

Abstract
Background
Expressed Sequence Tag (EST) sequences are generally single-strand, single-pass sequences, only 200–600 nucleotides long, contain errors resulting in frame shifts, and represent different parts of their parent cDNA. If the cDNAs contain translation initiation sites, they may be suitable for functional genomics studies. We have compared five methods to predict translation initiation sites in EST data: first-ATG, ESTScan, Diogenes, Netstart, and ATGpr.
Results
A dataset of 100 EST sequences, 50 with and 50 without, translation initiation sites, was created. Based on analysis of this dataset, ATGpr is found to be the most accurate for predicting the presence versus absence of translation initiation sites. With a maximum accuracy of 76%, ATGpr more accurately predicts the position or absence of translation initiation sites than NetStart (57%) or Diogenes (50%). ATGpr similarly excels when start sites are known to be present (90%), whereas NetStart achieves only 60% overall accuracy. As a baseline for comparison, choosing the first ATG correctly identifies the translation initiation site in 74% of the sequences. ESTScan and Diogenes, consistent with their intended use, are able to identify open reading frames, but are unable to determine the precise position of translation initiation sites.
Conclusions
ATGpr demonstrates high sensitivity, specificity, and overall accuracy in identifying start sites while also rejecting incomplete sequences. A database of EST sequences suitable for validating programs for translation initiation site prediction is now available. These tools and materials may open an avenue for future improvements in start site prediction and EST analysis.
Background
Expressed sequence tags
Complete sequences of the mouse and human genomes are available; completion of additional animal genomes is imminent. Effective methods for identifying genes, and the proteins they encode, have become increasingly important. Although most genes can be identified through the open reading frame (ORF) of the protein they encode, detection in eukaryotic genomic sequence is more difficult since these genes are fragmented into small exons (averaging 145 bp in human), extending across large regions (averaging 27 kb in human) [1].
Eukaryotic gene-discovery can be most effectively accomplished through direct sequencing of gene transcripts using cDNA libraries [2]. Because cDNAs represent processed mRNAs, intervening sequences have been removed, and ORFs can more easily be deduced. Due to cost and time constraints, most high-throughput cDNA sequencing efforts rely on end-sequences from cDNA clones that vary in length, and thus represent different portions of the mRNAs from which they derive.
These end sequences, called expressed sequence tags (ESTs), are generally single-strand, single-pass sequences, only 200–600 nucleotides long, contain errors leading to frame shifts, and represent different parts of the parent cDNA [3]. Comparison of ESTs to each other, and to genome sequence, is useful for gene discovery. Comparison of ESTs from different cDNA libraries may yield information about gene expression and alternative mRNA processing. Furthermore, ESTs can be used as 'tags' to identify genes and to probe the genome for matching sequences, such as in the construction of genome maps. As a result of their usefulness, large numbers of ESTs have been generated in both the public and private sectors; in 2001, ESTs made up more than 60% of all of the nucleotide sequence database entries [4].
ESTs also provide a resource for determining the complexity and quality of cDNA libraries, including identifying full-length cDNA clones suitable for isolation and functional analysis. A full-length cDNA should encompass all sequences from the CAP site to the poly (A) addition site. However, a cDNA comprising at least the entire ORF, from translation initiation site (TIS) to termination codon, is worthy of high accuracy re-sequencing and/or protein functional analysis. In fact, successful identification of the TIS alone leads to simple determination of the termination codon, if present. For this reason, most methods for determining the completeness of ESTs, and by extension the cDNAs from which they originate, focus on the TIS. This study reviews and compares – both qualitatively and quantitatively – the major computational methods and tools for identifying TISs and determining completeness of ESTs.
Identifying TISs in ESTs
The majority of eukaryotic mRNAs have one open reading frame and a single functional TIS, usually the AUG codon closest to the 5'-end [5]. The "scanning hypothesis" postulates that a 40S ribosomal subunit binds initially at the 5'-end of an mRNA and migrates linearly in a 3' direction until it reaches the first AUG codon [6-8]. If the first initiation codon lies in a suitable context (e.g., GCC [AG]CCatgG, Kozak's consensus) the 40S ribosomal subunit migrates no further, is joined by the 60S ribosomal subunit, and the complex initiates protein synthesis [5,6]. When the context is less than favourable, some protein synthesis may occur there, but most will start at the next downstream AUG codon [9].
Though Kozak's consensus has very good validity in vertebrate mRNAs [10], further analyses has revealed variation in the initiation context between different groups of eukaryotes [11]. Furthermore, despite the utility of Kozak's consensus in identification of TISs in mRNAs, EST data poses numerous problems that render the consensus sequence much less useful for it. The main problem involves the generality of the consensus sequence; while the absence of the pattern will usually exclude an ATG from being the initiation codon, the pattern is general enough to match many other ATG triplets in each sequence. In the case of an incomplete EST lacking the true initiation site, relying solely on Kozak's consensus would result in the false prediction of the most 5' Kozak consensus being the initiation site. Additional features are required to identify TISs in ESTs, such as the positioning of a Kozak's consensus sequence relative to a significant open reading frame.
Several computational tools have been developed to assist in this identification. Some methods, such as conditional probability matrices [12], consider only the nucleotides in the vicinity of ATGs. Other methods, such as NetStart [13], consider larger regions. ATGpr [14] considers a variety of factors. Still others, such as ESTScan [15] and Diogenes [16], though not specifically designed to identify TISs, perform very well in identifying open reading frames and might be expected to be useful for predicting EST completeness.
Methods evaluated
This study evaluates and compares five methods: first-ATG, ESTScan, Diogenes, Netstart, and ATGpr. These methods range from simple (choosing the first ATG) to complex (neural networks, discriminant functions). The methods were chosen on the bases of popularity, accessibility, and their collective ability to represent a variety of approaches to the problem of identifying TISs in EST data. Most are available on the web; their websites are listed in Table 1.
Table 1.
Programs Evaluated
| Program | Access | Available for download |
| First-ATG | Locally written using Microsoft Excel [27] | – |
| ATGpr [14] | http://www.hri.co.jp/atgpr/ | no |
| NetStart [13] | http://www.cbs.dtu.dk/services/NetStart/ | yes |
| Diogenes [16] | http://www.cbc.umn.edu/diogenes/diogenes.html | yes |
| ESTScan [15] | http://www.ch.embnet.org/software/ESTScan.html | yes |
First-ATG
Kozak, in 1989, reported that less than 10% of all eukaryotic mRNAs do not use the first ATG for the start codon [5]. If this remains true, it should therefore be possible to predict TIS with 90% accuracy by just selecting the first (most-5') ATG. However, this is only true for complete, error-free, mRNA sequences. The situation is very different with ESTs, which, as mentioned above, are partial, single-pass cDNA sequences. ESTs have more errors than genomic sequences and may represent different regions of the mRNA – in some cases lacking the true TIS. For these reasons, prediction of the TIS in an EST may benefit from consideration of TIS context. However, evaluating the simple method of choosing the first ATG can reveal the extent of the above problems. Furthermore, the first-ATG method serves as a meaningful baseline to use with more sophisticated methods.
ESTScan
Several programs distinguish between coding sequences and non-coding sequences based solely on the intrinsic properties of the nucleotide sequences, as opposed to using homology information. The most successful programs are GenScan [17] for genomic DNA and ESTScan [15] for ESTs. ESTScan is of particular interest for this study because of its potential to determine completeness of ESTs. ESTScan implements a fifth-order hidden Markov model that recognizes coding sequences by oligonucleotide frequencies. Additionally, ESTScan corrects for sequence errors, which could be an especially helpful feature for analyzing ESTs. Although ESTScan does not incorporate a model of the TIS, it does predict the beginning of the coding sequence. This prediction may not be very accurate – indeed, it may not even correspond to an ATG – but ESTScan's detection of coding sequences makes this program potentially useful for evaluating the EST completeness. An updated version is available [18].
Diogenes
Diogenes [16], developed at the University of Minnesota, is somewhat similar in purpose to ESTScan; it finds ORFs in short sequences. Diogenes identifies ORF candidates by scanning all six reading frames for stretches of sequence uninterrupted by stop codons. Various organism-specific statistical measures, such as codon frequency and ORF length, are then used to estimate the likelihood that these ORF candidates encode proteins. A quadratic discriminant statistic combining these various factors is reported as an overall score for the reliability of the final ORF prediction. Like ESTScan, Diogenes does not incorporate a model of the TIS. However, Diogenes also reports the predicted beginning of the coding sequence that may be useful for evaluating the EST completeness.
NetStart
NetStart [13], perhaps the most popular and accessible program for TIS prediction, analyzes a larger region – up to 100 bases upstream and 100 bases downstream of a putative start codon. NetStart uses an artificial neural network to predict the initiation site from this large fixed-length window around the potential start codon. Based on a training data set of conceptually-spliced mRNA derived from genomic sequences with known start sites, the neural network 'learned' on its own which features are indicative of a true TIS. This approach is especially appealing due to the complexity of translation initiation.
ATGpr
ATGpr [14] considers as many as six characteristics of the EST sequence in analyzing the context of a putative TIS:
• Positional triplet weight matrix around the ATG; the propensity for a particular triplet to be in a specific position relative to the ATG.
• Frequencies of in-frame hexanucleotides downstream of the ATG; favors longer reading frames with suitable hexanucleotide compositions.
• Hexanucleotide difference before and after the ATG; these regions correspond to the putative 5' untranslated region (UTR) and the putative open reading frame, respectively; the difference between these 50-nucleotide regions should be greater for real start codons.
• Likelihood of a signal peptide being present, based on the presence of hydrophobic 8-residue peptides within a 30 amino acid window downstream of the ATG.
• Presence of another upstream in-frame ATG, which decreases the likelihood of the ATG under analysis being the true initiation codon according to the ribosome scanning model of translation initiation [5].
• Upstream cytosine nucleotide presence; based on the observation that 5' untranslated regions of human genes are often rich in cytosine.
Each characteristic can distinguish true from false initiation sites. Reportedly, the most important features for correct predictions are the positional triplet weight matrix around the ATG and the hexanucleotide difference before and after the ATG [14]. A linear discriminant function is used to combine the statistical measures of these six features into a final score. Like NetStart, ATGpr was trained on conceptually-spliced mRNA derived from genomic sequences with known start sites.
A standard dataset for validation of TIS prediction
A major limitation of previous studies of methods for TIS prediction concerns the test datasets used. Several of the early computational methods for TIS and coding region prediction were evaluated before a large amount of EST data was available, and thus used instead mRNA or conceptually-spliced mRNA. Such datasets fail to capture the problems unique to EST data (described above). Furthermore, lack of consistency in data and types of data used for evaluating the different methods renders comparison problematic at best. Study of methods for TIS prediction would therefore benefit from a single dataset that is representative of the type of data seen in practical applications. This study benchmarks the key computational tools with a relevant dataset.
Results
The five methods described above were applied to dataset of 50 EST sequences with, and 50 without, translation initiation codons. In order to simulate the practical use of these methods in actual EST projects, only the top scoring ATG from each sequence is predicted to be the initiation codon, given that the corresponding score is above the threshold value under consideration.
Figure 1 contains an example of a query sequence and the start site predictions made by the various methods. The query sequence contains 672 nucleotides. The comment line indicates that the sequence was obtained from the 5' end of a human cDNA clone. The average number of ATGs per sequence in the dataset is approximately 8. In this example, the actual TIS at position 137 (underlined and bold) is not the first ATG of the sequence (underlined). In fact, the TIS is the second of this sequence's eleven ATGs. As expected, Diogenes and ESTScan failed to correctly predict the precise position of the translation initiation site; however, ESTScan's prediction is closer to the actual start site. Still, the low scores reported by Diogenes and ESTScan mean that under reasonable thresholds these two programs would incorrectly predict that the sequence does not contain a TIS. ATGpr and NetStart correctly identified the TIS with reasonably high scores.
Figure 1.

Sample query sequence and corresponding start site predictions.
Presence versus absence of start sites
Simply predicting whether or not EST sequences contain the TIS may be very useful for some EST projects. It can indicate which region of the gene is represented by the EST sequence as well as roughly assess the completeness of the EST's 5' end. Accordingly, this study evaluates the ability of ESTScan, Diogenes, Netstart, and ATGpr to predict the presence or absence of TIS.
Since sensitivity and specificity are of varying degrees of interest for different types of EST projects, ROC curves were plotted for the four methods across the entire observed range of threshold scores. ESTScan generally fails to discriminate between the presence and absence of translation initiation sites in the dataset (Figure 2). However, the high p-value (0.3408, Table 2) attests to problems in the evaluation of ESTScan's performance due in part to the program's scoring system. This high value is caused by the large number of zero-scoring results from ESTScan (40 out of 100 total predictions), from both sequences that contain actual initiation sites and sequences that do not. ESTScan's documentation states that sequences with scores of zero are considered noncoding. These results reveal a major drawback of using ESTScan for predicting the presence of TIS rather than for its more conventional use of detecting coding regions.
Figure 2.
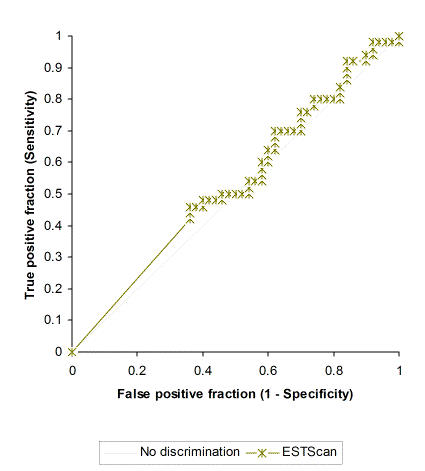
Prediction of presence versus absence of a translation initiation site: ROC curve of ESTScan across score thresholds. A positive test state represents the known presence of a TIS. A negative test state represents the known absence of a TIS. Statistical details in Table 2.
Table 2.
Statistical Details of ROC Curves. Analysis of 100 sequences, 50 with and 50 without, translation initiation sites, for ability to predict presence vs absence of translation inititiation sites (Figures 2 and 3).
| A. ROC curve values for each program. | ||||
| Curve | Area | SE | p | 95% CI of Area |
| ATGpr | 0.850 | 0.0378 | <0.0001 | 0.776 to 0.924 |
| NetStart | 0.715 | 0.0521 | <0.0001 | 0.613 to 0.817 |
| Diogenes | 0.706 | 0.0514 | <0.0001 | 0.605 to 0.806 |
| ESTScan | 0.524 | 0.0580 | 0.3408 | 0.410 to 0.638 |
| B. ROC curve values for comparisons between two programs. | ||||
| Contrast | Difference | p | ||
| NetStart v Diogenes | 0.009 | 0.8838 | ||
| NetStart v ATGpr | -0.136 | 0.0105 | ||
| Diogenes v ATGpr | -0.145 | 0.0072 | ||
| ESTScan v ATGpr | -0.327 | - | ||
| NetStart v ESTScan | 0.191 | - | ||
| Diogenes v ESTScan | 0.182 | - | ||
The other three programs perform much better on the dataset in terms of sensitivity and specificity (Figure 3). ATGpr, NetStart, and Diogenes are each able to discriminate between the presence and absence of translation initiation sites with reasonable sensitivity and specificity. Diogenes performs better than ESTScan; this is likely due to Diogenes' different scoring system as well as its inclusion of more factors in its predictions. NetStart's performance is slightly better than that of Diogenes. Unfortunately, because NetStart is based on neural networks, it is difficult to determine what factors contributed to the method's performance in predicting the presence or absence of start sites. ATGpr is the most effective method for discriminating between the presence and absence of translation initiation sites in the dataset (Figure 3). ATGpr's discriminative performance on the dataset is significantly better than those of NetStart and Diogenes (Table 2).
Figure 3.
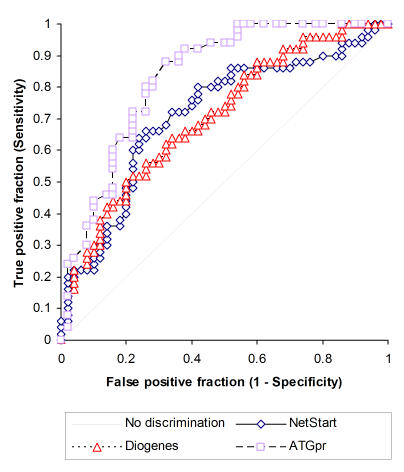
Prediction of presence versus absence of a translation initiation site: ROC curve of NetStart, Diogenes and ATGpr across score thresholds. A positive test state represents the known presence of a TIS. A negative test state represents the known absence of a TIS. Statistical details in Table 2.
Identification of start sites
The overall percentage accuracy of each of the four programs in identifying the locations of TISs, as well as their absence when appropriate, is shown in Figure 4. In other words, for each sequence, each program could predict either a position for the putative start site or the absence of a start site. The absence of a start site is predicted when the score of the predicted start site falls below the threshold score being considered. ATGpr is shown to be the most accurate method for identifying true TISs while rejecting sequences lacking true ones. ATGpr achieves a maximum accuracy of 76% at a threshold score of approximately 0.45. NetStart is the second most accurate method, achieving a maximum accuracy of 57% at a threshold score of approximately 0.7. Diogenes' and ESTScan's accuracies are quite low; these two programs fail to predict the precise locations of TISs since they do not explicitly model them. The overall accuracy of each of the methods approaches 50% at the highest threshold scores; at that point almost all sequence are predicted to lack TISs, which is true for half of the sequences in the dataset.
Figure 4.
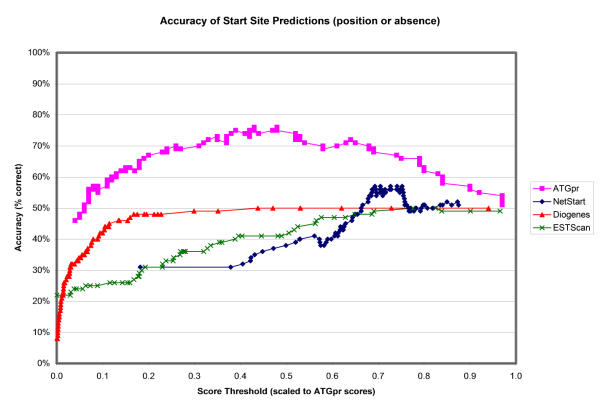
Accuracy of start site predictions: position or absence by score thresholds. The maximum accuracy for ATGpr is 74% at a score threshold of 0.48; the maximum accuracy for Netstart is 55% at a score threshold of approximately 0.7.
The overall percent accuracy of each program over the 50 sequences that contain true TISs is shown in Figure 5. No thresholds are used; the highest-scoring prediction for each sequence is considered for each method. Simply choosing the first ATG correctly identifies the TIS in 74% of the sequences that contain true TISs. This decrease from the theoretical 90% accuracy of this method (explained above) is most likely due to the frequent incompleteness of EST sequences. ATGpr performs extremely well on this limited dataset, correctly identifying the translation initiation site of 90% of the sequences. Surprisingly, NetStart is less accurate (60% correctly predicted) than the first-ATG method. Again, as expected, Diogenes' and ESTScan's accuracies are extremely low, respectively 0% and 2% correctly predicted.
Figure 5.
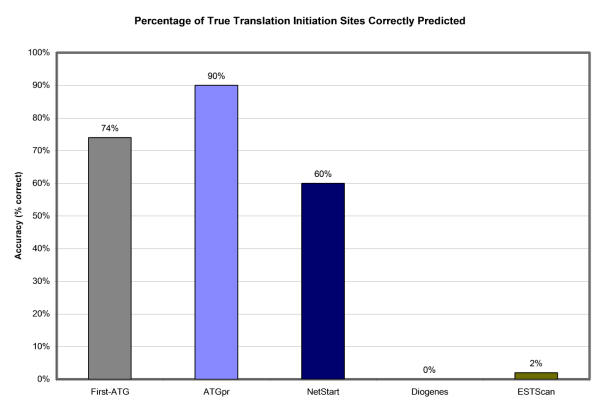
Percentage of true initiation sites correctly identified. The overall percent accuracy of each program over the 50 sequences that contain true start sites is shown. No cutoffs are used; the highest-scoring prediction for each sequence is considered for each method.
Discussion
Comparisons
The low accuracy of ESTScan points to the potential drawbacks of using this method for identifying TIS (or even their presence or absence) rather than for its more conventional use of detecting coding regions. However, the features considered by Diogenes were found to be sufficient to predict the presence or absence of start sites with moderate reliability.
Overall, ATGpr is shown to be effective in identifying TISs in EST sequences as well as in rejecting sequences that lack a true TIS. While an accuracy of 76% for prediction of true TISs leaves room for improvement, ATGpr achieves levels of sensitivity, specificity, and overall accuracy that are suitable for practical application. Furthermore, ATGpr's high accuracy in the dataset of sequences containing true TISs indicates that this method will become more useful as methods for generating 5'-complete ESTs improve.
Interestingly, ATGpr was found to be generally more effective than NetStart. Considering that ATGpr is based on a predetermined set of rules whereas NetStart utilizes artificial neural networks, ATGpr's favorable results indicate that improved understanding of the mechanism of translation initiation may lead to greater ability to identify translation initiation sites. Both programs might benefit from being retrained on newer, larger datasets, preferably consisting of ESTs instead of conceptually-spliced mRNA sequences.
Combined analysis
The main aim of this study was to compare and contrast the performance of several algorithms in identifying TISs in EST data. However, combined analysis of the results from all of the algorithms yields additional information. For example, analysis of an EST corresponding to the human MSMB locus (GenBank ID BF679106) resulted in identical predictions by firstATG, NetStart, and ATGpr (TIS at nucleotide position 34) that are consistent with annotation at ENSEMBL [19] and GenBank [20], but in disagreement with annotation at ProtEST [21] (TIS at position 232), the 'gold standard' used in this study.
In another example, an EST corresponding to the human RanBPM gene (Genbank ID AA311767) was one of the original 50 sequences with known TISs in the validation set. The consensus prediction by these same three algorithms (TIS at position 221) was in disagreement with annotation at ProtEST (TIS at position 336). The ProtEST annotation was consistent with experimental data [22] that reported RanBPM was a 55 kDa protein. A more recent analysis [23] revealed the molecular mass to be 90 kDa; the AA311767 EST corresponds to a 5'-truncated cDNA, so contains no TIS. The RanBPM EST was replaced in the final validation set. However, as these examples demonstrate, neither computational nor molecular approaches are completely accurate in predicting the presence or location of TISs in ESTs.
Disagreement between an annotated TIS location and predictions corroborated by more than one algorithm can suggest problems with annotation, incomplete ESTs, and/or cDNA truncation. Differing results from multiple algorithms could also theoretically be used to identify alternative start codons and upstream open reading frames. Important future work would be to automate the extraction of such data from combined or serial analysis using multiple algorithms.
Homology
In principle, many coding sequences (and thus also TISs) can be characterized through alignment with homologous proteins. Several programs are capable of aligning nucleotide sequences to protein sequences databases; BLASTX [24] and FASTX/FASTY [25] are among the most popular. However, there are several major limitations to this approach. First, aligning a nucleotide sequence to protein sequences is more prone to error than other types of alignment due to the multiple reading frames and possible false hits from 5' or 3' untranslated regions. On the other hand, searching for matches in a nucleotide database does not guarantee that the matched sequences represent the complete gene. Perhaps most importantly, approaches based on alignment rely on homologous proteins and therefore cannot be used to find novel genes.
Methods based solely on the analysis of intrinsic properties of nucleotide sequences therefore seem to be the most promising – and perhaps the only useful – approaches for identifying TIS in EST data. Since living cells' translation machinery is able to identify start sites without using homology information, it should in principle be possible for computer programs to do the same [26].
Still, homology can be used to ease the task of identifying TISs. Homology searches can be used early in an EST project to determine which ESTs correspond to previously identified genes. Having thus narrowed down the dataset, the scientist can then focus on the remaining, novel genes.
Also, homology can be used to increase the accuracy of TIS prediction, particularly in borderline cases. Nishikawa et al. [27] add similarity information to ATGpr score, slightly improving sensitivity and specificity. The program, ATGpr_sim, was not available for evaluation in this study.
Areas for improvement
Despite the discovery of Kozak's consensus sequence and its apparently important role in translation initiation, it is not truly understood how this consensus modulates ribosomal scanning of mRNAs. Specifically, it is not clear why the ribosome pauses at ATG sites characterized by Kozak's consensus. A better understanding of the requirements for ribosome scanning and – more importantly – of the context in which the ribosome pauses to initiate translation could lead to more reliable methods for identifying translation initiation sites. For example, mRNA secondary structure immediately downstream of the initiator ATG has been shown to play a role in translation initiation [28]. The context around initiation sites thus appears to be significantly more complex than current models. The superior performance of ATGpr in this project supports the notion of initiation sites being distinguished by a variety of features. ATGpr indeed bases its predictions on six types of sequence information. Even NetStart's neural networks apparently failed to capture the complexity of TISs.
Some of the complexities in transition initiation have recently been reviewed [29]. The most relevant to this project is the occurrence of multicistronic eukaryotic mRNAs. Though the majority of eukaryotic mRNAs have one TIS, some have more. In some of the multicistronic sequences, intercistronic distances are small (80 – 150 nucleotides) and upstream ORF(s) are short (< 30 nucleotides). Thus even short ESTs may have more than one valid TIS. It is important to develop methods to identify multiple TISs when they occur in ESTs, and to distinguish between TISs that initiate translation of short polypeptides and those that initiate much longer proteins.
Another area that deserves more attention is the 5' untranslated region (UTR). As described above, the ribosome binds the mRNA at the 5' cap region at the 5' terminus. This means that the entire length of the 5' UTR is passed over by the ribosome before it initiates protein synthesis. More detailed knowledge of the 5' UTR might provide insight as to why this region is passed over by the ribosome, possibly even clarifying why some first-ATGs are not true TISs.
Analysis and annotation of EST data would of course benefit from higher quality EST sequences, or even higher quality reference cDNA sequences. Oligo-capping [30] allows collection of full-length cDNA sequences by recognizing the cap structure and introducing an oligomer RNA at the 5' end of the mRNA. Comparison of ESTs to homologous sequences in oligo-capped cDNA libraries could vastly improve determination of the 5'-completeness of ESTs and thus improve EST analysis and annotation.
Conclusions
Gene identification is one of the major tasks of bioinformatics. As high throughput methods have facilitated complete genome sequencing, the importance of identifying coding regions has become more evident. Analyzing sequences from cDNAs is the most direct way to identify and characterize the coding regions. The structural annotation of genes in genomic sequences will therefore likely depend on cDNA analysis until/unless more efficient methods are developed. Accordingly, the number of novel cDNA and EST sequences is growing quite rapidly. Yet relatively few programs can reliably determine the completeness of EST sequences.
However, there has been recent rapid progress in the development of new methods for determining the 5'-completeness of EST sequences by identifying TISs. This project assesses the problem of EST analysis in the broader context of genomics and gene discovery, reviews the key concepts and relatively new methods for identifying translation initiation sites, as well as comparing the performance of these methods. Our analysis has confirmed that although detection of the presence and extent of an open reading frame is valuable, further information is required to accurately predict TISs in EST data. ESTScan and Diogenes did well in predicting that a sequence contains a CDS, the purpose for which these programs were developed. This capability is distinct from that required to identify start codons, as revealed by their poor performance in identifying the presence and position of TISs in the test set.
A successful method for identifying TISs has been identified in this paper. ATGpr demonstrated relatively high sensitivity, specificity, and overall accuracy in identifying start sites while also rejecting incomplete sequences. Including information on similarity to known protein sequences in later versions of ATGpr indicate that this method can provide more reliable information for annotating EST and cDNA sequences. Furthermore, advanced methods for generating ESTs, such as oligo-capping, which lead to full-length cDNAs, will improve EST databases, ultimately resulting in more reliable analysis and annotation of novel genes.
Methods
UniGene [31] is a system of GenBank sequences partitioned into non-redundant gene clusters. It contains the sequences of well-characterized genes as well as hundreds of thousands of EST sequences. UniGene Build #160: Homo sapiens was the initial sequence source. On February 16, 2003, it contained 111,064 clusters and 4,020,822 EST sequences.
The construction of the dataset is shown in Figure 6. Several filters were simultaneously applied to the data to ensure that the presence or absence of a TIS was known for every EST sequence used. Random selection, when needed, was carried out using a computer-based random number generator [32].
Figure 6.
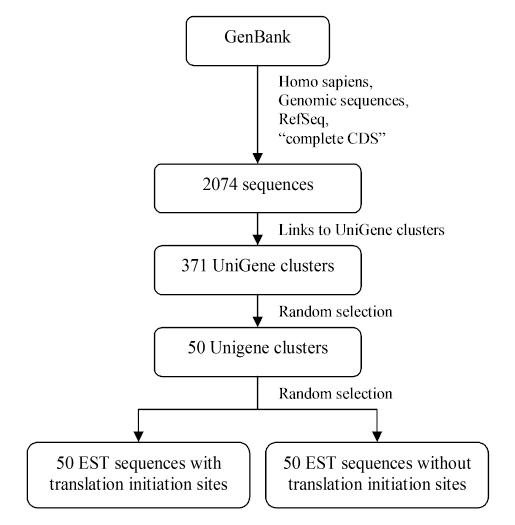
Construction of the EST dataset.
Human genomic sequences containing the annotation "complete CDS" (complete coding sequence) were selected as starting points for gene selection. A filter for RefSeq (NCBI Reference Sequence) entries was applied to ensure that the dataset was non-redundant, up-to-date, and composed of valid entries [33]. The resultant 2074 sequences were filtered to include only those with links to UniGene clusters. Of these 371 UniGene clusters, 50 clusters containing ProtEST links were randomly selected. ProtEST [21] provides protein matches for ESTs, and ensures that the matches exclude conceptual translations by using sequences only from Swissprot, PIR, PDB, and PRF. Finally, from this strict set of UniGene clusters, two 5'-EST sequences were selected randomly from each cluster: one containing the TIS and one lacking the TIS, confirmed by visual alignment with the reference sequence. The type of ESTs generated (5' versus 3') depends on the directionality of the primers used in vitro. A total of 100 EST sequences were used: 50 containing and 50 lacking the TIS.
The EST sequences were entered into the five programs: first-ATG, ESTScan, Diogenes, NetStart, and ATGpr. All of these methods except for first-ATG were accessed via their web sites (see Table 1). First-ATG was performed through Microsoft Excel [34] spreadsheet functions. Performance of each method was measured in terms of sensitivity and specificity of EST 5'-completeness predictions (in other words, presence versus absence of the TIS), and of percentage accuracy of predicting the position of the TIS or lack thereof. With the exception of the first-ATG method, all of the methods report a score along with the prediction. This permits users to employ custom thresholds. The statistical measures described above were calculated across all threshold scores. Statistical analyses were performed using Analyse-it statistical software for Microsoft Excel [35].
Authors' Contributions
AN created the dataset, chose the methods to be analyzed, and performed the analyses; SF conceived of the study and contributed expertise on molecular biology and eukaryotic translational control; LE coordinated and designed it. All authors read and approved the final manuscript.
Supplementary Material
TIS+50.txt, a FASTA format dataset of 50 EST sequences with translation initiation sites.
TIS-50.txt, a FASTA format dataset of 50 EST sequences without translation initiation sites.
Acknowledgments
Acknowledgements
We thank Eric Klee and Stephen Ekker for helpful discussions. This work was supported in part by NIH R01-GM63904.
Contributor Information
Afshin Nadershahi, Email: nade0043@umn.edu.
Scott C Fahrenkrug, Email: fahre001@tc.umn.edu.
Lynda BM Ellis, Email: lynda@tc.umn.edu.
References
- International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Adams MD, Kerlavage AR, Fleischmann RD, Fuldner RA, Bult CJ, Lee NH, Kirkness EF. Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence. Nature. 1995;377:3–174. [PubMed] [Google Scholar]
- Adams MD, Kelley JM, Gocayne JD, Dubnick M, Polymeropoulos MH, Xiao H, Merrill CR, Wu A, Olde B, Moreno RF, Kerlavage AR, McCombie WR, Venter JC. Complementary DNA sequencing: Expressed Sequence Tags and the human genome project. Science. 1991;252:1651–1656. doi: 10.1126/science.2047873. [DOI] [PubMed] [Google Scholar]
- Hatzigeorgiou A. Translation initiation start prediction in human cDNAs with high accuracy. Bioinformatics. 2002;18:343–350. doi: 10.1093/bioinformatics/18.2.343. [DOI] [PubMed] [Google Scholar]
- Kozak M. The scanning model for translation: an update. J Cell Biol. 1989;108:229–241. doi: 10.1083/jcb.108.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–292. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- Kozak M. Possible role of flanking nucleotides in recognition of the AUG initiator codon by eukaryotic ribosomes. Nucleic Acids Res. 1981;9:5233–52. doi: 10.1093/nar/9.20.5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. An analysis of 5'-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987;15:8125–48. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Translation of insulin-related polypeptide from messenger RNAs with tandemly reiterated copies of the ribosome binding site. Cell. 1983;34:971–978. doi: 10.1016/0092-8674(83)90554-8. [DOI] [PubMed] [Google Scholar]
- Cavener DR, Ray SC. Eukaryotic start and stop translation sites. Nucleic Acids Res. 1991;19:3185–92. doi: 10.1093/nar/19.12.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavener DR. Comparison of the consensus sequence flanking translational start sites in Drosophila and vertebrates. Nucleic Acids Res. 1987;15:1353–61. doi: 10.1093/nar/15.4.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL. A Method for Identifying Splice Sites and Translational Start Sites in Eukaryotic mRNA. Computer Applications in the Biosciences (CABIOS) 1997;13:365–376. doi: 10.1093/bioinformatics/13.4.365. [DOI] [PubMed] [Google Scholar]
- Pedersen AG, Nielen H. Neural Network Prediction of Translation Initiation Sites in Eukaryotes: Perspectives for EST and Genome analysis. In: Gasterland T, Karp P, Karplus K, Ouzounis C, Sander C, Valencia A, editor. In Proceedings of the Fifth International Converence on Intelligent Systems for Molecular Biology: 1997; Menlo Park. Vol. 5. American Association for Artificial Intelligence; 1997. pp. 226–233. [PubMed] [Google Scholar]
- Salamov AA, Nishikawa T, Swindells MB. Assessing protein coding region integrity in cDNA sequencing projects. Bioinformatics. 1998;14:384–390. doi: 10.1093/bioinformatics/14.5.384. [DOI] [PubMed] [Google Scholar]
- Iseli C, Jongeneel CV, Bucher P. In Proceedings of the Fifth International Converence on Intelligent Systems for Molecular Biology: 1997; Menlo Park. Vol. 7. American Association for Artificial Intelligence; 1999. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences; pp. 138–148. [PubMed] [Google Scholar]
- Diogenes: reliable ORF-finding in short genomic sequences http://web.ahc.umn.edu/diogenes/
- Burge C, Karlin S. Prediction of Complete Gene Structures in Human Genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- ESTScan2 http://www.ch.embnet.org/software/ESTScan2.html
- Clamp M, Andrews D, Barker D, Bevan P, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, Durbin R, Eyras E, Gilbert J, Hammond M, Hubbard T, Kasprzyk A, Keefe D, Lehvaslaiho H, Iyer V, Melsopp C, Mongin E, Pettett R, Potter S, Rust A, Schmidt E, Searle S, Slater G, Smith J, Spooner W, Stabenau A, Stalker J, Stupka E, Ureta-Vidal A, Vastrik I, Birney E. Ensembl 2002: accommodating comparative genomics. Nucl Acids Res. 2003;31:38–42. doi: 10.1093/nar/gkg083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucl Acids Res. 2003;31:23–27. doi: 10.1093/nar/gkg057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ProtEST – Protein matches for ESTs http://www.ncbi.nlm.nih.gov/UniGene/ProtEST/
- Nakamura M, Masuda H, Horii J, Kuma K, Yokoyama N, Ohba T, Nishitani H, Miyata T, Tanaka M, Nishimoto T. A novel centrosomal protein, RanBPM, when overexpressed, causes ectopic microtubule nucleation, similar to g-tubulin. J Cell Biol. 1998;143:1041–1052. doi: 10.1083/jcb.143.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitani H, Hirose E, Uchimura Y, Nakamura M, Umeda M, Nishii K, Mori N, Nishimoto T. Full-sized RanBPM cDNA encodes a protein possessing a long stretch of proline and glutamine within the N-terminal region, comprising a large protein complex. Gene. 2001;272:25–33. doi: 10.1016/S0378-1119(01)00553-4. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1006/jmbi.1990.9999. [DOI] [PubMed] [Google Scholar]
- Pearson WR. Flexible sequence similarity searching with the FASTA3 program package. Methods Mol Biol. 2000;132:185–219. doi: 10.1385/1-59259-192-2:185. [DOI] [PubMed] [Google Scholar]
- Zien A, Rätsch S, Mika S, Schölkopf B, Lengauer T, Müller KR. Engineering support vector machine kernels that recognize translation initiation sites. Bioinformatics. 2000;16:799–807. doi: 10.1093/bioinformatics/16.9.799. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, Ota T, Isogai T. Prediction whether a human cDNA sequence contains initiation codon by combining statistical information with protein sequences. Bioinformatics. 2000;16:960–967. doi: 10.1093/bioinformatics/16.11.960. [DOI] [PubMed] [Google Scholar]
- Kozak M. Circumstances and mechanisms of inhibition of translation by secondary structure in eukaryotic mRNAs. Mol Cell Biol. 1989;9:5134–5142. doi: 10.1128/mcb.9.11.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 2002;229:1–34. doi: 10.1016/S0378-1119(02)01056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama K, Sugano S. Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides. Gene. 1994;138:171–174. doi: 10.1016/0378-1119(94)90802-8. [DOI] [PubMed] [Google Scholar]
- Pontius JL, Wagner L, Schuler GD. In The NCBI Handbook. Chapter 20-21. Bethesda: National Library of Medicine (US), National Center for Biotechnology Information; 2002. UniGene: A Unified View of the Transcriptome.http://www.ncbi.nlm.nih.gov/books/bookres.fcgi/handbook/ch21d1.pdf [Google Scholar]
- Research Randomizer http://www.randomizer.org/
- Pruitt KD, Tatusova T, Ostell JM. The Reference Sequence (RefSeq) Project. In The NCBI Handbook Bethesda: National Library of Medicine (US), National Center for Biotechnology Information. 2002;Chapter 17-18 http://www.ncbi.nlm.nih.gov/books/bookres.fcgi/handbook/ch18d1.pdf [Google Scholar]
- Microsoft Twin Cities. 8300 Norman Center Drive, Suite 950, Bloomington, Minnesota 5 USA. http://office.microsoft.com/excel/
- Analyse-It Software, Ltd. PO Box 103, Leeds LS27 7WZ, England, United Kingdom. http://www.analyse-it.com/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TIS+50.txt, a FASTA format dataset of 50 EST sequences with translation initiation sites.
TIS-50.txt, a FASTA format dataset of 50 EST sequences without translation initiation sites.