

Abstract
Objective
On the luminal surface of injured arteries, platelet activation and leukocyte-platelet interactions are critical for the initiation and progression of arterial restenosis. The transcription factor nuclear factor kappa B (NF-κB) is a critical molecule in platelet activation. Here, we investigated the role of the platelet NF-κB pathway in forming arterial neointima after arterial injury.
Methods and Results
We performed carotid artery wire injuries in LDL receptor-deficient (LDLR–/–) mice with a platelet-specific deletion of IκB kinase beta (IKKβ) (IKKβfl/fl/PF4cre/LDLR–/–) and in control mice (IKKβfl/fl/LDLR–/–). The size of the arterial neointima was 61% larger in the IKKβfl/fl/PF4cre/LDLR–/– mice compared to the littermate control IKKβfl/fl/LDLR–/– mice. Compared to the control mice, the IKKβfl/fl/PF4cre/LDLR–/– mice exhibited more leukocyte adhesion at the injured area. The extent of GPIbα shedding after platelet activation was compromised in the IKKβ-deficient platelets. This effect was associated with a low level of the active form of A Disintegrin And Metalloproteinase 17 (ADAM17), the key enzyme involved in mediating GPIbα shedding in activated IKKβ-deficient platelets.
Conclusions
Platelet IKKβ deficiency increases the formation of injury-induced arterial neointima formation. Thus, NF-κB-related inhibitors should be carefully evaluated for use in patients after an arterial intervention.
Keywords: restenosis, arterial injury, platelets, leukocytes, NF-κB
Introduction
Neointimal hyperplasia following percutaneous interventions, such as balloon angioplasty or stenting, is the principal cause of arterial restenosis.1 Platelet deposition and subsequent leukocyte-platelet interactions on the injured luminal surface are critical in the repair process after arterial damage.2, 3, 4, 5 Although platelets are anucleate cells, they contain nearly all of the NF-κB family members.6 NF-κB plays a complex role in platelet activation. NF-κB inhibitors impair platelet aggregation mediated by ADP, collagen and thrombin and reduce ATP release, thromboxane B2 formation and P-selectin expression stimulated by thrombin.7 However, arachidonic acid-induced platelet activation is not affected by NF-κB inhibitors.7 The common pathway that regulates the activation of NF-κB is based on the degradation of IκB from the NF-κB complex, a process initiated by IκB phosphorylation. The latter is catalyzed by a multisubunit protein kinase called IκB kinase (IKK).8 IKKβ is more active than other IKK subunits in catalyzing IκB phosphorylation.9 The loss of IKKβ dramatically inhibits NF-κB activation, resulting in embryonic lethality in mice.10
We were interested in determining whether the inhibition of platelet NF-κB activation could suppress neointima formation after arterial injury. Using floxed IKKβ (IKKβfl/fl) mice, we generated mice with IKKβ deficiency specifically in platelets by breeding PF4-Cre (PF4cre) mice with these floxed mice and breeding the resulting mice with LDLR–/– mice. The role of platelet IKKβ in arterial neointima formation was evaluated by comparing the size of the neointima and leukocyte-platelet interactions in the injured arteries. Furthermore, the underlying mechanisms contributing to the change in the leukocyte-platelet interactions were explored.
Methods
The PF4-cre mice 11 and the floxed-IKKβ mice 12, provided by the groups of Drs. Radek C. Skoda and Michael Karin, were bred. A mouse 384 single nucleotide polymorphism panel (markers spread across the genome at approximately 7-Mbp intervals; Charles River Laboratories International, Inc., Wilmington, MA) was used to characterize the genetic background of the breeders. Polymorphic markers demonstrated that the IKKβfl/flPF4cre breeders were 98.17% C57BL/6. The LDLR-/- mice (002207) from the Jackson Lab were bred with IKKβfl/flPF4cre to generate IKKβfl/flPF4creLDLR-/- mice and their littermate IKKβfl/flLDLR-/- mice. The breeding scheme is detailed in Supplemental Figure I.
The mice were subjected to a guide wire injury in the carotid artery, and the size of the arterial neointima was examined using a method previously described.3, 13, 14 Leukocyte interactions with the injured arteries were studied by immunostaining the arteries with specific leukocyte markers or directly observing the injured arteries in vivo by intravital epifluorescence microscopy.3 All of the animal experiments and care were approved by the Georgia Health Sciences University Animal Care and Use Committee, in accordance with AAALAC guidelines. The data are presented as the mean ± SEM and were analyzed by either one-way ANOVA followed by a Bonferroni correction post-hoc test or Student's t-test to evaluate two-tailed levels of significance. The null hypothesis was rejected at P < 0.05.
Results
Platelet IKKβ deficiency increases neointimal formation in LDLR–/– mice
IKKβfl/fl/PF4cre/LDLR–/– mice and their littermate control IKKβfl/fl/LDLR–/– mice were fed a Western diet for two weeks, followed by carotid artery wire injury. Four weeks later, the arteries were excised and processed for analysis. The two groups of mice were identical in body weight, blood cholesterol levels, and number of peripheral leukocytes (Supplemental Tables I and II). The number of platelets in the IKKβfl/fl/PF4cre/LDLR–/– mice exhibited a decreasing trend, but this trend did not reach statistical significance when compared to that in IKKβfl/fl/LDLR–/– mice (Supplemental Table I). Surprisingly, the size of the neointima lesions was 61% larger in the IKKβfl/fl/PF4cre/LDLR–/– mice compared to the IKKβfl/fl/LDLR–/– mice (Figure 1A). The size of the media was also increased in the IKKβfl/fl/PF4cre/LDLR–/– mice compared to the IKKβfl/fl/LDLR–/– mice (Figure 1A). Notably, the ratio of the intima to the media (I/M) was markedly elevated in the IKKβfl/fl/PF4cre/LDLR–/– mice compared to the IKKβfl/fl/LDLR–/– mice (Supplemental Figure II). Macrophages in the injured carotid arteries were stained with F4/80 and found to be significantly increased in the IKKβfl/fl/PF4cre/LDLR–/– mice compared to the control mice (Figure 1B).
Figure 1.
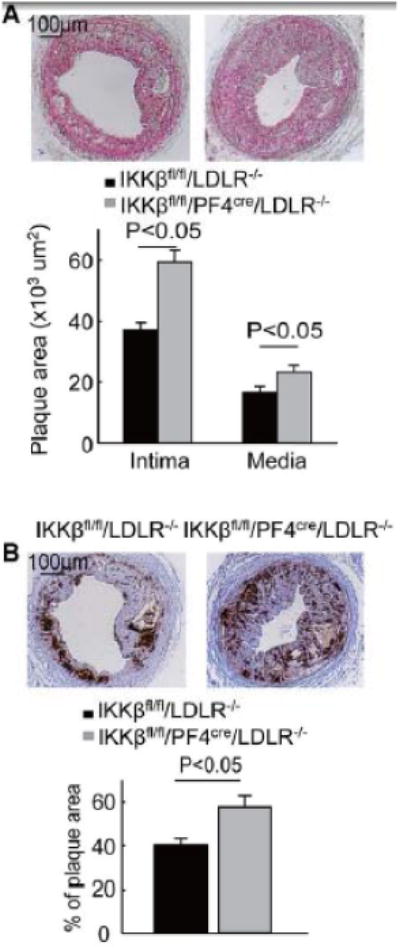
IKKβ deficiency in platelets augments injury-induced arterial neointima formation in LDLR–/– mice. A, Movat pentachrome staining of the arterial neointima 4 weeks after injury; the size of the neointima and media were measured (n = 12 for each group). B, Immunostaining (with anti-F4/80) of the infiltrating macrophages in the arterial neointima. Twelve cross sections of each mouse carotid artery were analyzed. The percent positive area was calculated by dividing the positive area by the measured lesion area. Twelve injured carotid arteries were included for each group.
To exclude the effect of IKKβ deficiency in other PF4-expressing cells on the formation of arterial neointima,15 we used bone marrow transplantation to generate LDLR–/– chimeric mice that lacked IKKβ only in their platelets and the control mice that retained IKKβ in their platelets. The carotid arteries of both groups were injured with a guide wire. Four weeks after the injury, we observed that the neointimal lesions were 64% larger in the LDLR–/– mice that received bone marrow from the IKKβfl/fl/PF4cre/LDLR–/– mice compared to the control mice. The arterial neointimas from the LDLR–/– mice that received bone marrow from IKKβfl/fl/PF4cre/LDLR–/– mice also stained more strongly with a macrophage marker (Supplemental Figure III A and B).
Platelet IKKβ deficiency increases leukocyte adhesion to injured carotid arteries and platelet-leukocyte aggregates
To evaluate the leukocyte interactions with the injured arteries, arterial cross-sections were immunostained with specific markers for platelets, neutrophils, and monocytes. One hour after the carotid arterial injury, platelets and leukocytes covered the denuded luminal surface (Figure 2A and 2B), and almost no monocytes were observed on the denuded luminal surface (data not shown). The number of platelets covering the injured area was not significantly different between the IKKβfl/fl/PF4cre/LDLR–/– and IKKβfl/fl/LDLR–/– mice (Figure 2A). However, considerably more neutrophils were adhered to the injured area of the carotid arteries in the IKKβfl/fl/PF4cre/LDLR–/– mice compared to the IKKβfl/fl/LDLR–/– mice (Figure 2B). Seven days after the wire injury, more infiltrating macrophages were observed in the injured carotid arteries of the IKKβfl/fl/PF4cre/LDLR–/– mice than in the IKKβfl/fl/LDLR–/– mice (Figure 2C).
Figure 2.
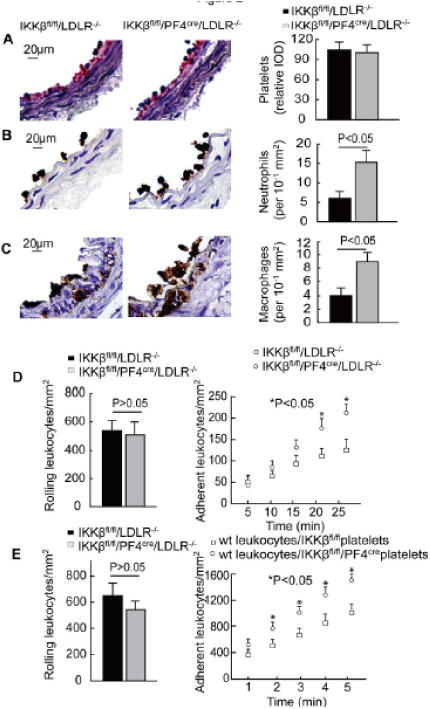
IKKβ deficiency in platelets increases leukocyte interactions with the injured arteries. A to C, The carotid arteries collected at 1 hour after wire injury were stained for platelets (A) and neutrophils (B), and the carotid arteries collected 7 days after the wire injury were stained for macrophages (C) (n = 5). D, Leukocyte rolling and adhesion in the injured carotid arteries within the first 25 min after injury (n = 5). E, Leukocyte rolling and adhesion on the activated IKKβfl/fl or IKKβfl/fl/PF4cre platelets in the microflow chambers (n = 5).
Next, we examined the interactions of the leukocytes with the injured mouse carotid arteries in vivo by using intravital epifluorescence microscopy. The circulating leukocytes, which were labeled with rhodamine 6G, rolled on and adhered to the injured vessel wall after the wire injury. The number of leukocytes rolling on the arterial wall did not differ between the IKKβfl/fl/PF4cre/LDLR–/– and IKKβfl/fl/LDLR–/– mice (Figure 2D). During the early stage after the arterial injury, the number of leukocytes adhering to the arterial wall was not markedly different between the 2 groups. Twenty minutes later, more leukocytes had adhered to the arterial walls of the IKKβfl/fl/PF4cre/LDLR–/– mice compared to the IKKβfl/fl/LDLR–/– mice (Figure 2D).
To further define the binding affinity of the IKKβ-deficient and control platelets to leukocytes, an ex vivo micro-flow chamber was used.3, 14 The chamber was coated with thrombin-activated IKKβ-deficient and control platelets, which were isolated from IKKβfl/fl/PF4cre or IKKβfl/fl mice. Then, whole blood from the wild-type mice was allowed to flow through the chamber. Interestingly, the number of rolling wild-type leukocytes on the chamber coated with activated IKKβ-deficient platelets was low, but these levels did not reach significance when compared to the chamber coated with control platelets. However, the number of adhering leukocytes was much greater in the chamber coated with IKKβ-deficient platelets compared to the chamber coated with control platelets (Figure 2E).
To evaluate the role of platelet IKKβ deficiency in mediating platelet-leukocyte interactions in the circulation, we examined the mouse blood leukocyte population by flow cytometry. Both platelet-neutrophil and platelet-monocyte aggregates were increased 4 to 5 times in the IKKβfl/fl/PF4cre/LDLR–/– mice compared to the IKKβfl/fl/LDLR–/– mice (Supplemental Figure IV). Within those aggregates, the level of GPIbα expression was remarkably higher in IKKβfl/fl/PF4cre/LDLR–/– mice (Supplemental Figure IV). Leukocyte Mac-1 (αMβ2, CD11b/CD18) and platelet GPIbα are critically involved in the formation of leukocyte-platelet aggregates.16-18 This result suggests that platelet IKKβ deficiency enhances platelet-leukocyte aggregation due to the elevated level of GPIbα on IKKβ-deficient platelets.
Platelet IKKβ deficiency decreases platelet activation, secretion and aggregation
To evaluate the role of IKKβ deficiency in platelet function, platelets from IKKβfl/fl/PF4cre mice and their IKKβfl/fl littermates were stimulated with thrombin. Flow cytometry revealed that there was no significant difference in P-selectin expression between the resting IKKβ-deficient platelets and the control platelets (Figure 3A). However, P-selectin expression induced with 0.1 U/ml thrombin was diminished in the IKKβ-deficient platelets compared to the control platelets (Figure 3A). In addition, Lumi-Aggregometer-based experiments demonstrated that IKKβ-deficient platelets exhibited reduced ATP release and aggregation compared to control platelets when stimulated with thrombin (Figure 3B). Electron microscopy also indicated that the release of α-granules and platelet aggregation were attenuated in the IKKβ-deficient platelets compared to the control platelets after stimulation with thrombin for 10 minutes (Figure 3C).
Figure 3.
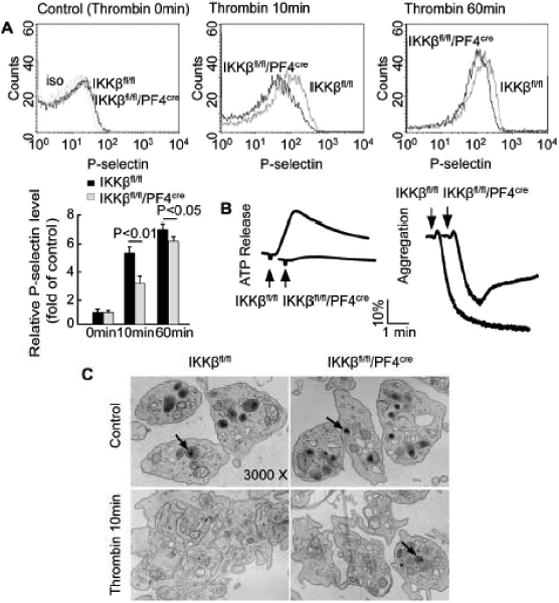
IKKβ deficiency in platelets decreases platelet activation, secretion and aggregation. The washed platelets from the IKKβfl/fl/PF4cre and the IKKβfl/fl mice were stimulated with or without thrombin. A, Platelet P-selectin expression was analyzed by flow cytometry after stimulation with 0.1 U/ml thrombin for 0, 10, and 60 min at 22°C, and the relative expression of P-selectin (mean fluorescence intensity) was compared (n = 5). B, ATP release and platelet aggregation were examined in a Lumi-Aggregometer after the platelets were incubated with 0.025 U/ml thrombin. The data represent 5 independent experiments. C, Platelet aggregation and α-granule release (arrowhead) were evaluated by electron microscopy after incubation with 0.1 U/ml thrombin for 10 min at 22°C. The data represent 5 independent experiments.
IKKβ deficiency attenuates GPIbα shedding in platelets
Platelet GPIbα plays an important role in the interaction between platelets and leukocytes.16-18 We examined the level of GPIbα on IKKβ-deficient platelets. Platelets were isolated from IKKβfl/fl/PF4cre mice and their littermate IKKβfl/fl mice. Flow cytometry (Figure 4A) and western blot analyses (Figure 4B) demonstrated that there was no significant difference in GPIbα expression between the resting IKKβ-deficient platelets and the control platelets. Stimulation with thrombin caused glycoprotein shedding from the surface of the platelets. Indeed, thrombin treatment led to rapid GPIbα shedding from the IKKβfl/fl platelets in a time-dependent manner. However, IKKβ deficiency significantly attenuated the extent of GPIbα shedding (Figure 4A and 4B). Flow cytometry demonstrated slow shedding of GPV in IKKβ-deficient platelets compared to control platelets (Supplemental Figure VA). Interestingly, stimulation with thrombin did not change the surface levels of GPVI (Supplemental Figure VB), GPIX (Supplemental Figure VC) or αIIbβ3 (Supplemental Figure VD) on the platelets. ADP (5 μM) and collagen IV (20 μg/ml) also caused GPIbα shedding, but no difference was observed between the IKKβ-deficient and control platelets (data not shown). No difference was observed in the protein levels of GPV, GPVI, GPIX or αIIbβ3 on the membrane of the platelets following stimulation with ADP or collagen (data not shown). These results demonstrate that platelet IKKβ plays a critical role in GPIbα and GPV shedding following thrombin stimulation.
Figure 4.
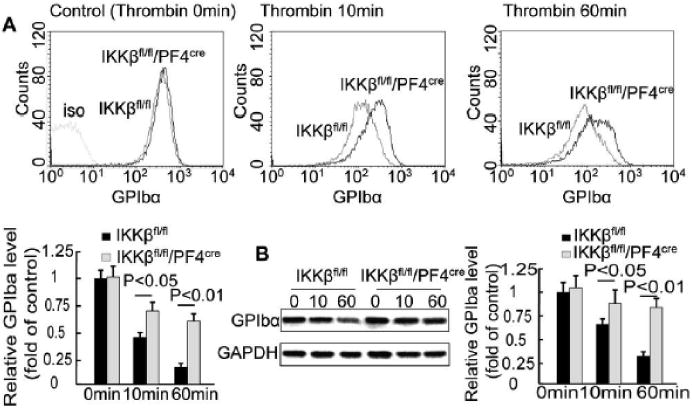
IKKβ deficiency inhibits thrombin-induced GPIbα shedding in platelets. Platelets isolated from IKKβfl/fl/PF4cre and IKKβfl/fl mice were stimulated with 0.1 U/ml thrombin. A, Representative flow cytometric histograms and the relative expression of GPIbα at different times after stimulation (n = 5). B, An immunoblot of platelet GPIbα at different times after stimulation (n = 5).
Blocking the GPIbα binding site on Mac-1 eliminates increased interactions between IKKβ-deficient platelets and leukocytes
To further determine whether the increased leukocyte adhesion to the injured arteries was due to the reduced GPIbα shedding in IKKβfl/fl/PF4cre/LDLR–/– mice, we utilized an anti-M2 antibody that selectively blocks GPIbα binding site on Mac-1 in mouse injury models.18 The carotid arteries collected from mice at one hour after the carotid arterial injury were examined by histology. The number of platelets covering the injured area was not significantly different between the IKKβfl/fl/PF4cre/LDLR–/– and the IKKβfl/fl/LDLR–/– mice treated with the control IgG or the anti-M2 antibody (Figure 5A). However, treatment with the anti-M2 antibody significantly reduced neutrophil adherence to the injured area of the carotid arteries in both the IKKβfl/fl/PF4cre/LDLR–/– mice and the IKKβfl/fl/LDLR–/– mice compared to mice treated with control IgG (Figure 5B). More importantly, the increased leukocyte adhesion observed in the control IgG-treated IKKβfl/fl/PF4cre/LDLR–/– mice compared to the control IgG-treated IKKβfl/fl/LDLR–/– mice was abolished when these mice were treated with the anti-M2 antibody (Figure 5B). The same results were observed when we directly examined leukocyte interactions with the injured arteries by intravital epifluorescence microscopy (Figure 5C). In addition, in an ex vivo micro-flow chamber, the rolling of leukocytes on the activated IKKβ-deficient platelets or the control platelets did not differ significantly. In line with the in vivo data for leukocyte adhesion, the adhesion of the IgG-treated leukocytes to the activated IKKβ-deficient platelets in this ex vivo model was much greater than that observed in the activated control platelets. However, treatment with the anti-M2 antibody eliminated the increased leukocyte adhesion on the activated IKKβ-deficient platelets (Figure 5D).
Figure 5.
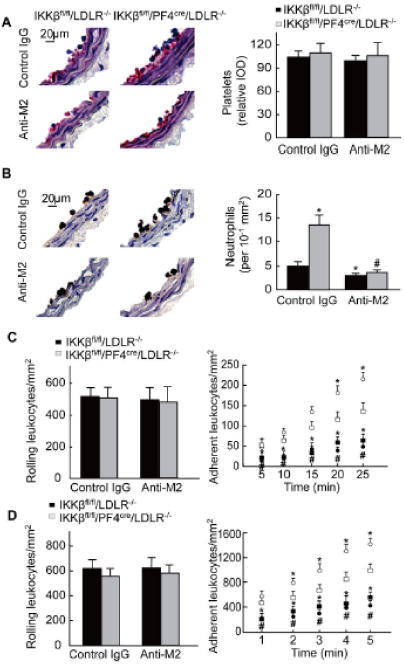
Blocking the GPIbα binding site on Mac-1 prevents the increase in leukocyte interactions with injured arteries in IKKβfl/fl/PF4cre/LDLR–/– mice. A to B, Immunostaining of platelets and neutrophils on carotid arteries collected 1 hour after wire injury (n = 5). *P < 0.05 vs. IKKβfl/fl/LDLR–/– mice treated with control IgG; #P < 0.05 vs. IKKβfl/fl/PF4cre/LDLR–/– mice treated with control IgG. C, Leukocyte rolling and adhesion in the carotid arteries within the first 25 min after injury (n = 5). White squares: IKKβfl/fl/LDLR–/– mice treated with control IgG; white circles: IKKβfl/fl/PF4cre/LDLR–/– mice treated with control IgG; black squares: IKKβfl/fl/LDLR–/– mice treated with anti-M2 antibody; black circles: IKKβfl/fl/PF4cre/LDLR–/– mice treated with anti-M2 antibody. *P < 0.05 vs. IKKβfl/fl/LDLR–/– mice treated with control IgG; #P < 0.05 vs. IKKβfl/fl/PF4cre/LDLR–/– mice treated with control IgG. D, Leukocyte rolling and adhesion on the activated IKKβ-deficient and control platelets in the micro-flow chambers within the first 5 minutes (n = 5). White squares: wild type mice were treated with control IgG, and their blood was passed through a micro-flow chamber coated with control platelets; white circles: wild type mice were treated with control IgG, and their blood was passed through a micro-flow chamber coated with IKKβ-deficient platelets; black squares: wild type mice were treated with anti-M2 antibody, and their blood was passed through a micro-flow chamber coated with control platelets; black circles: wild type mice were treated with anti-M2 antibody, and their blood was passed through a micro-flow chamber coated with IKKβ-deficient platelets. *P<0.05 vs. wild type mice were treated with the control IgG and blood was passed through a micro-flow chamber coated with the control platelets; #P<0.05 vs. wild type mice treated with control IgG and blood was passed through a micro-flow chamber coated with IKKβ-deficient platelets.
IKKβ deficiency suppresses ADAM17 maturation in platelets
The sheddase ADAM17 is critically involved in platelet surface receptor shedding.19, 20 To test the involvement of ADAM17 in our system, we stimulated IKKβ-deficient and control platelets with thrombin and detected immature (or pro-) and mature (or active) forms of ADAM17 using western blot analysis. The pro-form of ADAM17 was present in the resting and activated platelets (Figure 6A). The active form of ADAM17 was detectable in the resting control platelets and transiently increased by 2.7-fold during platelet activation. This finding is consistent with earlier studies.20, 21 IKKβ-deficient platelets had a similar level of active ADAM17 in the resting platelets but a much lower level of active ADAM17 during platelet activation compared to the control platelets (Figure 6A). The level of phosphorylated p38 MAPK has been shown to play a critical role in ADAM-17 activation.22-24 Thus, we tested the effect of IKKβ deficiency on p38 MAPK phosphorylation. Thrombin remarkably increased p38 MAPK phosphorylation in the IKKβfl/fl platelets, but no significant increase in p38 MAPK phosphorylation was observed in the IKKβ-deficient platelets after 30 min of thrombin stimulation (Figure 6B). These data indicate that IKKβ is a key molecule in ADAM17 maturation via the regulation of p38 MAPK phosphorylation.
Figure 6.
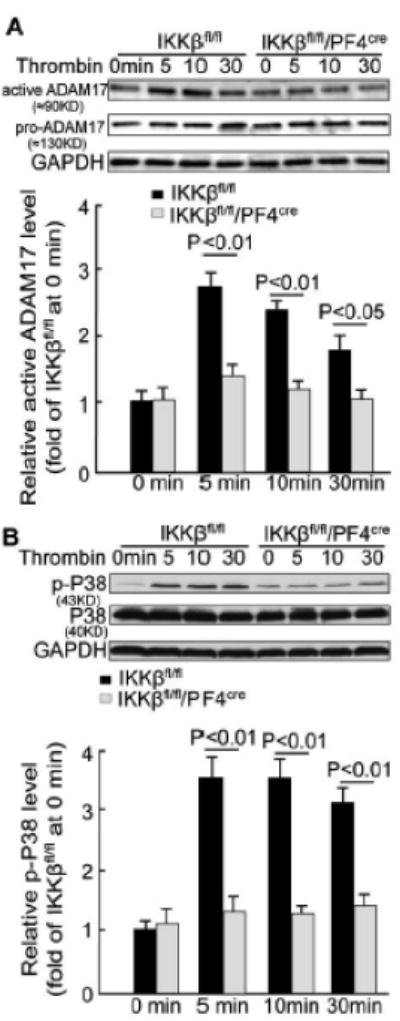
IKKβ deficiency suppresses ADAM17 maturation in platelets. Platelets from IKKβfl/fl/PF4cre and IKKβfl/fl mice were stimulated with 0.1 U/ml thrombin. A, Western blot analysis showing the levels of the immature (or pro) and mature (or active) ADAM17 in the platelets (n = 5). B, Western blot analysis showing the level of phosphorylated p38 MAPK in the platelets (n = 5).
Discussion
The initiation and progression of the neointima formation that underlies restenosis involves the adhesive interactions of platelets with leukocytes and leukocytes with the denuded vessel wall.4, 5 NF-κB is expressed in platelets and plays a critical role in platelet activation.6, 7 Here, we showed, in vivo, that platelet IKKβ deficiency increases neointima formation after arterial injury due to enhanced leukocyte-platelet interactions. Furthermore, we found that IKKβ deficiency inhibits ADAM17 maturation, resulting in delayed GPIbα shedding in platelets.
Wire injury-induced neointima formation in the mouse carotid artery is a widely used model to mimic the pathology of arterial neointima formation in patients with arterial restenosis.25 An excessive leukocyte-platelet interaction and leukocyte infiltration after artery injury has been shown to exaggerate the repair mechanisms and augment neointima formation.26, 27 After endothelial denudation by mechanical injury, platelets immediately adhere to and accumulate on the injured luminal surface of arteries.2, 3 P-selectin and many glycoproteins on activated platelets mediate leukocyte rolling and localization and further support leukocyte recruitment.28, 29 The binding of platelet P-selectin to its leukocyte ligand PSGL-1 initiates leukocyte recruitment to the activated platelets.30 The interaction of P-selectin and PSGL-1 rapidly activates the leukocyte integrin Mac-1.31-33 The binding of leukocyte Mac-1 to platelet GPIbα subsequently strengthens the firm adhesion and transmigration of leukocytes to sites of platelet deposition.16-18 The loss of Mac-1/GPIbα binding leads to reduced leukocyte accumulation after arterial injury and further results in the inhibition of neointima thickening.18 In this study, increased neointima formation in the IKKβfl/fl/PF4cre/LDLR–/– mice was accompanied by an increase in leukocyte adhesion to the injured area. This increased neointima was eliminated by neutrophil depletion (Supplemental Figure VI), indicating that this phenotypic change in neointima formation is due to increased leukocyte adhesion. Furthermore, this increased leukocyte adhesion was abrogated by a blocking antibody that specifically inhibits the binding of leukocyte Mac-1 with platelet GPIbα.18 These results demonstrate that high levels of GPIbα on activated platelets contribute to the increased neointima formation in IKKβfl/fl/PF4cre/LDLR–/– mice.
The dynamic change in GPIbα on activated platelets occurs mainly through ADAM17-mediated GPIbα shedding. ADAM17 is a sheddase on the cell surface that cleaves a variety of substrates, such as heparin-binding epidermal growth factor, transforming growth factor α, tumor necrosis factor receptor, epidermal growth factor receptor, vascular cell adhesion molecule-1 and L-selectin.34 Following cellular activation, the proform (134 kDa) of ADAM17 is proteolyzed to yield the mature (active) form (98 kDa). The latter active form cleaves its substrates.35 ADAM-17 activation in leukocytes occurs through p38 and/or the ERK MAPK pathway.36, 37 ADAM17 is a major sheddase involved in platelet GPIbα shedding.19 ADAM17 inactivation led to about a 90% reduction in GPIbα shedding in platelets. Brill et al. reported that oxidative stress activates ADAM-17 in platelets in a p38-dependent fashion.23 Through a similar mechanism, IKKβ deficiency inhibited ADAM-17 maturation, resulting in delayed shedding of platelet GPIbα. This result is consistent with the observation that, in oral squamous cell carcinoma cells, NF-κB inhibition suppresses ADAM-17 maturation.38
Platelet IKKβ deficiency did not cause a significant change in platelet adhesion and accumulation on the injured arteries. In this study, we found that IKKβ deficiency delayed GPIbα and GPV shedding. GPIbα and GPV are able to bind vWF and collagen, respectively.39, 40 High levels of GPIbα and GPV may enhance platelet adhesion to the subendothelial area of the injured arteries.39, 40 However, many other molecules that are important for platelet adhesion to the subendothelial area, including GPVI, GPIX and αIIbβ3, did not differ between the IKKβ deficient and the control platelets (Supplemental Figure V).40-42 In addition, studies from other groups using NF-κB inhibitors have shown that broad NF-κB inhibition causes a defect in platelet adhesion and spreading.6, 7 All of these factors may explain why decreased shedding of GPIbα and GPV does not result in an increase in platelet adhesion on the injured arteries.
NF-κB is a double-edged sword for activated platelet-mediated pathologies. NF-κB is a key regulator of inflammation, immunity, apoptosis and cell proliferation in all types of nucleated cells.43 NF-κB affects the progression of inflammatory diseases such as myocardial ischemia, bronchial asthma, arthritis, and cancer.44 The suppression of NF-κB activation has been shown to inhibit the expression of adhesion molecules and the release of chemokines and cytokines by various inflammatory cells, eventually suppressing the progression of inflammation.45, 46, 47 However, NF-κB activation during the late stage of inflammation is associated with the resolution of inflammation and anti-inflammatory gene expression.48 A few reports have indicated that blockade of the NF-κB pathway accelerates the pathology of inflammatory diseases. For example, Kanter et al. revealed that the inhibition of NF-κB activation in macrophages increased atherosclerosis in LDLR–/– mice.49 Zaph et al. reported that deficient NF-κB activation in the intestinal epithelium is associated with increased inflammation in vivo.50 Consistent with its effect on inflammation, NF-κB activation initiates platelet activation. This process is evident by the reduced level of P-selectin and the suppressed release of granules and ATP in IKKβ-deficient platelets. However, NF-κB activation appears to be necessary for activated platelets to shed their glycoproteins, and the inactivation of NF-κB led to sustained activation of platelets, as demonstrated by much higher levels of GPIbα and GPV on the surface of the activated IKKβ-deficient platelets compared to the wild-type platelets.
NF-κB has been shown to be involved in the transcriptional regulation of more than 150 genes, a significant proportion of which exhibit proinflammatory properties.51 Therefore, approaches to specifically inhibit the NF-κB pathway are under active development as possible therapeutic interventions. In this study, we demonstrated that platelet IKKβ deficiency enhances leukocyte-platelet interactions, resulting in aggregated neointima formation after arterial injury. The underlying mechanism is that NF-κB inactivation delays ADAM17-mediated-GPIbα shedding, thus strengthening the interaction of leukocytes with platelets. Because the inhibition of the NF-κB pathway is likely to be a promising therapeutic strategy and because tissue-specific therapies are not currently available, the application of NF-κB inhibitors for diseases that have pathologies in which activated platelets are extensive participants, such as arterial injury, sepsis and thrombosis, should be carefully evaluated.
Supplementary Material
Acknowledgments
Sources of Funding: This work was supported by grants from the American Diabetes Association (1-10-BS-76 to Y.H., 1-10-JF-54 to C.W.), the American Heart Association (10GRNT4400005 to Y.H., 12BGIA9050003 to C.W), the National Institutes of Health (HL080569, HL095556, and HL108922to Y.H., HL57506 MERIT Award to DIS), the Joint Research Fund for Overseas Chinese Young Scholars (31028008, to Y.H. and C.Z.), and the National Key Basic Research Program of China (2012CB910402 to Y.H.).
Footnotes
Disclosures: The authors declare that they have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davies MG, Hagen PO. Pathobiology of intimal hyperplasia. Br J Surg. 1994;81:1254–1269. doi: 10.1002/bjs.1800810904. [DOI] [PubMed] [Google Scholar]
- 2.Coller BS. Binding of abciximab to alpha v beta 3 and activated alpha m beta 2 receptors: With a review of platelet-leukocyte interactions. Thromb Haemost. 1999;82:326–336. [PubMed] [Google Scholar]
- 3.Wang H, Zhang W, Tang R, Hebbel RP, Kowalska MA, Zhang C, Marth JD, Fukuda M, Zhu C, Huo Y. Core2 1-6-n-glucosaminyltransferase-i deficiency protects injured arteries from neointima formation in apoe-deficient mice. Arterioscler Thromb Vasc Biol. 2009;29:1053–1059. doi: 10.1161/ATVBAHA.109.187716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schober A, Weber C. Mechanisms of monocyte recruitment in vascular repair after injury. Antioxid Redox Signal. 2005;7:1249–1257. doi: 10.1089/ars.2005.7.1249. [DOI] [PubMed] [Google Scholar]
- 5.Manka DR, Wiegman P, Din S, Sanders JM, Green SA, Gimple LW, Ragosta M, Powers ER, Ley K, Sarembock IJ. Arterial injury increases expression of inflammatory adhesion molecules in the carotid arteries of apolipoprotein-e-deficient mice. J Vasc Res. 1999;36:372–378. doi: 10.1159/000025676. [DOI] [PubMed] [Google Scholar]
- 6.Spinelli SL, Casey AE, Pollock SJ, Gertz JM, McMillan DH, Narasipura SD, Mody NA, King MR, Maggirwar SB, Francis CW, Taubman MB, Blumberg N, Phipps RP. Platelets and megakaryocytes contain functional nuclear factor-kappab. Arterioscler Thromb Vasc Biol. 2010;30:591–598. doi: 10.1161/ATVBAHA.109.197343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malaver E, Romaniuk MA, D'Atri LP, Pozner RG, Negrotto S, Benzadon R, Schattner M. Nf-kappab inhibitors impair platelet activation responses. J Thromb Haemost. 2009;7:1333–1343. doi: 10.1111/j.1538-7836.2009.03492.x. [DOI] [PubMed] [Google Scholar]
- 8.Huxford T, Huang DB, Malek S, Ghosh G. The crystal structure of the ikappabalpha/nf-kappab complex reveals mechanisms of nf-kappab inactivation. Cell. 1998;95:759–770. doi: 10.1016/s0092-8674(00)81699-2. [DOI] [PubMed] [Google Scholar]
- 9.Li J, Peet GW, Pullen SS, Schembri-King J, Warren TC, Marcu KB, Kehry MR, Barton R, Jakes S. Recombinant ikappab kinases alpha and beta are direct kinases of ikappa balpha. J Biol Chem. 1998;273:30736–30741. doi: 10.1074/jbc.273.46.30736. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka M, Fuentes ME, Yamaguchi K, Durnin MH, Dalrymple SA, Hardy KL, Goeddel DV. Embryonic lethality, liver degeneration, and impaired nf-kappa b activation in ikk-beta-deficient mice. Immunity. 1999;10:421–429. doi: 10.1016/s1074-7613(00)80042-4. [DOI] [PubMed] [Google Scholar]
- 11.Tiedt R, Schomber T, Hao-Shen H, Skoda RC. Pf4-cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood. 2007;109:1503–1506. doi: 10.1182/blood-2006-04-020362. [DOI] [PubMed] [Google Scholar]
- 12.Pasparakis M, Courtois G, Hafner M, Schmidt-Supprian M, Nenci A, Toksoy A, Krampert M, Goebeler M, Gillitzer R, Israel A, Krieg T, Rajewsky K, Haase I. Tnf-mediated inflammatory skin disease in mice with epidermis-specific deletion of ikk2. Nature. 2002;417:861–866. doi: 10.1038/nature00820. [DOI] [PubMed] [Google Scholar]
- 13.An G, Wang H, Tang R, Yago T, McDaniel JM, McGee S, Huo Y, Xia L. P-selectin glycoprotein ligand-1 is highly expressed on ly-6chi monocytes and a major determinant for ly-6chi monocyte recruitment to sites of atherosclerosis in mice. Circulation. 2008;117:3227–3237. doi: 10.1161/CIRCULATIONAHA.108.771048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H, Zhang W, Tang R, Zhu C, Bucher C, Blazar BR, Geng JG, Zhang C, Linden J, Wu C, Huo Y. Adenosine receptor a2a deficiency in leukocytes increases arterial neointima formation in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol. 2010;30:915–922. doi: 10.1161/ATVBAHA.109.202572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ravid K, Beeler DL, Rabin MS, Ruley HE, Rosenberg RD. Selective targeting of gene products with the megakaryocyte platelet factor 4 promoter. Proc Natl Acad Sci U S A. 1991;88:1521–1525. doi: 10.1073/pnas.88.4.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV, Ballantyne CM, Zhang L, Furman MI, Berndt MC, Lopez JA. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin mac-1 (cd11b/cd18) J Exp Med. 2000;192:193–204. doi: 10.1084/jem.192.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ehlers R, Ustinov V, Chen Z, Zhang X, Rao R, Luscinskas FW, Lopez J, Plow E, Simon DI. Targeting platelet-leukocyte interactions: Identification of the integrin mac-1 binding site for the platelet counter receptor glycoprotein ibalpha. J Exp Med. 2003;198:1077–1088. doi: 10.1084/jem.20022181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Sakuma M, Chen Z, Ustinov V, Shi C, Croce K, Zago AC, Lopez J, Andre P, Plow E, Simon DI. Leukocyte engagement of platelet glycoprotein ibalpha via the integrin mac-1 is critical for the biological response to vascular injury. Circulation. 2005;112:2993–3000. doi: 10.1161/CIRCULATIONAHA.105.571315. [DOI] [PubMed] [Google Scholar]
- 19.Bergmeier W, Piffath CL, Cheng G, Dole VS, Zhang Y, von Andrian UH, Wagner DD. Tumor necrosis factor-alpha-converting enzyme (adam17) mediates gpibalpha shedding from platelets in vitro and in vivo. Circ Res. 2004;95:677–683. doi: 10.1161/01.RES.0000143899.73453.11. [DOI] [PubMed] [Google Scholar]
- 20.Rabie T, Strehl A, Ludwig A, Nieswandt B. Evidence for a role of adam17 (tace) in the regulation of platelet glycoprotein v. J Biol Chem. 2005;280:14462–14468. doi: 10.1074/jbc.M500041200. [DOI] [PubMed] [Google Scholar]
- 21.Zhu L, Bergmeier W, Wu J, Jiang H, Stalker TJ, Cieslak M, Fan R, Boumsell L, Kumanogoh A, Kikutani H, Tamagnone L, Wagner DD, Milla ME, Brass LF. Regulated surface expression and shedding support a dual role for semaphorin 4d in platelet responses to vascular injury. Proc Natl Acad Sci U S A. 2007;104:1621–1626. doi: 10.1073/pnas.0606344104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Canault M, Duerschmied D, Brill A, Stefanini L, Schatzberg D, Cifuni SM, Bergmeier W, Wagner DD. P38 mitogen-activated protein kinase activation during platelet storage: Consequences for platelet recovery and hemostatic function in vivo. Blood. 2010;115:1835–1842. doi: 10.1182/blood-2009-03-211706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brill A, Chauhan AK, Canault M, Walsh MT, Bergmeier W, Wagner DD. Oxidative stress activates adam17/tace and induces its target receptor shedding in platelets in a p38-dependent fashion. Cardiovasc Res. 2009;84:137–144. doi: 10.1093/cvr/cvp176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duerschmied D, Canault M, Lievens D, Brill A, Cifuni SM, Bader M, Wagner DD. Serotonin stimulates platelet receptor shedding by tumor necrosis factor-alpha-converting enzyme (adam17) J Thromb Haemost. 2009;7:1163–1171. doi: 10.1111/j.1538-7836.2009.03476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindner V, Fingerle J, Reidy MA. Mouse model of arterial injury. Circ Res. 1993;73:792–796. doi: 10.1161/01.res.73.5.792. [DOI] [PubMed] [Google Scholar]
- 26.Libby P, Ganz P. Restenosis revisited--new targets, new therapies. N Engl J Med. 1997;337:418–419. doi: 10.1056/NEJM199708073370608. [DOI] [PubMed] [Google Scholar]
- 27.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 28.Kuijper PH, Gallardo Torres HI, Houben LA, Lammers JW, Zwaginga JJ, Koenderman L. P-selectin and mac-1 mediate monocyte rolling and adhesion to ecm-bound platelets under flow conditions. J Leukoc Biol. 1998;64:467–473. doi: 10.1002/jlb.64.4.467. [DOI] [PubMed] [Google Scholar]
- 29.Weber C, Springer TA. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of mac-1 with fibrinogen bound to alphaiibbeta3 and stimulated by platelet-activating factor. J Clin Invest. 1997;100:2085–2093. doi: 10.1172/JCI119742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McEver RP. Adhesive interactions of leukocytes, platelets, and the vessel wall during hemostasis and inflammation. Thromb Haemost. 2001;86:746–756. [PubMed] [Google Scholar]
- 31.Diacovo TG, Roth SJ, Buccola JM, Bainton DF, Springer TA. Neutrophil rolling, arrest, and transmigration across activated, surface-adherent platelets via sequential action of p-selectin and the beta 2-integrin cd11b/cd18. Blood. 1996;88:146–157. [PubMed] [Google Scholar]
- 32.Cerletti C, Evangelista V, de Gaetano G. P-selectin-beta 2-integrin cross-talk: A molecular mechanism for polymorphonuclear leukocyte recruitment at the site of vascular damage. Thromb Haemost. 1999;82:787–793. [PubMed] [Google Scholar]
- 33.Evangelista V, Manarini S, Sideri R, Rotondo S, Martelli N, Piccoli A, Totani L, Piccardoni P, Vestweber D, de Gaetano G, Cerletti C. Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent cd11b/cd18 adhesion: Role of psgl-1 as a signaling molecule. Blood. 1999;93:876–885. [PubMed] [Google Scholar]
- 34.Seals DF, Courtneidge SA. The adams family of metalloproteases: Multidomain proteins with multiple functions. Genes Dev. 2003;17:7–30. doi: 10.1101/gad.1039703. [DOI] [PubMed] [Google Scholar]
- 35.Schlondorff J, Becherer JD, Blobel CP. Intracellular maturation and localization of the tumour necrosis factor alpha convertase (tace) Biochem J. 2000;347(Pt 1):131–138. [PMC free article] [PubMed] [Google Scholar]
- 36.Rizoli SB, Rotstein OD, Kapus A. Cell volume-dependent regulation of l-selectin shedding in neutrophils. A role for p38 mitogen-activated protein kinase. J Biol Chem. 1999;274:22072–22080. doi: 10.1074/jbc.274.31.22072. [DOI] [PubMed] [Google Scholar]
- 37.Soond SM, Everson B, Riches DW, Murphy G. Erk-mediated phosphorylation of thr735 in tnfalpha-converting enzyme and its potential role in tace protein trafficking. J Cell Sci. 2005;118:2371–2380. doi: 10.1242/jcs.02357. [DOI] [PubMed] [Google Scholar]
- 38.Takamune Y, Ikebe T, Nagano O, Shinohara M. Involvement of nf-kappab-mediated maturation of adam-17 in the invasion of oral squamous cell carcinoma. Biochem Biophys Res Commun. 2008;365:393–398. doi: 10.1016/j.bbrc.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 39.Moog S, Mangin P, Lenain N, Strassel C, Ravanat C, Schuhler S, Freund M, Santer M, Kahn M, Nieswandt B, Gachet C, Cazenave JP, Lanza F. Platelet glycoprotein v binds to collagen and participates in platelet adhesion and aggregation. Blood. 2001;98:1038–1046. doi: 10.1182/blood.v98.4.1038. [DOI] [PubMed] [Google Scholar]
- 40.Massberg S, Gawaz M, Gruner S, Schulte V, Konrad I, Zohlnhofer D, Heinzmann U, Nieswandt B. A crucial role of glycoprotein vi for platelet recruitment to the injured arterial wall in vivo. J Exp Med. 2003;197:41–49. doi: 10.1084/jem.20020945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gruner S, Prostredna M, Schulte V, Krieg T, Eckes B, Brakebusch C, Nieswandt B. Multiple integrin-ligand interactions synergize in shear-resistant platelet adhesion at sites of arterial injury in vivo. Blood. 2003;102:4021–4027. doi: 10.1182/blood-2003-05-1391. [DOI] [PubMed] [Google Scholar]
- 42.Smyth SS, Reis ED, Zhang W, Fallon JT, Gordon RE, Coller BS. Beta(3)-integrin-deficient mice but not p-selectin-deficient mice develop intimal hyperplasia after vascular injury: Correlation with leukocyte recruitment to adherent platelets 1 hour after injury. Circulation. 2001;103:2501–2507. doi: 10.1161/01.cir.103.20.2501. [DOI] [PubMed] [Google Scholar]
- 43.Barnes PJ, Karin M. Nuclear factor-kappab: A pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 44.Suzuki J, Ogawa M, Muto S, Itai A, Isobe M, Hirata Y, Nagai R. Novel ikb kinase inhibitors for treatment of nuclear factor-kb-related diseases. Expert Opin Investig Drugs. 2011;20:395–405. doi: 10.1517/13543784.2011.559162. [DOI] [PubMed] [Google Scholar]
- 45.Baldwin AS., Jr Series introduction: The transcription factor nf-kappab and human disease. J Clin Invest. 2001;107:3–6. doi: 10.1172/JCI11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uwe S. Anti-inflammatory interventions of nf-kappab signaling: Potential applications and risks. Biochem Pharmacol. 2008;75:1567–1579. doi: 10.1016/j.bcp.2007.10.027. [DOI] [PubMed] [Google Scholar]
- 47.Traber KE, Okamoto H, Kurono C, Baba M, Saliou C, Soji T, Packer L, Okamoto T. Anti-rheumatic compound aurothioglucose inhibits tumor necrosis factor-alpha-induced hiv-1 replication in latently infected om10.1 and ach2 cells. Int Immunol. 1999;11:143–150. doi: 10.1093/intimm/11.2.143. [DOI] [PubMed] [Google Scholar]
- 48.Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA. Possible new role for nf-kappab in the resolution of inflammation. Nat Med. 2001;7:1291–1297. doi: 10.1038/nm1201-1291. [DOI] [PubMed] [Google Scholar]
- 49.Kanters E, Pasparakis M, Gijbels MJ, Vergouwe MN, Partouns-Hendriks I, Fijneman RJ, Clausen BE, Forster I, Kockx MM, Rajewsky K, Kraal G, Hofker MH, de Winther MP. Inhibition of nf-kappab activation in macrophages increases atherosclerosis in ldl receptor-deficient mice. J Clin Invest. 2003;112:1176–1185. doi: 10.1172/JCI18580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zaph C, Troy AE, Taylor BC, Berman-Booty LD, Guild KJ, Du Y, Yost EA, Gruber AD, May MJ, Greten FR, Eckmann L, Karin M, Artis D. Epithelial-cell-intrinsic ikk-beta expression regulates intestinal immune homeostasis. Nature. 2007;446:552–556. doi: 10.1038/nature05590. [DOI] [PubMed] [Google Scholar]
- 51.Pahl HL. Activators and target genes of rel/nf-kappab transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.