

Abstract
Background
Information obtained by DNA microarray technology gives a rough snapshot of the transcriptome state, i.e., the expression level of all the genes expressed in a cell population at any given time. One of the challenging questions raised by the tremendous amount of microarray data is to identify groups of co-regulated genes and to understand their role in cell functions.
Results
MiCoViTo (Microarray Comparison Visualization Tool) is a set of biologists' tools for exploring, comparing and visualizing changes in the yeast transcriptome by a gene-centric approach. A relational database includes data linked to genome expression and graphical output makes it easy to visualize clusters of co-expressed genes in the context of available biological information. To this aim, upload of personal data is possible and microarray data from fifty publications dedicated to S. cerevisiae are provided on-line. A web interface guides the biologist during the usage of this tool and is freely accessible at http://www.transcriptome.ens.fr/micovito/.
Conclusions
MiCoViTo offers an easy-to-read picture of local transcriptional changes connected to current biological knowledge. This should help biologists to mine yeast microarray data and better understand the underlying biology. We plan to add functional annotations from other organisms. That would allow inter-species comparison of transcriptomes via orthology tables.
Keywords: microarray, functional categories, Saccharomyces cerevisiae, clustering
Background
Although the genome is mostly invariant in each cell of an organism, genes can have different expression patterns related to environmental conditions or developmental programs. For a given genome, different transcriptome states can be observed, depending on complex networks regulating adaptation and homeostasis. Fundamental questions about the topology of networks as protein interactome [1], metabolome [2] or transcriptional regulation networks [3,4] have been previously addressed. Interfaces like KEGG [5], Biocyc [6] or GenMapp [7] help biologists to put these results into a cellular context by mapping expression states onto metabolic network representations. Moreover, to visualize and edit networks, different tools like Cytoscape [8] or Osprey [9] are available. To understand the biology of the studied systems better, the trend is clearly towards the aggregation of multiple sources of biological information.
This article focuses on the analysis of transcriptome states. Numerous microarray gene expression datasets are now available, giving the opportunity to get a picture of the transcriptome state in various cellular conditions. Various methods to mine compendia of transcriptome states and to try to understand their biological meaning already exist. One of the most successful is based on the assumption that genes having similar expression profiles across a set of conditions are likely to be involved in the same biological process [10]. Many approaches, including multivariate analysis, hierarchical clustering and SOM have been used to find patterns of expression and to create biologically relevant clusters (see [11] for review). By analysing whole microarray datasets, these "global" clustering approaches have already led to the formulation of interesting testable predictions [12]. However, it has been previously emphasized that classical clustering methods could be inefficient on large number of biologically unrelated datasets [13]. Indeed, in response to environmental changes, gene expression is modified only in a fraction of the transcriptome and the signal is hence diluted over the whole dataset. Few methods have been proposed to find these regulatory sub-signatures [13,14], and in fine, the most reliable way to go deeper into the data to capture interesting trends is to be an expert in the field. That is why tools allowing the biologist to mine microarray results, in order to find such expression modifications in a sub-set of genes related to his area of expertise, are highly desirable. Such "gene-centric" clustering analysis that distinguishes differentially-expressed genes in specific parts of the transcriptome (centred around a "seed gene") should overcome some drawbacks of global analysis approaches.
The ideal tool would be gene-centric, and would provide understandable outputs mapping biological knowledge onto results. We present here MiCoViTo, a user-friendly tool for identifying and visualizing groups of genes having similar expression in two sets of microarray experiments representing two distinct transcriptome states.
Implementation
Principle
A given transcriptome state can be represented as a network where genes are joined pairwise by a weighted link proportional to a similarity measure between their corresponding expression profiles. The basic idea is therefore to compare the immediate transcriptome neighbourhood of a given gene (referred to hereafter as the seed gene) in two sets of microarray experiments describing two distinct transcriptome states. By neighbourhood, we mean genes whose expression profiles are closely related to that of the seed according to a chosen metric. By relaxing step by step the distance criterion, several neighbourhood levels, corresponding to larger and larger parts of the transcriptome state, can be defined.
Thus, for each set of microarray experiments to be compared, each gene is assigned to the neighbourhood level reflecting the distance between its expression profile and the expression profile of the seed gene. In a second step, all neighbourhood level intersections are computed and arranged in a table (Figure 1, A). In this table, the upper left groups are genes having similar expression in both experiments compared to the seed profile while the upper right and the lower left groups include genes that have very close expression profiles in only one set of experiments. Only the upper left part of the table is of interest, as we do not want to consider genes that are co-expressed with the seed gene in none of the two transcriptome states. Therefore, the lower right part of the table is not analysed and is coloured in grey (Figure 1, A).
Figure 1.
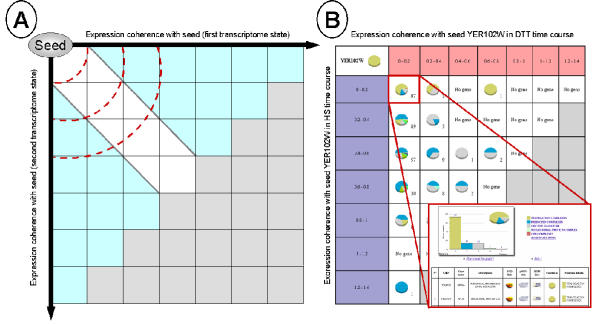
MiCoViTo principle A) Neighbourhood comparison around a seed in two transcriptome states. The location of genes gives an idea of their expression compared to the seed gene. The X axis represents the neighbourhood levels computed in the first transcriptome state, the Y axis represents the neighbourhood levels computed in the second transcriptome state. Regions that are closer to the origin reflect more closely co-expressed genes (delimited by red dash lines). The white region is the area containing genes correlated in both microarray datasets while the blue area contains genes that are co-expressed only in one of the conditions. As we do not want to consider genes that are far away from the seed gene in both transcriptome states, the lower right part of the table is not analysed (grey region). B) Result of the comparison of S. cerevisiae transcriptome state time courses during a 30°C to 37°C temperature shift and during exposure to the reducing agent dithiotreitol (DTT) [29] available in the online tutorial. The comparison was performed using a structural constituent of a ribosome subunit (RPS8B/YER102W gene) as seed and Pearson distance as the distance metric. The step value used to define neigbourhood levels is 0.2 (see intervals 0–0.2, 0.2–0.4, ..., 1.2–1.4). Each group of genes located in a neighbourhood intersection is given in the form of a clickable pie chart constructed according to the MIPS functional classification catalogue [15].
Note that MiCoViTo has been designed so that different types of comparisons can be performed, according to the given biological question. Neighbourhood comparison of one seed in two transcriptome states is described above (Figure 1). But, using the same principle, more elaborate comparisons can be performed like neighbourhood comparison of two distinct seeds in the same transcriptome state (Figure 2, A).
Figure 2.
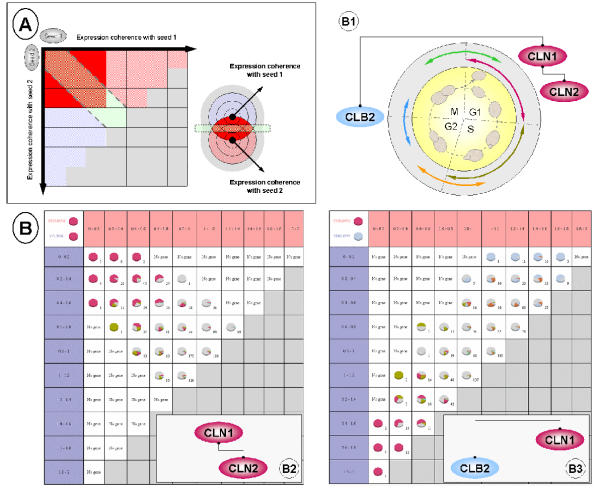
Example of study A) Neighbourhood comparison using two distinct seeds and a single transcriptome state. The results depend on the relative position of the seed genes in the overall transcriptome. This example shows the best-case scenario: two genes close to each other, with a circular gradient of expression coherence. This leads to the definition of zone of interest, represented here in red, which contains genes that are close to both seed genes. These genes are the other partners of the cluster defined by seeds 1 and 2. B) Results of the neighbourhood comparison of the two distincts seeds CLN1/CLN2 (B2) and CLN1/CLB2 (B3) in the Cho et al. yeast cell cycle datasets [23]. CLN1 and CLN2 are both involved in the G1 phase whereas CLB2 is involved in the G2/M transition (B1). Each induced group of genes is represented in the form of a pie chart showing gene distribution in each cell cycle phase (color code is provided in B1 diagram, red: G1; dark green: S; orange: S/G2; blue: G2/M; light green: M/G1).
Visualization and mining of isolated clusters
In order to capture the biological meaning of gene clusters generated by MiCoViTo, it is necessary as a first step to visualize them in the context of current biological knowledge. Latter options for studying promising clusters are to map list of genes onto the metabolic pathways, to look for co-regulation in another expression dataset or to look for common cis-regulatory elements in promoters. Using a relational database system storing yeast MIPS functional information and external links, MiCoViTo provides access to all this information.
For each neighbourhood intersection constructed as detailed above, a pie chart representing the gene distribution in one of the MIPS catalogues [15] (functional classification, protein class, EC number, PROSITE motifs, mutant phenotype and complexes catalogue) is displayed (Figure 1, B). When gene listing for one cluster is requested, additional information is available including direct links to the individual gene description pages of the SGD [16] and MIPS [15]. Furthermore, full gene lists can be posted directly to other online tools like KEGG for metabolism mapping [5], RSA tools for the discovery cis-regulatory motifs [17], SGD for Gene Ontology term mapping [16] or yMGV database for expression mining [18].
Datasets available
Microarray data standards are now available [19] and public repositories have been constructed [20,21]. The community effort will ensure that more and more clean and formatted datasets become available for this kind of study. Here we use data from yMGV [18], the largest available yeast expression database.
More than fifty previously-published microarray datasets are provided, giving a fair coverage of possible yeast transcriptome states (a listing is available in the web site). But users can also upload files onto the site to confront their results with published data or use MiCoViTo to compare two personal datasets.
Technical information
MiCoViTo is composed of three parts: a set of programs for microarray data preprocessing and for comparing expression profiles, a web-interface and a relational database. All the softwares used to power MiCoViTo are freely distributed under an open source licence. Data pre-processing options such as pre-scaling or pre-centering expression profiles have been written in PERL while distance computation routines (Pearson distance, Squared Pearson distance and Euclidean distance) have been written in C in order to minimize computation time. The interface has been written in PHP. MIPS and SGD information is stored in a PostgreSQL relational database. Microarray data are stored in flat files. All graphical outputs are dynamically generated using the JpGraph PHP library [22].
Results
As an illustration, we used MiCoViTo to compare genes co-expressed during the cell cycle (Cho el al. dataset [23]) for two pairs of cyclin: CLN1/CLN2 and CLN1/CLB2 (Figure 2, B1). Results depend mainly on the proximity of seed expressions. Indeed, two genes close one to another like CLN1 and CLN2 (both implicated in the G1 phase), lead to the identification of numerous correlated genes also implicated in the G1 phase (Figure 2, B2) like PCL1 [24], MCD1 [25] or RNR1 [26]. But if CLN1 is compared to a gene implicated in another phase of the cycle like CLB2 (G2/M transition), no correlation at all is observed (Figure 2, B3). Moreover, the upper right and the lower left groups show genes implicated in the G2/M transition and the G1 phase like CLB2 and CLN1 respectively.
Discussion
MiCoViTo aims to help biologists achieve an intuitive understanding of their own data using an approach based on the comparison of sets of microarray experiments. The user-friendly web interface is designed to be accessible to those with no particular technical skill.
The major drawback of visualization tools based on biologists' knowledge and intuition is that it does not give a computer readable output and cannot be applied systematically to find interesting expression patterns. Particularly, seed choice is a crucial point of our approach because it precisely defines which part of the transcriptome to focus on. The gene-centric approach presented in this paper empowers biologists to focus on a particular sub-part of the transcriptome. In order to assist seed selection process, online lists of candidates for each set of microarray experiments are proposed. Those candidates are ranked according to their neighbourhood density (density is the number of genes co-expressed with seed according to a given expression distance threshold). Genes with a large neighbourhood density are those whose nearest neighbourhoods contain a lot of genes, which means that their expression profiles are similar to the expression of many other genes. Such information may be used as a starting point when no prior information about the transcriptome state topology is available.
As initiated in a recent paper [27], one possible future direction is to use this approach to compare transcriptomes from different organisms captured in the same state. This will be possible if we can find functional annotation in the same format for two organisms and if we are able to define pairs of seeds having inter-organism correspondence. An annotation project like GO [28] addresses the first question. To define relevant inter-organism seeds, two ways appear realistic. The first one is to assume that sequence-based conservation implies function conservation. This is not always the case, but one can expect MiCoViTo results to be incoherent if seeds are not functionally related. Alternatively, one can start from pairs of genes previously described as orthologous. S. pombe could provide a good benchmark system to test this approach, since GO annotations are available and microarray datasets describing states previously studied in S. cerevisiae are publicly available. Moreover well-characterised S. cerevisiae/S. Pombe orthologous genes have been listed by the Sanger Center (Valerie Wood, personnal communication).
Conclusions
MiCoViTo allows users to compare and visualize different transcriptome states in a gene-centric way. The generated clusters of genes are mapped onto existing biological knowledge to gain a higher level view of the ongoing transcriptional changes. Upload of personal data onto the site is possible but not mandatory since a compendium of more than fifty yeast microarray datasets is available.
At present, this tool is restricted to S. cerevisiae, but a natural future direction will be to incorporate data originating from different species as well as orthology tables to allow comparison of seeds from different organisms.
Availability and requirements
MiCoViTo is available at http://www.transcriptome.ens.fr/micovito/. The source code and database scheme are freely distributed to academic users upon request to the authors.
A more detailed description of MiCoViTo including a step by step tutorial can be found online.
List of abbreviations
MiCoViTo: Microarray Comparison Visualization Tool; DTT: dithitreitol; MIPS: Munich Information center for Protein Sequences; RSA tools: Regulatory Sequence Analysis Tools; SGD: Saccharomyces Genome Database; TF: transcription factor; yMGV: yeast Microarray Global Viewer.
Authors' contributions
GL conceived, implemented MiCoViTo and drafted the manuscript, PMsuggested the first version of the visualization interface, contributed todiscussions and drafted the manuscript, PV and CJ are GL's Ph.D. advisors, they both contributed to discussions, SV provided help with algorithms and coordinated the project. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
The authors want to thanks Stéphane Le Crom, Frédéric Devaux and Serge Hazout for helpful discussions and Boris Barbour for English correction. The MiCoViTo project was funded by the Programme Bioinformatique Inter-EPST-CNRS 2003, GL is supported by a MENRT, PM is supported by the French Therapeutical Research Association (AFRT) and the PhRMA foundation Center of Excellence in Integration of Genomics and Informatics (CEIGI), SV is supported by Hoechst Marion Roussel – Aventis grant number FRHMR2/9908.
Contributor Information
Gaëlle Lelandais, Email: lelandais@biologie.ens.fr.
Philippe Marc, Email: pmarc@genetics.med.harvard.edu.
Pierre Vincens, Email: vincens@biologie.ens.fr.
Claude Jacq, Email: jacq@biologie.ens.fr.
Stéphane Vialette, Email: vialette@biologie.ens.fr.
References
- Bu D, Zhao Y, Cai L, Xue H, Zhu X, Lu H, Zhang J, Sun S, Ling L, Zhang N, Li G, Chen R. Topological structure analysis of the protein-protein interaction network in budding yeast. Nucleic Acids Res. 2003;31:2443–2450. doi: 10.1093/nar/gkg340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Tombor B, Albert R, Oltvai ZN, Barabasi AL. The large-scale organization of metabolic networks. Nature. 2000;407:651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- Guelzim N, Bottani S, Bourgine P, Kepes F. Topological and causal structure of the yeast transcriptional regulatory network. Nat Genet. 2002;31:60–63. doi: 10.1038/ng873. [DOI] [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, Zeitlinger J, Jennings EG, Murray HL, Gordon DB, Ren B, Wyrick JJ, Tagne JB, Volkert TL, Fraenkel E, Gifford DK, Young RA. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Kawashima S, Nakaya A. The KEGG databases at GenomeNet. Nucleic Acids Res. 2002;30:42–46. doi: 10.1093/nar/30.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp PD, Riley M, Paley SM, Pellegrini-Toole A. The MetaCyc Database. Nucleic Acids Res. 2002;30:59–61. doi: 10.1093/nar/30.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlquist KD, Salomonis N, Vranizan K, Lawlor SC, Conklin BR. GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat Genet. 2002;31:19–20. doi: 10.1038/ng0502-19. [DOI] [PubMed] [Google Scholar]
- Ideker T, Ozier O, Schwikowski B, Siegel AF. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 2002;18 Suppl 1:S233–40. doi: 10.1093/bioinformatics/18.suppl_1.s233. [DOI] [PubMed] [Google Scholar]
- Breitkreutz BJ, Stark C, Tyers M. Osprey: a network visualization system. Genome Biol. 2003;4:R22. doi: 10.1186/gb-2003-4-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quackenbush J. Computational analysis of microarray data. Nat Rev Genet. 2001;2:418–427. doi: 10.1038/35076576. [DOI] [PubMed] [Google Scholar]
- Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM. Systematic determination of genetic network architecture. Nat Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- Gasch AP, Eisen MB. Exploring the conditional coregulation of yeast gene expression through fuzzy k-means clustering. Genome Biol. 2002;3:RESEARCH0059. doi: 10.1186/gb-2002-3-11-research0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Church GM. Biclustering of expression data. Proc Int Conf Intell Syst Mol Biol. 2000;8:93–103. [PubMed] [Google Scholar]
- Mewes HW, Frishman D, Guldener U, Mannhaupt G, Mayer K, Mokrejs M, Morgenstern B, Munsterkotter M, Rudd S, Weil B. MIPS: a database for genomes and protein sequences. Nucleic Acids Res. 2002;30:31–34. doi: 10.1093/nar/30.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwight SS, Harris MA, Dolinski K, Ball CA, Binkley G, Christie KR, Fisk DG, Issel-Tarver L, Schroeder M, Sherlock G, Sethuraman A, Weng S, Botstein D, Cherry JM. Saccharomyces Genome Database (SGD) provides secondary gene annotation using the Gene Ontology (GO) Nucleic Acids Res. 2002;30:69–72. doi: 10.1093/nar/30.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helden J. Regulatory sequence analysis tools. Nucleic Acids Res. 2003;31:3593–3596. doi: 10.1093/nar/gkg567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Crom S, Devaux F, Jacq C, Marc P. yMGV: helping biologists with yeast microarray data mining. Nucleic Acids Res. 2002;30:76–79. doi: 10.1093/nar/30.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, Aach J, Ansorge W, Ball CA, Causton HC, Gaasterland T, Glenisson P, Holstege FC, Kim IF, Markowitz V, Matese JC, Parkinson H, Robinson A, Sarkans U, Schulze-Kremer S, Stewart J, Taylor R, Vilo J, Vingron M. Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nat Genet. 2001;29:365–371. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazma A, Parkinson H, Sarkans U, Shojatalab M, Vilo J, Abeygunawardena N, Holloway E, Kapushesky M, Kemmeren P, Lara GG, Oezcimen A, Rocca-Serra P, Sansone SA. ArrayExpress--a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 2003;31:68–71. doi: 10.1093/nar/gkg091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JpGraph - An 00 Graph library for PHP4 http://www.aditus.nu/jpgraph
- Cho RJ, Campbell MJ, Winzeler EA, Steinmetz L, Conway A, Wodicka L, Wolfsberg TG, Gabrielian AE, Landsman D, Lockhart DJ, Davis RW. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell. 1998;2:65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- Ogas J, Andrews BJ, Herskowitz I. Transcriptional activation of CLN1, CLN2, and a putative new G1 cyclin (HCS26) by SWI4, a positive regulator of G1-specific transcription. Cell. 1991;66:1015–1026. doi: 10.1016/0092-8674(91)90445-5. [DOI] [PubMed] [Google Scholar]
- Guacci V, Koshland D, Strunnikov A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell. 1997;91:47–57. doi: 10.1016/S0092-8674(01)80008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge SJ, Davis RW. Two genes differentially regulated in the cell cycle and by DNA-damaging agents encode alternative regulatory subunits of ribonucleotide reductase. Genes Dev. 1990;4:740–751. doi: 10.1101/gad.4.5.740. [DOI] [PubMed] [Google Scholar]
- Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP, Werner-Washburne M. The genomics of yeast responses to environmental stress and starvation. Funct Integr Genomics. 2002;2:181–192. doi: 10.1007/s10142-002-0058-2. [DOI] [PubMed] [Google Scholar]