorption simulation. AAPS PharmSciTech. 2011;12:165–71. doi: 10.1208/s12249-010-9573-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simulations Plus, Manual GastroPlus™, California, EUA; 2010.
- 12.Wei H, Löbenberg R. Biorelevant dissolution media as predictive tool for glyburide a class II drug. Eur J Pharm Sci. 2006;29:45–52. doi: 10.1016/j.ejps.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 13.Okumu A, Dimaso M, Löbenberg R. Computer simulations using GastroPlus™ to justify a biowaiver for etoricoxib solid oral drug products. Eur J Pharm Biopham. 2009;72:91–8. doi: 10.1016/j.ejpb.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 14.Kovacevic I, Parojeie J, Homsek I, Tubie-Grozdanis M, Langguth P. Justification of biowaiver for carbamazepine, a low soluble high permeable compound, in solid dosage forms based on IVIVC and gastrointestinal simulation. Mol Pharm. 2009;6:40–7. doi: 10.1021/mp800128y. [DOI] [PubMed] [Google Scholar]
- 15.Okumu A, DiMaso M, Löbenberg R. Dynamic dissolution testing to establish in vitro/in vivo correlations for montelukast sodium, a poorly soluble drug. Pharm Res. 2008;25:2778–85. doi: 10.1007/s11095-008-9642-z. [DOI] [PubMed] [Google Scholar]
- 16.Maggiolo F. Efavirenz: a decade of clinical experience in the treatment of HIV. J Antimicrob Chemother. 2009;64:910–28. doi: 10.1093/jac/dkp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kasim NA, Whitehouse M, Ramachandran C, Bermejo M, Lennerna H, Hussain AS, et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol Pharm. 2004;1:85–96. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- 18.Lindenberg M, Kopp S, Dressman JB. Classification of orally administered drugs on the world model list of essential medicines according to the biopharmaceutics classification system. Eur J Pharm Biopharm. 2004;58:265–78. doi: 10.1016/j.ejpb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 19.Müller CE. Prodrug approaches for enhancing the bioavailability of drugs with low solubility. Chem Biodivers. 2009;6:2071–83. doi: 10.1002/cbdv.200900114. [DOI] [PubMed] [Google Scholar]
- 20.International Standard Organization. General requirements for the competence of the material producers. ISO Guide 34, 2009.
- 21.The United States Pharmacopoeia and National Formulary. 34th ed. Rockville, MD: USP Convention Inc.; 2011.
- 22.ICH, validation of analytical procedures: text and methodology (Q2R1), in International Conference on Harmonization; November 2005.
- 23.Azarmi S, Roa W, Löbenberg R. Current perspectives in dissolution testing of conventional and novel dosage forms. Int J Pharm. 2007;328:12–21. doi: 10.1016/j.ijpharm.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Brazilian Pharmacopoeia. 5th ed. Brazilian Health Surveillance Agency; 2010.
- 25.Lu ATK, Frisella ME, Johnson KC. Dissolution modeling: factors affecting the dissolution rates of polydisperse powders. Pharm Res. 1993;10:1308–14. doi: 10.1023/A:1018917729477. [DOI] [PubMed] [Google Scholar]
- 26.Mithani SD, Bakatselou V, TenHoor CN, Dressman JB. Estimation of the increase in solubility of drugs as a function of bile salt concentration. Pharm Res. 1996;10:164–7. doi: 10.1023/a:1016062224568. [DOI] [PubMed] [Google Scholar]
- 27.Mahapatra S, Thakur TS, Joseph S, Varughese S, Desiraju GR. New solid state forms of the anti-HIV drug efavirenz. Conformational flexibility and Z' issues. Cryst Growth Des. 2010;10:3191–202. doi: 10.1021/cg100342k. [DOI] [Google Scholar]
- 28.Kesisoglou F, Wu Y. Understanding the effect of API properties on bioavailability through absorption modeling. AAPS J. 2008;10:516–25. doi: 10.1208/s12248-008-9061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Desai D, Rinaldi F, Kothari S, Paruchuri S, Li D, Lai M, et al. Effect of hydroxypropyl cellulose (HPC) on dissolution rate of hydrochlorothiazide tablets. Int J Pharm. 2006;308:40–5. doi: 10.1016/j.ijpharm.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 30.Gorman EA, Rhodes CT, Rudnic EM. An evaluation of croscarmellose as a tablet disintegrant in direct compression systems. Drug Dev Ind Pharm. 1982;8:397–410. doi: 10.3109/03639048209022108. [DOI] [Google Scholar]
- 31.Rowel RC, Sheskey PJ, Quinn ME. Handbook of pharmaceutical excipients. 6th ed. London: Pharmaceutical Press; 2009.
- 32.Brown CK, Chokshi HP, Nickerson B, Reed RA, Rohrs BR, Shah PS. Acceptable analytical practices for dissolution testing of poorly soluble compounds. Pharm Technol. 2004;28:56–65. [Google Scholar]
- 33.Takano R, Furumoto K, Shiraki K, Takata N, Hayashi Y, Aso Y, et al. Rate-limiting steps for oral absorption for poorly water-soluble drugs in dogs; prediction from miniscale dissolution test and a physiologically-based computer simulation. Pharm Res. 2008;25:2334–44. doi: 10.1007/s11095-008-9637-9. [DOI] [PubMed] [Google Scholar]
- 34.Takano R, Sugano K, Higashida A, Hayashi Y, Machida M, Aso Y, et al. Oral absorption of poorly water-soluble drugs: computer simulation of fraction absorbed in humans from miniscale dissolution test. Pharm Res. 2006;23:1144–56. doi: 10.1007/s11095-006-0162-4. [DOI] [PubMed] [Google Scholar]
- 35.He Z, Zhong D, Chen X, Liu X, Tang X, Zhao L. Development of medium for nimodipine tablets based on bioavailability evaluation. Eur J Pharm Sci. 2004;21:487–91. doi: 10.1016/j.ejps.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 36.Pabla D, Akhlaghi F, Zia H. A comparative pH-dissolution profile study of selected commercial levothyroxine products using inductively couple plasma mass spectrometry. Eur J Pharm Biopharm. 2009;72:105–10. doi: 10.1016/j.ejpb.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 37.Rekić D, Röshammar D, Mukonzo J, Ashton M. In silico prediction of efavirenz and rifampicin drug-drug interaction considering weight and CYP 2B6 phenotype. Br J Clin Pharmacol. 2011;71:536–43. doi: 10.1111/j.1365-2125.2010.03883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nanzigu S, Eriksen J, Makumbi F, Lanke S, Mahindi M, Kiguba R, et al. Pharmacokinetics of the nonnucleoside reverse transcriptase inhibitor efavirenz among HIV-infected Ugandans. HIV Med. 2012;13:193–201. doi: 10.1111/j.1468-1293.2011.00952.x. [DOI] [PubMed] [Google Scholar]
- 39.Villani P, Regazzi MB, Castelli F, Viale P, Torti C, Seminari E, et al. Pharmacokinetics of efavirenz (EFV) alone and in combination therapy with nelfinavir (NFV) in HIV-I infected patients. Br J Clin Pharmacol. 1999;48:712–5. doi: 10.1046/j.1365-2125.1999.00071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cristofoletti R, Nair A, Abrahamsson B, Groot DW, Kopp S, Langguth P, Polli JE, Shah VP, Dressman JB. Biowaiver monographs for immediate release solid oral dosage forms: efavirenz. J Pharm Sci. 2013;102:318–29. doi: 10.1002/jps.23380. [DOI] [PubMed] [Google Scholar]
- 41.DiCenzo R, Forrest A, Squires KE, Hammer SM, Fischl MA, Wu H, et al. Indinavir, efavirenz, and abacavir pharmacokinetics in human immunodeficiency virus-infected subjects. Antimicrob Agents Chemother. 2003;47:1929–35. doi: 10.1128/AAC.47.6.1929-1935.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pfister M, Labbé L, Hammer SM, Mellors J, Bennett KK, Rosenkranz S, et al. Population pharmacokinetics and pharmacodynamics of efavirenz, nelfinavir, and indinavir: adult AIDS clinical trial group study 398. Antimicrob Agents Chemother. 2003;47:130–7. doi: 10.1128/AAC.47.1.130-137.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kocic I, Homseka I, Dacevic M, Grbic S, Parojcic J, Vucicevic K, et al. A case study on the in silico absorption simulations of levothyroxine sodium immediate-release tablets. Biopharm Drug Dispos. 2012;33:146–59. doi: 10.1002/bdd.1780. [DOI] [PubMed] [Google Scholar]
- 44.Wei H, Dalton C, Di Maso M, Kanfer I, Löbenberg R. Physicochemical characterization of five glyburide powders: a BCS based approach to predict oral absorption. Eur J Pharm Biopharm. 2008;69:1046–56. doi: 10.1016/j.ejpb.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 45.Jantratid E, Prakongpan S, Amidon GL, Dressman JB. Feasibility of biowaiver extension to biopharmaceutics classification system class III drug products cimetidine. Clin Pharmacokinet. 2006;45:385–99. doi: 10.2165/00003088-200645040-00004. [DOI] [PubMed] [Google Scholar]