

Abstract
A vapour generation (VG) procedure has been described for determination of Cd by ICP-MS. Volatile species of Cd were generated on-line by interacting acidic sample solution containing potassium hexacyanochromate(III), K3Cr(CN)6, with sodium borohydride (NaBH4). The hexacyanochromate(III) complex was generated on-line by reacting 0.04 mol L−1 chromium(III) nitrate and 0.16 mol L−1 potassium cyanide (KCN) solutions in water. The resulting suspension of chromium(III) hydroxide, Cr(OH)3, was fed continuously to acidic stream of sample solution in the presence of excess KCN. The experimental conditions were optimized for effective generation of volatile species of Cd. Optimum signals were obtained from reaction of sample solutions in 4% v/v HCl with 2% m/v NaBH4 solution. Presence of K3Cr(CN)6 improved the efficiency of Cd vapour generation substantially affording 15-fold higher sensitivity. This phenomenon was thought to occur through formation of reactive intermediates evolved from interaction of [Cr(CN)6]3− with NaBH4 that react with Cd(II) to increase the yield volatile Cd species. Under the optimum conditions, no significant interferences were observed from the transition metals, including Cu and Ni, up to 1.0 μg mL−1 levels. Among the hydride forming elements, Bi, Pb, Sb and Sn depressed the signals above 0.1 μg mL−1. The detection limits (3s) were 6.2 and 5.2 ng L−1 for 110Cd and 111Cd isotopes, respectively. The method was successfully applied to determination of Cd by ICP-MS in several certified reference materials, including Nearshore seawater (CASS-4), Bone ash (SRM 1400), Dogfish liver (DOLT-4) and Mussel tissue (SRM 2976).
1. Introduction
Vapour generation (VG) has always been considered as a potential tool for determination of cadmium by atomic spectroscopy because of the toxicological importance of this ubiquitous element even at trace levels. The first report for the generation of cadmium vapour was published by Cacho et al.1 more than two decades ago. Since then extensive research has been made to elucidate the mechanism of this process,2–5 and to improve the efficiency by means of different treatments.6–15 Nevertheless, the mechanism underlying the formation volatile species of Cd is not fully understood yet. In the earlier reports it was proposed that Cd vapour, Cd0(g), formed upon decomposition of unstable cadmium hydride (CdH2) that was produced from the reaction of Cd(II) in acidic solution with sodium borohydride (NaBH4) or potassium borohydride (KBH4).2,3 More recently, however, Feng et al.5 found that the reaction of NaBH4 with Cd(II) in acidic solution produced Cd vapour initially. The formation of the CdH2 species was therefore attributed to the interaction of Cd vapour with hydrogen atom (e.g., nascent hydrogen) evolved from the decomposition of NaBH4 in acid solution. This latter assumption was eventually disproved by D’Ulivo et al.,16,17 who found that the formation of nascent hydrogen was not kinetically plausible as to support the formation of CdH2. The formation of volatile hydrides was proposed to arise from direct action of NaBH4 with a suitable form the metal ion in the solution.
Unlike common hydride forming elements, such as As, Bi, Sb and Se), the efficiency of Cd vapour generation is poor and highly dependent on the acidity of sample, NaBH4 concentration and transition metal chemistry of the samples. Over the years, various vapour generation systems have been interfaced to spectrometric techniques, including electrothermal atomic absorption spectrometry (ETAAS),3,8,9 atomic fluorescence spectrometry (AFS),6,10 inductively coupled plasma atomic emission (ICP-AES)18 and mass spectrometry (ICP-MS)4,12,19 to achieve better sensitivity and detection limits. Electrochemical hydride generation approaches have also been reported more recently as an alternative way for determination of Cd by atomic absorption spectroscopy.20,21 However, the issues associated with the chemical interferences remained to be the major hurdle in Cd vapour generation regardless of the technique utilized.8 Sanz-Medel et al.2 first generated the Cd vapour in a vesicle of didodecyldimethylammonium bromide (DDAB) for CV-AAS with a detection limit of 80 ng L−1. Later, they extended this vesicular vapour generation to FI-HG-ICP-MS and achieved a detection limit determination of 7 ng L−1 for 50 μL sample. The enhancement was about four-fold compared with conventional FI-ICP-MS but no information was reported regarding the chemical interferences.4 Guo and Guo6 reported severe interferences from Au(III), Cu(II), Bi(III), Ni(II) and Pb(II) on Cd vapour generation in aqueous media using KBH4. Sensitivity was improved by adding thiourea and Co(II) into the samples. The authors attributed this effect to the catalytic activity of thiourea and Co(II) on the formation of Cd vapour. Others have also confirmed the effects of thiourea and Co(II).3,9,19 Nevertheless, the thiourea-Co(II) combination was far from alleviating the depressive effects of Cu, Pb and Ni completely.
An overview of the existing literature for the chemical interferences and performances of various masking and catalytic agents have been published in a recent paper.8 The findings show inconsistency and highly variable performances depending on the chemical conditions, types of samples and instrumentation. More recently, sodium iodate (NaIO3) was reported to improve the formation of Cd vapour in acidic solutions affording about 10-fold enhancement in sensitivity.10 Though a detection limit of 10 ng L−1 was achieved by AFS, the performance of this procedure is still unknown in the presence of the interfering metal ions. Alternatively, different gas-liquid separators,7 quartz-tube traps,11 and matrix elimination approaches via coprecipitation,6 cloud point extraction,14 and solid phase preconcentration15,22 have been developed. Yet, the effects of the interfering elements, especially those of Cu, Ni and Pb, could not be totally eliminated. Consequently, standard additions method was applied in most determinations.3,11,9,13 The existing information clearly points to the fact that vapour generation has not become a mature and preferred tool for Cd determination, which is due mainly to the difficulties associated with the generation and transport of the unstable volatile Cd species, and the unresolved severe chemical interferences.
Potassium cyanide has been realized as an effective masking agent in alleviating the depressive effects of transition metals in chemical vapour generation.6,8,23,24 It also forms complexes with transition metal ions, such as Cr(III), Cu(II), Fe(II) and Fe(III).25,26 The Fe(II) and Fe(III) complexes, Fe[(CN)6]4− and Fe[(CN)6]3− respectively, have been known as stable species for more than a century. The latter has also been proven by far the most effective single agent (as potassium salt) to date, both in terms of efficiency and tolerance to chemical interferences, in the generation of Pb hydride (PbH4, plumbane).27–32 In view of the interesting chemical and catalytic features of transition metal cyanide complexes, we examined the performance of chromium(III) cyanide complexes in an effort to develop a sensitive and robust chemical vapour generation methodology for determination of Cd. In this paper, we described an innovative chemical vapour generation approach based on the interaction of hexacyanochromate(III) complex - [Cr(CN)6]3− - that was generated on-line and mixed with acidic solution. Volatile Cd species were generated by reaction with alkaline sodium borohydride solution and transported rapidly to argon plasma through a peltier-cooled spray chamber. The effects of sample acidity and the concentrations of chemical reagents (NaBH4, KCN, and Cr(NO3)3) were examined on vapour generation. Manifold parameters, including flow rates of sample and carrier gas, and the length of the tubings were optimized. The tolerance of the method to chemical interferences of transition metals and hydride forming elements was investigated. The method was applied to the determination of Cd in various certified reference materials by ICP-MS.
2. Experimental
2.1 Reagents and solutions
Double deionized water with minimum resistivity of 18 MΩ cm obtained from a Barnstead E-Pure system fed by a reverse-osmosis system (SpectraPure) attached to was used throughout. A 1.0 μg mL−1 multielement standard solution of As, Bi, Cd, Co, Cr, Cu, Fe, Ge, K, Mg, Mn, Na, Mn, Ni, Pb, Sb, Se, Sn, and Zn was prepared from a 1000 μg mL−1 standard solution each element (SPEX Certiprep, Metuchen, NJ) and stored in 1% v/v HCl/1% HNO3 (Trace metal grade, BDH Chemicals). All experimental solutions and calibration standards were prepared by one-stage dilution from these stock standard solutions. Chromium nitrate (Cr(NO3).9H2O, ACS certified) was obtained from Fisher Scientific, Pittsburgh, PA (Lot No: 931745). Potassium cyanide (KCN, Bioultra ≥98%, Lot No: BCBG2070V) and sodium borohydride (NaBH4, ≥98%, Lot No: 31396JJ) were obtained from Sigma Aldrich, St. Louis, MO. Chromium nitrate and potassium cyanide solutions were prepared with appropriate concentrations in water. Sodium borohydride solution was prepared daily in 0.1% m/v sodium hydroxide (NaOH) solution (Fisher Scientific). Solutions of the other trace elements, including Ca, Co, Cu, Fe, K, Mg, Mn, Na, Ni, and Zn, were either prepared from high-purity salts or from 1000 μg mL−1 stock solutions (Spex Certiprep) as appropriate. Hydrogen peroxide (H2O2, 99.999%, Sigma Aldrich, Batch No: 09327LE) was used with HNO3 in digestion of tissue samples.
2.2 Instrumentation
A Varian 820MS ICP-MS instrument (Varian, Australia) was used throughout the experiments. The instrument was equipped with a peltier-cooled double-pass glass spray chamber, one-piece low flow ball-and-socket connection quartz torch, and standard Ni sampler and skimmer cones. Samples were introduced manually. The instrument was optimized in nebulization mode for sensitivity, doubly charged ions (<2%) and oxides (<3%) with 5 μg L−1 solution of 138Ba, 25Mg, 115In, 140Ce, 208Pb before vapour generation measurements. Data collection was achieved by ICP-MS Expert software package (version 2.2 b126). The operating parameters of the instrument are summarized in Table 1. Major isotopes (112Cd and 114Cd) of Cd suffer from isobaric interferences of tin, 112Sn and 114Sn.4 In addition, Sn forms hydride (SnH4) very efficiently compared with Cd such that small concentrations of Sn in solution could confound the accuracy for Cd when the most abundant isotopes are used. Thus, determinations were performed with less abundant 110Cd and 111Cd isotopes that were not affected from interferences of Sn.
Table 1.
Operating conditions for Varian 820MS ICP-MS and vapour generation system
|
ICP-MS
| |
| RF Power (kW) | 1.4 |
| Plasma Ar flow (L min−1) | 18 |
| Auxiliary Ar flow (L min−1) | 1.8 |
| Nebulizer Ar flow (L min−1) | 1.2 |
| Sheath Ar flow (L min−1) | 0.1 |
| Sampling depth (mm) | 7 |
| Pump rate (rpm) | 10 |
| Stabilization time (sec) | 40 |
| Spray chamber temperature (°C) | 2 |
| Scan mode | Peak hopping |
| Dwell time (ms) | 50 |
| Points/peak | 1 |
| Scans/peak | 6 |
| Scans/replicate | 10 |
| Isotopes measured | 110Cd and 111Cd |
|
| |
|
Vapour generation
| |
| Sample acidity/flow rate | 4% v/v HCl/1.0 mL min−1 |
| Cr(NO3)3 concentration/flow rate | 0.04 mol L−1/0.5 mL min−1 |
| KCN concentration/flow rate | 0.16 mol L−1/0.5 mL min−1 |
| NaBH4 concentration/flow rate | 2% m/v/1.0 mL min−1 |
2.3 Vapour generation setup and operation
The schematic diagram of the chemical vapour generation manifold is illustrated in Fig. 1. The stand-alone spray chamber with 100 mL inner volume was used as gas-liquid separator. The nebulizer housing was fitted with a T-adaptor through which the sample line was inserted into the spay chamber (Fig. 1).
Fig. 1.
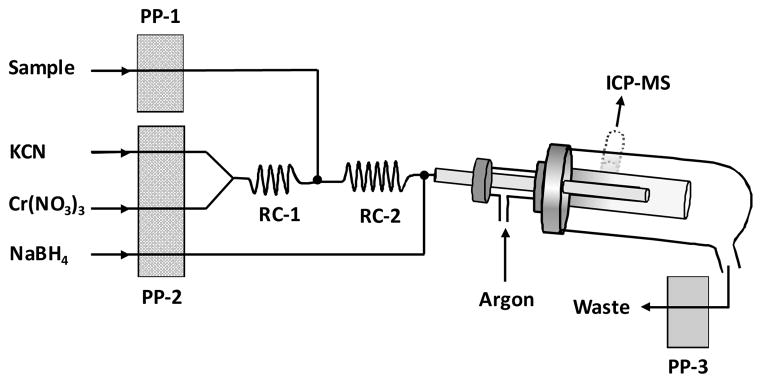
Schematic diagram of the chemical vapour generation manifold. The stand-alone double pass glass spray chamber was used as gas-liquid separator. The spray chamber was peltier-cooled to 2 °C. RC-1 and RC-2 are 10- and 15-cm long tygon tubings (1.4 mm. i.d), respectively.
Carrier gas flow was controlled by nebulizer argon gas introduced through the T-inlet. Tygon peristaltic pump tubings were used. Sample and NaBH4 lines were of red-red stop (1.14 mm i.d.) and those for Cr(NO3)3 and KCN were black-black stop (0.76 mm i.d.). The waste line was purple-white stop tygon tubing (2.79 mm i.d.,) running on a separate peristaltic pump (Ismatec). The reaction lines, RC-1 and RC-2, were made of 10-and 15-cm long tygon tubing (1.4 mm i.d.). The transfer line inserted through the spray chamber was 15-cm long PTFE tubing (1.6 mm i.d). All other connecting lines were made of PTFE (0.8 mm i.d.).
Both 0.04 mol L−1 Cr(NO3)3 and 0.16 mol L−1 KCN solutions were pumped at 0.5 mL min−1 and reacted on-line through RC-1 line. The reaction medium was highly alkaline (ca. pH 10–11) due to excess KCN resulting in formation a suspension of chromium hydroxide, Cr(OH)3. This suspension was fed and dissolved by reacting on-line with the sample solution in 4% v/v HCl along the RC-2 line to yield CrCl3. The hexacyanochromate(III) complex, [Cr(CN)6]3−, is produced by the reaction of the CrCl3 in the presence of excess CN− (e.g., KCN) in acidic solution. The mixture containing the Cd(II) in dilute HCl was reacted 2% m/v NaBH4 running at 1.0 mL min−1. The resulting volatile species of Cd were swept into the argon plasma. The instrument was run in the vapour generation settings for about 30 min daily before collecting the data.
2.4 Optimization of vapour generation conditions
Initial experiments were performed with potassium hexacyanochromate(III), K3Cr(CN)6, solutions produced by batch synthesis in the laboratory by mixing solutions of Cr(NO3)3 (purple) and KCN (colorless) at stiochiometric combinations. The preliminary studies showed the evidence for the generation of Cd vapour; however, the results were sporadic due to instability of the complex. To overcome this obstacle, we opted for on-line approach to generate [Cr(CN)6]3− in-line freshly.
In on-line studies, first the effect of the acidity of the sample solution was examined by varying HCl concentration of 10 μg L−1 multi-element solutions from 0 to 10% v/v HCl. In this experiment, 0.02 mol L−1 Cr(III) reacted with 0.12 mol L−1 KCN solution on-line using the manifold in its initial configuration (RC-1 = 10 cm and RC-2 = 30 cm). The concentration of NaBH4 was kept constant at 2% m/v throughout until its optimization later. Next, a series of studies were carried out at the optimum HCl concentration to elucidate the effects of Cr(III) and KCN. The concentration of Cr(III) was varied from 0 to 0.1 mol L−1 while that of KCN was increased from 0 to 0.2 mol L−1.
The lengths of RC-1 and RC-2 lines were the determining variables for the formation of reaction precursors, viz. Cr(OH)3 and [Cr(CN)6]3−, that mediate the vapour generation efficiency with HCl concentration. The former was varied formation 2 to 20 cm while the latter was investigated between 5 and 30 cm. The length of the NaBH4 line was also examined from 10 to 20 cm PTFE tubing to improve the Cd vapour generation.
In the last stage of the optimization, sodium borohydride concentration was examined from 0.5 to 4% m/v to determine its effect on Cd signals. The flow rate for sample solution was examined between 0.5 and 2.5 mL min−1. The flow rates of other solutions, Cr(III), KCN and NaBH4, were kept constant throughout as in Table 1. The carrier argon flow rate was also optimized for the maximum signals by varying the flow rate of nebulizer gas from 0.8 to 1.4 L min−1.
2.5 Matrix effects
The chemical generation of Cd vapour is highly susceptible to depressive effects of transition metals (Cu and Ni) and hydride forming elements (Pb and Se). Thus, the effects of a number of matrix elements, including As(III), Bi(III), Co(II), Cu(II), Fe(III), Mn(II), Ni(II), Pb(II), Sb(III), Se(IV), Sn(II) and Zn(II) were investigated for 10 μg L−1 Cd(II) (as multi-element stock solution) containing different concentrations of each element. Effects for Ca(II), K(I) Mg(II), Na(I) were examined for 1000 μg mL−1 solutions that contained 10 μg L−1 Cd(II). The detection limits were calculated as the concentration of analyte equivalent to three times the standard deviation of the blank signal (cps) (n=13). Calibration was performed with 0, 0.02, 0.05, 0.1, 0.2, 0.5, 1.0 and 2.0 μg L−1 aqueous standard solutions in 4% v/v HCl.
2.6 Sample preparation and method validation
A number certified standard reference materials (CRMs) with different matrix composition were analyzed for method validation purposes. These CRMs include Nearshore seawater (CASS-4) and Dogfish liver (DOLT-4) from National Research Council Canada, and Bone ash (SRM 1400) and Mussel tissue (SRM 2976) obtained from National Institutes of Standards and Technology, Gaithersburg, MD. Bone ash (SRM 1400) is purely calcium phosphate produced from calcinations of bone containing high levels of Al, Fe, Pb and Zn. Major matrix components in mussel tissue and dogfish liver were As, Cu, Fe and Zn at high μg g−1 levels.
The seawater CRM was used directly from the bottle. A 10 mL solution was taken and acidified to 4% v/v HCl with concentrated HCl. For bone ash, about 50 mg sub-samples were dissolved and then digested in 1 mL concentrated HNO3 in 4-mL PTFE tubes (Savillex) at 120 °C using Digiprep Cube digestion system (SCP Science, Champlain, NY) as described elsewhere.32 The solutions were first heated to dryness. Then, 1 mL deionized water added and evaporated to dryness to get rid of residual HNO3. The final residue was dissolved and diluted to 10 mL with 4% v/v HCl. Digestions for the mussel tissue and dogfish liver were performed similarly. Approximately, 25 mg sub-samples were digested with 2-mL concentrated HNO3 and 1 mL H2O2 in 60-mL screw-capped PTFE tubes (Savillex) at 140 °C for 2 h. At the completion, caps were opened and additional 1 mL H2O2 was added to hot solution to oxidize the organics. Clear solutions were obtained with this procedure. The contents were heated to dryness as for the bone ash samples, and then redissolved with 2 mL water and heated again to remove all residual HNO3 and H2O2. At the end, contents were dissolved in 4% v/v HCl and diluted to 10 mL.
3. Results and discussion
3.1 Synthesis of hexacyanochromate(III) and preliminary studies
The synthesis of potassium hexacyanochromate(III) is described in literature33 where the synthesis is based on Cr(OH)3 produced from the reaction of Cr(III) with ammonia solution. The Cr(OH)3 is then converted to chromium acetate, Cr(C2H3O2)3, by reacting with acetic acid (HC2H3O2). The reaction between Cr(C2H3O2)3 and KCN yields the light yellow crystals of hexacyanochromate(III) (K3Cr(CN)6) according to the sequence given below.
| (1) |
| (2) |
| (3) |
In off-line studies, K3Cr(CN)6 was produced as described above. Cr(C2H3O2)3 was dissolved in warm KCN solution yielding a pale yellow solution of hexacyanochromate(III). However, the vapour generation attempts with these solutions were problematic. The sensitivity degraded gradually during optimization of HCl concentration when measurements were repeated with several hours differential. Further, the results were not consistent among daily measurements. This was because of the instability of the [Cr(CN)6]3− complex in water and strong mineral acids. In water, K3Cr(CN)6 hydrolyzes to precipitate Cr(III) hydroxide, while it decomposes in acidic solutions.25,33 In our studies, the K3Cr(CN)6 solution developed suspending particles and cloudy green color over the course of the time, presumably Cr(OH)3 from hydrolysis of the complex. Consequently, the sensitivity dropped during measurements with 2 to 3-h old solutions. These results suggested that it would be difficult to achieve quantitative Cd vapour generation in batch or off-line mode with K3Cr(CN)6 solution. The issue was successfully avoided by means of on-line approach.
3.2 On-line generation of hexacyanochromate(III) and its effect on Cd vapour generation
The reaction mechanism proposed for the formation of hexacyanochromate(III) in on-line method is very analogous to the batch method with some modifications as outlined below.
| (4) |
| (5) |
| (6) |
| (7) |
The aqueous solution of KCN is highly alkaline (pH ~ 11). Thus, the reaction with Cr(NO3)3 resulted in the formation of suspension (slurry) of Cr(OH)3 through reactions 4 and 5 along the RC-1 line. This chemistry was also verified with a separate batch synthesis that grayish green Cr(OH)3 formed in a solution of KCN and Cr(NO3)3. Differently from the batch method, chromium chloride (CrCl3) was generated as the suspension of Cr(OH)3 reacted with dilute HCl (rxn 6), which was instantaneously converted to K3Cr(CN)6 upon reaction with KCN along the RC-2 coil (rxn 7). Possible instability of the [Cr(CN)]3−did not have any significant effect on the signal stability and the repeatability of the measurements as it merged with alkaline NaBH4 in a few seconds.
The signal profiles for Cd vapour generation are illustrated in Fig. 2 with and without Cr(III) + KCN combination. As indicated, the multi-element standard solution also contained severely interfering elements, such as Bi(III), Cu(II), and Pb(II) at 10 μg L−1. Initially, deionized water was run through both Cr(III) and KCN lines to determine the efficiency with traditional Cd vapour generation. The signals were very low, which could be attributed to the depression from the interfering elements. In the presence of KCN, signals were enhanced to a certain extent within a broader acidity range from 2 to 7% v/v HCl. KCN has been used for alleviating the interferences of Cu(II) and a number of transition metals (Au, Co, Cu, Pb, and Ni) in chemical vapour generation of Cd6 and As.23 Although its effectiveness in masking the interferences of numerous other transition metals in Cd vapour generation is not known, this improvement could be ascribed to partial masking of the interferences, especially from Bi(III) and Pb(II).
Fig. 2.

Vapour generation profile for 10 μg L−1 Cd(II) in multielement solution as a function of HCl concentration with and without hexacyanochromate(III) complex. Cr(III) = 0.02 mol L−1, KCN = 0.12 mol L−1; NaBH4 = 2% m/v, RC-1 = 10 cm; RC-2 = 30 cm.
The increase in Cd signals was remarkable when Cr(III) and KCN solutions were mixed on-line (see Fig. 2). The enhancement was about 4-fold better compared with 0.12 M KCN and 15-fold higher than that for the direct nebulization suggesting a different mechanism of action mediated by a new substance of Cr(III) and CN−. This result evidently points to the formation of hexacyanochromate(III) complex in light of the proposed mechanism. In addition, the complex was stable in slightly acidic medium in the presence of KCN as depicted by increasing signals upon interaction with NaBH4 which affects the vapour generation in acidic solutions only. The optimum range exhibited relatively robust profile ranging from 2 to 5% v/v HCl. Presence of KCN could have also contributed to the broadening since the optimum acidity range was the largest for water + 0.12 M KCN configuration. Unlike the scenario with KCN, the decrease at and above 6% v/v HCl was sharp which was thought to be due to the inefficient formation as well as rapid decomposition of the complex in more acidic conditions. Additional data for the HCl concentration suggested that 4% v/v HCl was relatively better than 3% v/v HCl in terms of daily precision of measurements. Thus, solutions were acidified to 4% v/v HCl in subsequent experiments to optimize the remaining variables of the method.
The enhancement in Cd signals also demonstrates that reaction of hexacyanochromate(III) complex with NaBH4 facilitated the generation of volatile Cd species, as ferricyanide, Fe[(CN)6]3−, did for plumbane (PbH4) in acidic solutions.31,32 Mechanistically, this phenomenon cannot be explained by the reduction of Cd2+ to Cd0 (E0 = −0.403 V), because Cr[(CN)6]3− is more stable in solution than [Cr(CN)6]4− (E0 = −1.28 V).25 An alternative and rather more feasible explanation is the formation of reactive intermediates from the reaction of [Cr(CN)6]3− with NaBH4, which react effectively with Cd(II) to increase the yield of volatile species of Cd (e.g., CdH2). Support for this explanation is based on a recent report by D’Ulivo et al.31 who observed similar enhancement in the generation of plumbane (PbH4) regardless of the sequence [Fe(CN)6]3− was interacted with either Pb(II) or NaBH4 first. They concluded that [Fe(CN)6]3− was critical in formation of “borane complex intermediates” that react with Pb(II) for generation of PbH4 efficiently.
3.3 Effects of Cr(III) and KCN concentrations
The effects of the concentration of Cr(III) and KCN solutions are shown in Fig. 3. The results were gathered by univariate method. Initially, Cr(III) concentration was kept as 0.02 mol L−1. In this case, steep increase occurred in Cd signals with up to 0.04 mol L−1 KCN. Signals continued to increase gradually up to 0.12 mol L−1 and then leveled off. Apparently, excess KCN had no deleterious effects on signals. On the contrary, higher concentration was advantageous for better stabilization of [Cr(CN)6]3− as well as reducing possible interferences from the transition metals.
Fig. 3.

The effects of Cr(III) and KCN concentrations on the vapour generation for 10 μg L−1 Cd(II) in multielement solution. Sample acidity = 4% HCl; NaBH4 = 2% m/v, RC-1 = 10 cm; RC-2 = 30 cm.
For optimization of Cr(III) concentration, the concentration of KCN was increased to 0.16 mol L−1. Likewise, increasing Cr(III) concentration resulted in steep increase in Cd signals up to 0.04 mol L−1 and then leveled off slightly. Optimum concentration was 0.04 mol L−1. Unlike KCN, excess of Cr(III) was not desirable as it also increased the suspension of Cr(OH)3 which adsorbed on the walls of the RC-1 coil. In conclusion of this experiment, the HCl concentration was reexamined optimum concentrations of Cr(III) and KCN. It was found that the working range slightly shifted to higher acidity (2–6% v/v HCl) compared with the initial conditions (2–5%, Fig. 1), which was likely due to the increased KCN concentration in the system. Yet, 4% v/v HCl appeared to be the optimum medium for chemical generation of Cd vapour.
3.4 Optimization of lengths of reaction lines
It should be noted that suspensions of Cr(OH)3 formed visibly and instantaneously upon mixing of Cr(III) and KCN solutions. Tubings with narrow internal diameter (e.g., 0.8 mm) were not suitable due to the risk of flow restriction through the line. Tygon tubing with 1.4 mm i.d. afforded smooth flow of the suspension and hence was chosen as the optimum tubing throughout. The lengths of the RC-1 and RC-2 lines were studied at the optimum conditions (0.04 mol L−1 Cr(III) and 0.16 mol L−1 KCN). The results are illustrated in the Fig. 4A. In general, the length of RC-1 line was very influential on the signals (e.g., vapour generation efficiency) since it determined the influx of Cr(OH)3 into the sample line to react with HCl. A length of 5 to 10 cm was sufficient to attain desired sensitivity and stability. In general 10-cm line provided better sensitivity and precision. Signals decreased with increasing length of RC-1 line, which was ascribed to the loss of Cr(OH)3 along the line. As mentioned earlier, the reaction of Cr(III) and KCN yielded a suspension or slurry of Cr(OH)3, which developed noticeable adsorption on the walls of RC-1 line over the course of the time. It appears that this adsorption was higher with longer lines to lead to significant reduction in Cr(OH)3 before interacting with sample solution.
Fig. 4.
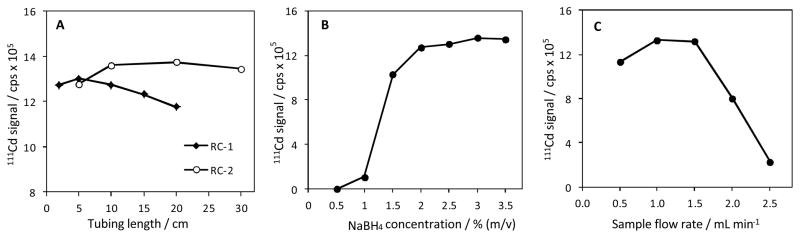
Effects of various experimental parameters on signals during vapour generation with 10 μg L−1 Cd(II) by 4% v/v HCl-hexacyanochromate(III) system. A = Lengths of RC-1 and RC-2 reaction lines; B = NaBH4 concentration; C = Sample flow rate.
The effect of RC-2 line was marginal when its length was increased from 5 to 30 cm. Signals were better and relatively constant for 10 cm and above. Likewise, the length of RC-2 line was reduced from 30 to 15 cm. In case of NaBH4 (2% m/v), the signals did not show any differences between 10 to 20-cm long PTFE tubing (1.6 mm i.d.). A 15-cm long tubing was chosen to be optimum and suitable length to ensure completeness of reaction and successful delivery of species into the spray chamber.
3.5 Effects of NaBH4 concentration and flow rates of sample and carrier argon gas
The effect of NaBH4 is shown in Fig. 4B for a series of NaBH4 solutions prepared in 0.1% NaOH solution. Often, vapour generation of Cd demands higher concentrations of NaBH4 (ca. 3–6% m/v).10–19 In this study, a solution of 2% m/v NaBH4 was sufficient for efficient generation of Cd vapour and optimum signals. NaBH4 concentration was increased to as high as 4% without any instability in plasma. However, higher concentrations did not afford any significant increase in signals.
Maximum sample flow rate for the sample solution was around 1.5 mL min−1 (Fig. 4C). Signals increased with increasing flow rate up 1.5 mL min−1. In general, analyte signal increases to a certain extent and then flattens in vapour generation procedures. Here, on the contrary, Cd signals steeply declined with further increases in flow rate of the sample solution. This phenomenon is related with the formation of the hexacyanochromate(III) in the medium. Apparently, the declining signals imply that reaction time was not long enough for successful formation of the complex in the line before interacting with the reductant. Thus, flow rate of the sample solution was kept constant at 1 mL min−1. In the last step of the optimization, the nebulizer gas (carrier as) flow rate was varied from 0.8 to 1.4 L min−1 to affect the signal intensity. Optimum gas flow rate was found to be around 1.2 L min−1. Signals declined with flow rates at higher flow rates, which was due to the change in the sampling depth in the plasma.
3.6 Analytical performance and chemical interferences
Under the optimum conditions, the detection limits (3s) were 6.2 and 5.7 ng L−1 for 110Cd and 111Cd isotopes. The detection limits were limited by the background signals ranging between 3,000 to 7,000 cps for 4% v/v HCl solution. Relative standard deviations (RSD) varied between 0.6% and 5.6% for replicate measurements (n = 6) of the standard solutions (0.02 to 2 μg L−1). Calibration curves were linear within 0 to 2 μg L−1 (r2 = 0.993 – 0.998). The procedure afforded an enhancement of at least 15-fold in sensitivity compared with the direct nebulization, which was also better than those reported previously (e.g., 4- to 10-fold) by different methods.4,10 No memory effects were observed between replicate measurements and among samples. A washout of 20 s with 4–5% HCl (at 4 mL min−1) was sufficient for cleaning up the system between samples.
The interferences from transition metals and hydride forming elements are summarized in Table 2. The values are given as percent relative response obtained from 10 μg L−1 Cd(II) solution containing the interfering element to that without the interfering element. For transition metals, including Co, Cu, Fe, Mn, Ni and Zn, no significant depression was observed from any of the elements when test concentration ranged from 0.1 to 1.0 μg mL−1. Responses varied between 99% (Ni) to 108% (Mn). Severe depression has been reported for as low as 0.1 μg mL−1 Cu(II) using thiourea-Co(II)6,9 and quartz traps.11 In this study, Cu(II) had no significant influence up to 1.0 μg mL−1. For the hydride forming elements, interferences were noted from Bi(III), Pb(II), Sb(III), and Sn(II), whereas As(III) and Se(IV) had no significant interference. These results were also consistent with previous reports.6–9,11 However, this procedure appeared to be more tolerant to these elements such that the effects of the interfering elements were alleviated at 0.1 μg mL−1 levels. Alkaline and alkaline earth elements (Ca, Mg, K, and Na) did not cause any interference in the presence of as high as 1000 μg mL−1 levels. Relative signals were 98% (Ca) and 103% (K).
Table 2.
Effects of transition metals and hydride forming elements on Cd vapour generation
| Element | Concentration/μg mL−1 | Relative signal/% |
|---|---|---|
| As(III) | 0.5 | 104 ± 6 |
| Bi(III) | 0.1 | 84 ± 3 |
| 0.5 | 60 ± 5 | |
| Co(II) | 1.0 | 99 ± 4 |
| Cu(II) | 1.0 | 99 ± 3 |
| Fe(III) | 1.0 | 105 ± 3 |
| Mn(II) | 1.0 | 108 ± 5 |
| Ni(II) | 1.0 | 102 ± 2 |
| Pb(II) | 0.1 | 94 ± 4 |
| 0.2 | 82 ± 4 | |
| 0.5 | 50 ± 3 | |
| Sb(III) | 0.1 | 93 ± 3 |
| 0.2 | 77 ± 5 | |
| 0.5 | 64 ± 4 | |
| Se(IV) | 0.5 | 103 ± 2 |
| Sn(II) | 0.1 | 86 ± 2 |
| 0.2 | 67 ± 4 | |
| 0.5 | 12 ± 3 | |
| Zn(II) | 1.0 | 101 ± 3 |
3.7 Analysis of samples
The results obtained from the analysis of certified reference materials (CRMs) are summarized in Table 3. The seawater (CASS-4) was analyzed by adjusting the acidity of sub-sample to 4% v/v HCl. Bone ash (SRM 1400) was digested in concentrated HNO3 as described in Section 2.6 and then acidified to 4% v/v HCl. Both samples were characterized by high total dissolved solids concentration. For instance, bone ash (SRM 1400) is mainly calcium phosphate for which analysis solutions of contained about 2000 μg mL−1 Ca, 2750 μg mL−1 PO4, 3.3 μg mL−1 Fe and 2.6 μg mL−1 Al along with about 0.9 μg mL−1 Zn and 0.045 μg mL−1 Pb. The results for Cd from these samples were consistent with the certified and indicative values. No significant interference was observed in highly saline seawater matrix nor did elevated levels of Ca, PO4, Al and Fe affect the accuracy.
Table 3.
The results for Cd from analysis of certified reference materials by chemical vapour generation ICP-MS. Results are given as mean ± standard deviation of five replicate analyses for each sample. Values in parenthesis are “information only”
| Sample | Isotope | Found | Certified value |
|---|---|---|---|
| Nearshore seawater (CASS-4)/μg L−1 | 110Cd | 0.024 ± 0.004 | 0.026 ± 0.003 |
| 111Cd | 0.024 ± 0.004 | ||
| Bone ash (SRM 1400)/μg g−1 | 110Cd | 0.026 ± 0.002 | (0.03) |
| 111Cd | 0.024 ± 0.002 | ||
| Dogfish liver (DOLT-4)/μg g−1 | 110Cd | 23.1 ± 1.1 | 24.3 ± 0.8 |
| 111Cd | 23.2 ± 0.9 | ||
| Mussel tissue (SRM 2976)/μg g−1 | 110Cd | 0.78 ± 0.02 | 0.82 ± 0.16 |
| 111Cd | 0.75 ± 0.01 |
The dogfish liver (DOLT-4) and mussel tissue (SRM 2976) are predominantly organic with high levels of iron. Thus, H2O2 was used to assist in mineralization during HNO3 digestions. Determinations were made in 2-fold diluted solutions for SRM 2976 and 50-fold diluted solutions for DOLT-4 to reduce the Cd concentration to the range of calibration standards. Unlike the seawater and bone ash samples, recoveries for these samples were low in initial preparations, about 75% for mussel tissue and 45% dogfish liver. A careful examination of the sample preparation procedures and compositional differences between calibration standards showed that the inaccuracy was caused by residual H2O2 in their analysis solutions. Trace amounts of H2O2 in the acidic sample solutions resulted in either inefficient formation of the hexacyanochromate(III) complex or accelerated its decomposition before interacting with NaBH4. In following analysis, a portion of the stock sample solutions were reheated dried at 120 °C for about 15–20 min to completely remove H2O2 and HNO3. Such treatment allowed accurate results to be achieved with the optimized conditions by using external calibration method (Table 3).
4. Conclusion
In this study, we have developed a new method and utilized potassium hexacyanochromate(III) for the first time for determination of Cd by chemical vapour generation ICP-MS. Aqueous solutions of potassium hexacyanochromate(III) were unstable diminishing its potential use in off-line vapour generation applications due the constantly degrading efficacy. These difficulties were overcome successfully by opting for online approach. Despite its instability, the results demonstrated that hexacyanochromate(III) could be generated in slightly acidic solutions through short interaction of Cr(OH)3 with dilute HCl in the presence of excess KCN. The results also demonstrated that hexacyanochromate(III) plays a key role in the generation of volatile species of Cd. At this point, it is proposed that hexacyanochromate(III) promotes the generation of volatile species through formation of reactive species upon reaction with NaBH4. Further studies should be conducted to elucidate the underlying mechanism of this process.
Additionally, the method is simple and offers accurate determination of Cd in variety of samples and salt matrices by ICP-MS. In comparison to previous vapour generation methods, this approach is virtually insensitive to interferences of common transition metals at μg mL−1 (ppm) levels. It is also relatively more tolerant sub-ppm concentrations of hydride forming elements (Bi, Sb, Sn, and Pb). The interferences of hydride forming elements, however, do not possess any limitation to the capability of the method since these elements are often present at μg L−1 or sub-μg L−1 levels in most samples. Interfacing to other atomic spectroscopy techniques, such as flame atomic absorption (FAAS) and inductively coupled plasma (ICP-AES) would greatly enhance the detection power of these techniques for Cd in analysis of complex samples from sediments to biological materials.
Acknowledgments
This project was supported by grants from the National Center for Research Resources (5 G12 RR013459-15), the National Institute on Minority Health and Health Disparities (8 G12 MD007581-15) and the Research Initiative for Scientific Enhancement (2 R25 GM067122) from the National Institutes of Health. The views expressed herein are those of authors and do not necessarily represent the official views of the NIH and any of its sub-agencies. The authors also acknowledge financial support from the Turkish Higher Education Council to Dr. Vedat Yilmaz during the course of this project.
Notes and References
- 1.Cacho J, Beltran I, Nerin C. J Anal At Spectrom. 1989;4:661. [Google Scholar]
- 2.Sanz-Medel A, Valdes-Hevia y Temprano MC, Bordel Garcia N, Fernandez de la Campa MR. Anal Chem. 1995;67:2216. [Google Scholar]
- 3.Matusiewicz H, Kopras M, Sturgeon RE. Analyst. 1997;122:331. doi: 10.1039/a606747f. [DOI] [PubMed] [Google Scholar]
- 4.Infante HG, Sanchez MLF, Sanz-Medel A. J Anal At Spectrom. 1998;13:899. [Google Scholar]
- 5.Feng Y-L, Sturgeon RE, Lam JW. Anal Chem. 2003;75:635. doi: 10.1021/ac020529+. [DOI] [PubMed] [Google Scholar]
- 6.Guo X, Guo X. Anal Chim Acta. 1995;310:377. [Google Scholar]
- 7.Vargas-Razo C, Tyson JF. Fresenius J Anal Chem. 2000;366:182. doi: 10.1007/s002160050036. [DOI] [PubMed] [Google Scholar]
- 8.Lampugnani L, Salvetti C, Tsalev DL. Talanta. 2003;61:683. doi: 10.1016/S0039-9140(03)00324-2. [DOI] [PubMed] [Google Scholar]
- 9.Chuachuad W, Tyson JF. Can J Anal Sci Spectrosc. 2004;49:362. [Google Scholar]
- 10.Li G, Wu L, Xin J, Hou X. J Anal At Spectrom. 2004;19:1010. [Google Scholar]
- 11.Korkmaz D, Demir C, Aydin F, Ataman OY. J Anal At Spectrom. 2005;20:46. [Google Scholar]
- 12.Chang Y-T, Jiang S-J. J Anal At Spectrom. 2008;23:140. [Google Scholar]
- 13.Alp O, Demiroz H, Ataman OY, Ertas N. Turk J Chem. 2012;36:247. [Google Scholar]
- 14.Manzoori JL, Abdolmohammad-Zadeh H, Amjadi M. Talanta. 2007;71:582. doi: 10.1016/j.talanta.2006.04.036. [DOI] [PubMed] [Google Scholar]
- 15.Sahan S, Sahin U. Talanta. 2012;88:701. doi: 10.1016/j.talanta.2011.11.069. [DOI] [PubMed] [Google Scholar]
- 16.D’Ulivo A, Baiocchi C, Pitzalis E, Onor M, Zamboni R. Spectrochim Acta, Part B. 2004;59:471. [Google Scholar]
- 17.D’Ulivo A. Spectrochim Acta, Part B. 2004;59:793. [Google Scholar]
- 18.Valdes-Hevia y Temprano MC, Fernandez de la Campa MR, Sanz-Medel A. J Anal At Spectrom. 1994;9:231. [Google Scholar]
- 19.Hwang T-J, Jiang S-J. J Anal At Spectrom. 1997;12:579. [Google Scholar]
- 20.Arbab-Zavar MH, Chamsaza M, Youssefi A, Aliakbari M. Anal Chim Acta. 2005;546:126. doi: 10.1016/j.aca.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 21.Arbab-Zavar MH, Chamsaz M, Youssefi A, Aliakbari M. Talanta. 2012;97:229. doi: 10.1016/j.talanta.2012.04.022. [DOI] [PubMed] [Google Scholar]
- 22.Pourreza N, Ghanem K. J Hazard Mater. 2010;78:566. doi: 10.1016/j.jhazmat.2010.01.122. [DOI] [PubMed] [Google Scholar]
- 23.Jamoussi B, Zafzouf M, Hassine B. Fresenius’ J Anal Chem. 1996;356:331. doi: 10.1007/s0021663560331. [DOI] [PubMed] [Google Scholar]
- 24.Pohl P, Zyrnicki W. Anal Chim Acta. 2002;468:71. [Google Scholar]
- 25.Chadwick BM, Sharpe AG. In: Advances in Inorganic Chemistry and Radiochemistry. Emeleus HJ, Sharpe AG, editors. Vol. 8. Academic Press; New York: 1966. pp. 84–162. [Google Scholar]
- 26.Cotton FA, Wilkinson G. Advanced Inorganic Chemistry – A Comprehensive Text. 2. Interscience Publishers; New York: 1966. [Google Scholar]
- 27.Ertas N, Arslan Z, Tyson JF. J Anal At Spectrom. 2008;23:223. [Google Scholar]
- 28.Elci L, Arslan Z, Tyson JF. J Hazard Mat. 2009;162:880. doi: 10.1016/j.jhazmat.2008.05.113. [DOI] [PubMed] [Google Scholar]
- 29.Chuachuad W, Tyson JF. J Anal At Spectrom. 2005;20:282. [Google Scholar]
- 30.Chen S, Zhang Z, Yu H, Liu W, Sun M. Anal Chim Acta. 2002;463:177. [Google Scholar]
- 31.D’Ulivo, Onor M, Spiniello R, Pitzalis E. Spectrochim Acta, Part B. 2008;63:835. [Google Scholar]
- 32.Afonso D, Arslan Z, Baytak S. J Anal At Spectrom. 2010;25:726. doi: 10.1039/b920280c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferneliues WC. Inorganic Syntheses. Vol. 2. McGraw-Hill Book Company, Inc; New York: 1946. pp. 203–207. [Google Scholar]