

Abstract
The thiazolidinedione, compound 1, has previously shown pan-inhibition of the phosphoinositide 3-kinase (PI3K) class I isoforms. We hypothesized the derivatization of the thiazolidinedione core of compound 1 could introduce isoform selectivity. We report the synthesis, characterization, and inhibitory activity of a novel series of 4-iminothiazolidin-2-ones for inhibition of the class I PI3K isoforms. Their synthesis was successfully achieved by multiple pathways described in this paper. Initial in vitro data of 28 analogues demonstrated poor inhibition of all class I PI3K isoforms. However, we identified an alternate target, the phosphodiesterases, and present preliminary screening results showing improved inhibitory activity.
Introduction
The thiazolidinediones (TZDs) are one of the most widely examined compound classes in medicinal chemistry. Several representatives have been used clinically while analogous compound families such as the rhodanines (2-thioTZD) and 2-iminothiazolidin-4-ones have been extensively studied in medicinal chemistry programs. In contrast, there has been very little research reported of the synthesis or biology of 4-iminothiazolidin-2-ones. Their first reported synthesis was in the 1960s (Fig. 1, structures I–III).[1–3] The body of published literature currently is encompassed in just 30 research articles. These, together with 10 other patents, embody 262 analogues of this structural class with a further 800 compounds commercially available. Biological applications for these compounds include a report by Kim et al. of the modest inhibition of GSK-3β by compound V,[4] and compound VII was described in a compound library of potential antimalarial leads.[5]
Fig. 1.

Literature examples of 4-iminothiazolidin-2-ones.
The thiazolidinedione 2 is a potent inhibitor of phosphoinositide 3-kinase (PI3K). Our previously reported evaluation of the inhibition of PI3K by thiazolidinedione and rhodanine derivatives suggested that elaboration through the 4-position might provide for interaction with non-conserved residues of the binding site and thus induce PI3K isoform selectivity.[6,7] We prepared a series of 5-arylidene-4-substituted iminothiazolidin-2-ones (Table 1) by multiple pathways described further. Although each pathway proved successful, we found that certain pathways produced trace amounts of a biologically active impurity giving false positives in screening assays.[8] While the majority of compounds assayed were inactive against PI3K, inhibitory activity was observed for an alternate enzyme family, the phosphodiesterases (PDEs).
Table 1.
Synthesized 4-iminothiazolidin-2-ones
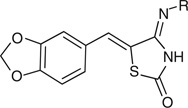 | |||
|---|---|---|---|
| Number | R | Yield [%] | Mp [°C] |
| 7a | -Ph | 100 | 268–270 |
| 7b | -PhCH2 | 97 | 221–223 |
| 7c | 67 | 213–215 | |
| 7d | 89 | 268 | |
| 7e | 73 | 269 | |
| 7f | 42 | 262–265 | |
| 7 g | 20 | 232–235 | |
| 7h | 100 | 108–110 | |
| 7i | L-Trp(OMe) | 8 | 89 |
| 7j | 17 | 169 | |
| 7k | 16 | 213–215 | |
| 7l | 38 | 195–198 | |
| 7m | 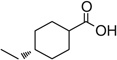 |
100 | 249 |
| 7n | D-Trp(OH) | 50 | 94 |
| 7o | 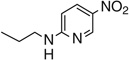 |
77 | 248 |
| 7p | 87 | 268 | |
| 7q | 57 | 244 | |
| 7r | 51 | 215–217 | |
| 7s | 42 | 212–214 | |
| 7t | 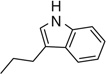 |
56 | 248–251 |
| 7u | 35 | 223–225 | |
| 7v | 41 | 226–228 | |
| 7w | 23 | 210 | |
| 7x | -Me | 74 | 285 |
| 7y | -Et | 59 | 230 |
| 7z | 34 | 228–230 | |
| 7aa | -L-Ala(OH) | 51 | 233 |
| 7ab | -L-Trp(OH) | 51 | n.d. |
n.d., not determined
Chemistry
The synthesis of arylidene-4-iminothiazolidin-2-ones was achieved from commercially available thiazolidinedione 1 by several possible routes shown in Scheme 1, using thionation, Knoevenagel condensation, and amine displacement reactions in any of three sequences.
Scheme 1.

Synthesis of 4-iminothiazolidin-2-ones. Reagents and conditions: (a) Piperonal (0.56 equiv), β-alanine (1 equiv), CH3CO2H, reflux, 3 h, 100 %. (b) 1,4-Dioxane, P2S5 (0.36 equiv), reflux, 2.5 h, 56 %. (c) MeI (5 equiv), N-methyl-2-pyrrolidinone, N,N-diisopropylethylamine (1.15 equiv), room temp., 2 h, 87 %. (d) NH2R (1–5 equiv), MeOH or EtOH, reflux, 18 h, 100 %. (e) 1 N NaOH/ethanol (1 : 2, 7.5 mL), reflux, 5 h, 39 %.
When following the general route of Komaritsa and Plevichuk,[1,3] (1→3→6→7), treatment of thiazolidinedione 1 with phosphorus pentasulfide gave isorhodanine 3 in 56% yield after recrystallization from 1,2-dichloroethane.[1,9] A single carbonyl peak was observed in the isorhodanine IR spectrum at 1700 cm−1, consistent with the literature value.[9]
Treatment of isorhodanine 3 with various amines gave the corresponding 4-iminothiazolidin-2-ones 6a–6f in good yields and purity, with products precipitating from the reaction mixture and then collected by filtration. The products were reacted on directly without further purification.
Compounds 6a–6f were treated with piperonal under Knoevenagel conditions to give the arylidene substituted compounds 7a–7f. The desired products precipitated from the acetic acid solvent upon cooling to give excellent yields and high purity. In samples 7e and 7f a trace contaminant was observed at a retention time of 8.9 min (Fig. 2). Although apparently negligible, the nature and importance of this contaminant was indeed significant as discussed later.
Fig. 2.

Analytical high-performance liquid chromatography trace at 254nm of compound 7f showing the impurity with retention time of 8.90 min.
An alternate synthesis of 4-iminothiazolidin-2-ones by pathway 1 → 3 → 4 → 5 → 7 proved most satisfactory. Condensation of piperonal with isorhodanine 3 to compound 4 (Scheme 1) gave a quantitative yield. The critical synthetic step proved to be the S-methylation of precursor 4 to give the intermediate compound 5 in 87% yield after precipitation from ethyl acetate. Compound 5 was an excellent substrate for substitution reactions with amines such as n-pentylamine affording the crystalline product 7f in 42% yield. In this case, no sign of the contaminant described above was observed.
Twenty-two compounds 7e–7z were prepared by the thiomethyl intermediate 5 in 16–100% yield as a precipitate from EtOH, shown in Table 2. Full details of the synthesis of all analogues are provided in the Supplementary material. We were also able to utilize a solid phase adaptation of the method to afford the carboxylic acid derivatives 7aa and 7ab (Scheme 2).
Table 2.
Phosphoinositide 3-kinase (PI3K) and phosphodiesterase (PDE) inhibition by selected 4-iminothiazolidin-2-ones
| Compound | IC50 × 10−6 [M] | |||||
|---|---|---|---|---|---|---|
| PI3Kα inhibition | PI3Kβ inhibition | PI3Kγ inhibition | PI3Kδ inhibition | PDE2 inhibition | PDE4 inhibition | |
| 2 | 0.05 | 0.6 | 0.04 | n.d. | n.d. | n.d. |
| 4 | 0.45 | 0.15 | 0.12 | 0.76 | n.d. | n.d. |
| 5 | 0.056 | >1 | 0.06 | 0.038 | n.d. | n.d. |
| 7p | >100 | >100 | >100 | >100 | 4.7 | 5.1 |
| 7x | 90 | >100 | >100 | >100 | >10 | >10 |
| 7z | >100 | >100 | >100 | >100 | >10 | >10 |
| 7aa | 19 | 94 | 14.5 | 62 | n.d. | n.d. |
| 7ab | 26 | 80 | 11 | 43 | n.d. | n.d. |
n.d., not determined; IC50, half maximum inhibitory concentration
Scheme 2.
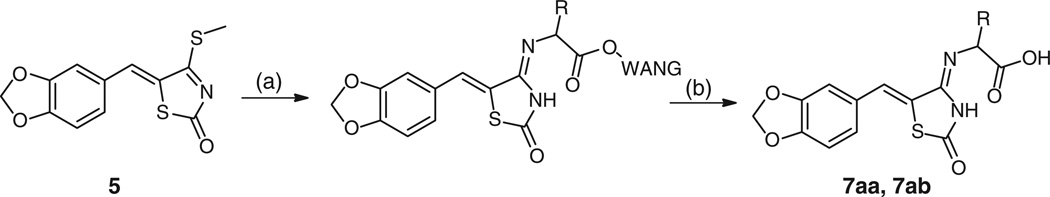
Synthesis of 4-iminothiazolidin-2-one amino acids from Wang resin. Reagents and conditions: (a) DMF, piperidine (20 %), Fmoc-R1-Wang resin. (ii) H2N-R1-Wang, DMF, 80°C, overnight. (b) TFA (95 %), H2O (5 %), 51 %.
We also commenced the third possible route (1 → 2 → 4) shown in Scheme 1.[3] It was abandoned due to difficult isolation of compound 4 from the precursor 2 despite attempts at purification. However, the investigation of this reaction led us to identify the contaminant found in 7e and 7f described above, as compound 2. Moreover, compound 2 was sometimes found as a by-product of the synthesis of compound 4 from isorhodanine 3. In the worst case, the base peak (electrospray ionization timeof- flight mass spectrometry, ESI-TOF MS) for the desired material (m/z 263.9733) was accompanied by another for compound 4 (m/z 247.9996, 55 %). It should also be noted that the corresponding thiorhodanine product was also identified as a small component in the mass spectrum (m/z 279.9279, 7 %). We identified compound 2 as the contaminant in the products 7e and 7f, by the match of high-performance liquid chromatography (HPLC) retention, parent MS adduct, and 1H NMR spectrum. The specific alkene δ7.72 and aromatic proton δ7.17–7.11 NMR signals were shown to match the authentic material compound 2.
We investigated the purity of the isorhodanine 3 samples, by IR and NMR spectroscopy, and attribute the contamination to trace thiazolidinedione 1, either retained in the second crop from the recrystallization of 3 or obtained upon storage, and carried through subsequent synthetic steps. The IR and NMR spectra of the various samples of isorhodanine 3 showed the apparent heterogeneity from isolation or storage. The presence of thiazolidinedione in samples was apparent as a shoulder at 1730 cm−1 in the carbonyl stretching region, whereas isorhodanine absorbs at 1700 cm−1.[9] The 1H NMR spectrum shows a resonance for the methylene protons at δ4.1 corresponding to the thiazolidinedione as compared to δ4.6 for isorhodanine.
From the perspective of our research this was an important result as compound 2 is a potent PI3K inhibitor (IC50 v. PI3Kα=50 × 10−9 M). Compounds 7e and 7f initially showed significant (IC50 ~ 5 × 10−6 M) inhibitory activity at PI3K isoforms due entirely to the residual contaminant 2 in the sample. We found that the same compounds synthesized by compound 5 as a key intermediate showed no inhibition of PI3K.
Results and Discussion
Of the 28 compounds synthesized, we found only two displayed modest inhibition of PI3K isoforms. Therefore, modification at the 4-position of the thiazolidinedione group severely disrupted enzyme-inhibitor binding. Even the very small N-methyl substitution effectively abolished the activity of 7x (IC50>90 × 10−6 M, Table 2). The exceptions were the amino acid derivatized analogues: 7aa (Gly) and 7ab (L-Trp) amino acid derivatives. These showed modest inhibition against certain PI3K isoforms with an IC50 range of 10–20 × 10−6 M (Table 2).
With the compounds in hand we were able to investigate their activity at other targets. Previously, Irvine et al. has described inhibition of PDE4 by arylidene rhodanine derivatives, so a sample set of 4-iminothiazolidin-2-ones were screened against both PDE4 and PDE2.[10] We tested compounds 7p, 7x, and 7z. Compounds 7x and 7z were virtually inactive, while the compound 7p showed robust inhibition of PDE2 and PDE4 with IC50 values 4.7 × 10−6 and 5.1 × 10−6 M. This compound showed no inhibition of PI3K isoforms. The activity of the analogue bearing the catechol ether moiety characteristic of PDE inhibitors suggests that the 3,4-dimethoxybenzyliminothiazolidinone portion is the key pharmacophore for this activity.[10] This is the first description of PDE inhibition by this compound class. As a derivation of the privileged thiazolidinedione structure, these compounds warrant broader investigation as bioactive ligands.
This work also provides a significant warning to the screening of compounds en masse without thorough consideration of compound purity. Commercial compound libraries are typically sold with purities advertised as >90% by HPLC and NMR analysis. A library of compounds derived by our first method could easily fall within such specifications.[8] Gonzalez and Polli report an impurity of less than 2.5% is a reasonable target, provided the impurity is less than 10-fold more potent than the test compound.[11] Fortunately for this work, the possible formation and activity of this impurity was recognized, which led to the adoption of a more suitable synthetic pathway.
The related rhodanine class has been identified as a group of Pan-Assay Interference compounds (PAINS) and it is within the realms of possibility that this class might also deliver ‘nuisance’ compounds.[8] Our work does not have the scope to confirm or deny this possibility, which would require a broad suite of screening, stability, and/or reactivity assays. However, we can confirm that active trace impurities, another characteristic of PAINS, may need to be considered when preparing compounds of this class. Our description of the outcomes of these syntheses will hopefully assist in future analyses.
Conclusion
In conclusion, we have established an expedient route to the synthesis of 4-iminothiazolidin-2-ones with potential for activity against a wide range of biological targets. While the 4-iminothiazolidin-2-ones were relatively poor inhibitors of the class I PI3K isoforms, the class is worthy of evaluation against the many other targets of thiazolidinediones, as exemplified by the observed inhibition of PDE4 and PDE2 by one representative compound.
Experimental
General
All chemical reagents acquired from Sigma–Aldrich, Fluka, Merck, BDH laboratories, CSL, Ajax Finechem, Merck Schuhardt, ChemSupply, Auspep, Prolabo, Lancaster, TCI, Matrix Scientific, Boron Molecular, Alfa Aesar, Chem-Impex, and May and Baker were used without further purification. 1H NMR spectra were recorded with either a 300MHz Varian widebore NMR spectrometer or a 400 MHz Bruker Ultrashield-Avance III NMR spectrometer. 13C NMR spectra were recorded with either a 600MHz Varian Oxford AS600-Unity Inova NMR spectrometer or a 300MHz Varian Widebore NMR spectrometer. Results were recorded as follows: chemical shift values are expressed in δ units acquired in either CDCl3 (7.26 ppm), (CD3)2SO (2.50 ppm) or CD3OD (3.31 ppm) as references, multiplicity (s=singlet, d=doublet, t=triplet, q= quartet, m=multiplet), integration and coupling constants (J) are reported in Hertz. Infrared spectra were recorded on a Scimitar Series Varian 800 FT-IR Spectrometer fitted with a PIKE Technologies MIRacle ATR and running Varian’s Resolutions software package version 4.0. Mass spectra were acquired in the positive and negative mode using an atmospheric pressure (ESI/atmospheric pressure chemical ionization, APCI) ion source on a Micromass Platform II ESI/APCI single quadrupole mass spectrometer with sample management facilitated by an Agilent 1100 series HPLC system using MassLynx version 3.5 software. High resolution MS analyses were collected on a Waters Micromass LCT Premier XE Orthogonal Acceleration Time-of -Flight Mass Spectrometer coupled to an Alliance 2795 Separation Module using MassLynx version 4.1 software. Analytical reverse phase (RP)-HPLC was obtained on a Waters Millenium 2690 system, with UV detection at 254 nm, with gradient elution through a Supelco C8 column (150 × 2.1 mm ID) 80–20% Buffer B (Buffer A: H2O, 0.1% TFA; Buffer B: 80% CH3CN, 0.1% TFA, 19.9% H2O or Buffer B: 80% CH3OH, 0.1%TFA, 19.9% H2O) over 10 min at 1.0 mL min−1. Preparative RP-HPLC was obtained on a Waters 600 HPLC system, with UV detection at 254 nm with gradient elution through a Phenomonex Luna C8 column (250 × 20mm ID), 20–90% Buffer B (Buffer A: H2O, 0.1% TFA; Buffer B: 80% CH3CN, 0.1% TFA, 19.9% H2O) over 15 min at 10 mL min−1. Melting point determination was performed uncorrected using a Mettler Toledo MP50 melting point apparatus.
General Procedure for the Preparation of 4-Thioxothiazolidin-2-one (Isorhodanine)
To a mixture of P2S5 (1.4 g, 6.1 mmol, 1 equiv) in dry 1,4-dioxane (10.0 mL) was added thiazolidinedione (2.0 g, 2.8 equiv), and the mixture heated to reflux for 3.5 h. The mixture was cooled, the product collected by filtration, and solvent removed under reduced pressure. The crude product was crystallized from dichloroethane to yield the title compound as a brown powder. The following compound 3 was prepared in this manner.
4-Thioxothiazolidin-2-one 3
Light brown powder (1.5 g, 67.9 %), mp 151–155°C, also 160–5°C; (lit. 145–50°C, 158–162°C).[1,9] δH (300.13 MHz, DMSO) 4.61 (s, 2H, CH2), m/z (ESI) 132.0 [M−H]−. νmax/cm−1 3347.3, 3112.7, 2950.2, 2910.7, 1701.0, 1464.9, 1383.8, 1170.1, 1119.1, 1008.4, 786.0, 749.6, 705.4, 561.9.
General Procedure for the Preparation of Monosubstituted 4-Iminothiazolidin-2-ones
To a solution of isorhodanine 3 (0.10 g, 0.75 mmol, 1 equiv) in MeOH (1.0 mL) was added aniline (0.070 g, 1 equiv), and the mixture was refluxed for 1.5 h. The mixture was cooled, the product collected by filtration, washed with MeOH, then dried to yield the product 6a. If solid not isolated, then product was reacted on as an oil in crude form. The following substituted 4-iminothiazolidin-2-ones 6a–6d were prepared in this manner.
4-(Phenylimino)thiazolidin-2-one 6a
Off-white crystals (0.075 g, 51.9 %), mp 208–211°C (lit 221– 222°C).[1] δH (300.13 MHz, DMSO) 10.95 (br s, 1H, NH), 7.73 (d, J 8.3, 2H, ArH), 7.40 (t, J 7.5, 2H, ArH), 7.17 (t, J 7.4, 1H, ArH), 4.52 (s, 2H, CH2). m/z (ESI, 20 V) 193.0 (100 %, M+•).
(Z)-4-(Benzylimino)thiazolidin-2-one 6b
White crystals (0.036 g, 33.2 %). δH (300.13 MHz, DMSO) 9.47 (br s, 1H, NH), 7.35 (m, 5H, ArH), 4.55 (d, J 7.9, 2H, SCH2), 4.27 (s, 2H, ArCH2). m/z (ESI, 20 V) 207.1 (100 %, M+•).
(Z)-4-(sec-Butylimino)thiazolidin-2-one 6c
Yellow oil (0.088 g, 97.8 %), δH (300.13 MHz, CDCl3) 5.73 (br s, 1H, NH), 4.20 (m, 3H, NCH, SCH2), 1.65 (m, 2H, CH2CH3), 1.27 (d, J 6.6, 3H, CHCH3), 0.97 (m, 3H, CH2CH3). m/z (ESI, 20 V) 173.1 (100 %, M+•).
(Z)-4-(Bicyclo[2.2.1]heptan-7-ylimino)thiazolidin-2-one 6d
Orange crystals (0.045 g, 50.0 %), HPLC: purity >85 %, 6.28 min.
General Procedure for the Preparation of Benzodioxol Thiazolidinediones and 4-Amino Substituted Benzodioxol Thiazolidinediones (Knoevenagel Condensation)
A mixture of piperonal (0.72 g, 4.79 mmol, 1 equiv), β-alanine (0.76 g, 1.8–2 equiv), and isorhodanine 3 (1.14 g, 1.8–2 equiv) in glacial acetic acid (20.0 mL) was heated to 100°C for 2–3 h. The reaction mixture was cooled, the product was collected by filtration, washed with acetic acid, and then water to yield the title compound as a yellow crystalline solid, unless otherwise indicated. The following benzodioxol thiazolidinedione 2, isorhodanine 4, and 4-amino substituted benzodioxol thiazolidinediones 7a–7d were prepared in this manner.
(Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene) thiazolidine-2,4-dione 2
Yellow powder (0.43 g, 99 %), mp 246–249°C (lit. 247–249°C).[12] δH (300.13 MHz, DMSO) 12.5 (br s, 1H, NH), 7.71 (s, 1H, CH), 7.14 (m, 3H, ArH), 6.13 (s, 2H, CH2). m/z (ESI 20 V) 248.1 (100 %, M−•). HPLC purity >95 %.
(Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-thioxothiazolidin-2-one 4
Red crystals (1.09 g, 100 %), Mp 264°C (dec.). δH (300.13 MHz, DMSO) 13.9 (br s, 1H, NH), 8.04 (s, 1H, CH), 7.27 (d, J 8.1, 1H, ArH), 7.21 (s, 1H, ArH), 7.10 (d, J 8.2, 1H, ArH), 6.15 (s, 2H, CH2). δC (75.5 MHz, DMSO) 195.14, 170.63, 150.02, 148.45, 136.43, 127.74, 127.63, 127.31, 109.46, 109.46, 102.33. m/z (ESI 20V) 264.1 (100 %, M−•). HPLC: purity >99 %, 10.36 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-(phenylimino)thiazolidin-2-one 7a
A solution of (Z)-4-(phenylimino)thiazolidin-2-one 6a (0.050 g, 0.26 mmol) in acetic acid (1 mL) was reacted with piperonal (0.021 g, 0.13 mmol) and after filtering afforded a yellow powder (0.057 g, 100 %). Mp 268–270°C. δH (300.13 MHz, DMSO) 10.67 (br s, 1H, NH), 8.05 (s, 1H, CH), 7.76 (d, J 8.1, 2H, ArH), 7.44 (t, J 7.9, 2H, ArH), 7.15 (m, 4H, ArH), 6.12 (s, 2H, CH2). m/z (ESI, 20V) 325.1 (100 %, M+•). m/z (HR-ESI) Calc. for C17H12N2O3S, [M+H]+ : 325.0647. Found 325.0640. HPLC purity 90 %, 8.20 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-(benzylimino)thiazolidin-2-one 7b
A solution of (Z)-4-(benzylimino)thiazolidin-2-one 6b (0.030 g, 0.15 mmol) in acetic acid (1 mL) was reacted with piperonal (0.013 g, 0.09 mmol) and after filtering afforded green crystals (0.029 g, 96.8 %). Mp 221–223°C. δH (300.13 MHz, DMSO) 9.75 (br s, 1H, NH), 7.81 (s, 1H, CH), 7.37 (d, J 8.1, 3H, ArH), 7.33 (s, 1H, ArH), 7.31 (dt, J 9.0, 4.5, 1H, ArH), 7.08 (s, 1H, ArH), 7.06 (d, J 11.0, 2H), 6.12 (s, 2H, CH2), 4.31 (t, J 5.1, 2H, NCH2); m/z (ESI, 20V) 339.3 (100 %, M+•). HPLC purity >94 %.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-(sec-butylimino)thiazolidin-2-one 7c
A solution of (Z)-4-(sec-butylimino)thiazolidin-2-one 6c (0.080 g, 0.46 mmol) in acetic acid (1.5 mL) was reacted with piperonal (0.040 g, 0.27 mmol) and after filtering afforded yellow crystals (0.054 g, 66.9 %). Mp 213–215°C. δH (300.13 MHz, DMSO) 8.93 (br s, 1H, NH), 7.80 (s, 1H, CH), 7.07 (d, J 9.9, 3H, ArH), 6.12 (s, 2H, CH2), 4.32 (m, 1H, NCH), 1.62 (m, 2H,CH2), 1.23 (d, J 6.6, 3H, CHCH3), 0.90 (t, J 7.4, 3H, CH2CH3). m/z (ESI, 20 V) 305.1 (100 %, M+•). m/z (HR-ESI) Calc. for C15H16N2O3S, [M+H]+ 305.0960. Found 305.0955. HPLC purity >96 %.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-(bicyclo[2.2.1]heptan-7-ylimino)thiazolidin-2-one 7d
A solution of (Z)-4-(bicyclo[2.2.1]heptan-7-ylimino) thiazolidin-2-one 6d (0.045 g, 0.21 mmol) in acetic acid (3 mL) was reacted with piperonal (0.018 g, 0.12 mmol) and after filtering afforded brown crystals (0.055 g, 88.9 %). Mp 268°C (decomp.). δH (300.13 MHz, DMSO) 8.71 (d, J 5.3, 1H, NH), 7.87 (s, 1H, CH), 7.07 (d, J 11.7, 3H, ArH), 6.12 (s, 2H, CH2), 3.83 (s, 1H, NCH), 2.33 (s, 2H, CH2), 1.74 (m, 1H, CH), 1.63 (m, 2H, CH2), 1.49 (t, J 10.1, 2H, CH2), 1.20 (d, J 9.9, 3H, CH, CH2). m/z (HR-ESI) Calc. for C18H18N2O3S, [M+H]+ : 343.1111. Found 343.1122. HPLC purity 95 %, 11.41 min.
General Procedure for the Preparation of (Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-(methylthio) thiazol-2(5H)-one 5
To a solution of 5-benzo[1,3]dioxol-5-ylmethylene-4-thioxothiazolidin-2-one 4 (0.50 g, 1.9 mmol, 1 equiv) and N,N-diisopropylethylamine (DIPEA, 0.27 g, 1.1 equiv) in N-methyl 2-pyrrolidinone (NMP, 20 mL) was added methyl iodide (1.27 g, 5 equiv) in N-methyl 2-pyrrolidinone (2.5 mL). The reaction mixture was stirred for 2 h at room temperature, diluted with ethyl acetate (45 mL), washed with brine, and then water. Solvent was removed to 50%volume, resulting in the product 5 collected by filtration as a yellow solid. The following thiomethyl benzodioxol 5 was prepared in this manner.
(Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-(methylthio)thiazol-2(5H)-one 5
Yellow powder, (0.49 g, 86.5 %). δH (300.13 MHz, DMSO) 7.81 (s, 1H, CH), 7.35 (dd, J 8.3, 1.6, 1H, ArH), 7.28 (d, J 1.6, 1H, ArH), 7.13 (d, J 8.2, 1H, ArH), 6.16 (s, 2H, CH2), 2.76 (s, 3H, SCH3). m/z (ESI 20 V) 280.2 (100 %, M+•). HPLC: purity >98 %, 11.05 min.
General Procedure for the Preparation of 4-Substituted Iminothiazolidin-2-ones
To a solution of 5-benzo[1,3]dioxol-5-ylmethylene-4-methylsulfanyl-thiazol-2-one 5 (0.030 g, 0.11 mmol, 1 equiv) was added an amine such as cyclopentylamine (0. 079 g, 5 equiv) in EtOH (4.0–9.0 mL) and the mixture refluxed at 85°C for 19–64 h. After cooling, the organic layer was reduced to 50 %, the product was collected by filtration and washed with chilled ethanol to yield the title compound as a yellow solid, unless otherwise indicated. The following 4-substituted iminothiazolidin-2-ones 7e–z were prepared in this manner.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-((4-hydroxyphenethyl)imino)thiazolidin-2-one 7e
Yellow powder (0.048 g, 72.5 %). Mp 269°C (decomp). δH (300.13 MHz, DMSO) 9.35 (br s, 1H, NH), 9.19 (s, 1H, OH), 7.71 (s, 1H, CH), 7.06 (dd, J 8.1, 5.4, 5H, ArH), 6.70 (d, J 8.3, 2H, ArH), 6.12 (s, 2H, CH2), 3.64 (t, J 7.5, 2H, NCH2), 2.83 (t, J 7.5, 2H, ArCH2). m/z (ESI, 20 V) 369.3 (100 %, M+•). m/z (HR-ESI) Calc. for C19H16N2O4S, [M−H]−: 367.0758. Found 367.0737. HPLC purity >95 %, 9.51 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-(cyclopentylimino)thiazolidin-2-one 7f
Yellow crystals (0.010 g, 41.9 %). Mp 262–265°C. δH (300.13 MHz, DMSO) 9.00 (br s, 1H, NH), 7.80 (s, 1H, CH), 7.06 (d, J 10.8, 3H, ArH), 6.11 (s, 2H, CH2), 4.32 (s, 1H, NCH), 1.98 (m, 2H, CH2), 1.67 (m, 6H, CH2). δC (75.5 MHz, DMSO) 176.64, 172.37, 148.63, 148.06, 128.43, 127.40, 126.42, 125.39, 109.05, 108.25, 101.85, 56.37, 31.74, 23.68. m/z (ESI, 20 V) F J.-A. Pinson et al. 317.1 (100 %, M+•). m/z (HR-ESI) Calc. for C16H16N2O3S, [M+H]+: 317.0954. Found 317.0966. HPLC purity >99 %, 10.60 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-((3-morpholinopropyl)imino)thiazolidin-2-one 7g
Yellow powder (0.011 g, 20.3 %). Mp 232–235°C. δH (300.13 MHz, DMSO) 8.21 (s, 1H, NH), 7.71 (s, 1H, CH), 7.06 (s, 1H, ArH), 7.04 (d, J 13.2, 2H, ArH), 6.13 (s, 2H, CH2), 3.53 (s, 4H, OCH2), 2.31 (s, 4H, CH2), 1.76 (m, 2H, CH2). m/z (HR-ESI) Calc. for C18H21N3O4S, [M+H]+ : 376.1331. Found 376.1329. HPLC purity >95 %, 8.80 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-((4-methylpiperazin-1-yl)imino)thiazolidin-2-one 7h
Yellow powder (0.060 g, 100 %). Mp 108–110°C. δH (300.13 MHz, DMSO) 7.56 (s, 1H, CH), 7.03 (m, 3H, ArH), 6.09 (s, 2H, CH2), 3.03 (t, J 6.3, 2H, CH2), 2.97 (t, J 3.6, 6H, CH2), 2.22 (s, 3H, NCH3). m/z (ESI, 20 V) 347.1 (100 %, M+•). HPLC purity >94 %, 6.58 min.
(S)-Methyl 2-((Z)-((Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-2-oxothiazolidin-4-ylidene)amino)-3-(1H-indol-2-yl)propanoate 7i
Yellow powder (0.005 g, 7.8 %). Mp 89°C (decomp). m/z (ESI, 20 V) 450.0 (100 %, M+•). HPLC purity >99%, 10.79 min.
(4Z,5Z)-4-((3-(1H-Imidazol-1-yl)propyl)imino)-5-(benzo[d][1,3]dioxol-5-ylmethylene)thiazolidin-2-one 7j
Yellow crystals (0.011 g, 16.6 %). Mp 169°C (decomp.). δH (300.13 MHz, DMSO) 7.69 (d, J 8.3, 1H, ArH), 7.60 (s, 1H, CH), 7.12 (m, 4H, ArH), 6.88 (d, J 8.8, 1H, ArH), 6.09 (s, 2H, CH2), 4.02 (m, 2H, NCH2), 2.08 (m, 2H, NCH2), 1.76 (m, 2H, CH2). m/z (ESI, 20 V) 357.0 (100 %, M+•). m/z (HR-ESI) Calc. for C17H16N4O3S, [M+H]+: 357.1016. Found 357.1025. HPLC purity >90 %, 6.30 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-((2-morpholinoethyl)imino)thiazolidin-2-one 7k
Yellow crystals (0.011 g, 16.2 %). Mp 213–215°C. δH (300.13 MHz, DMSO) 7.71 (s, 1H, CH), 7.06 (d, J 11.8, 3H, ArH), 6.12 (s, 2H, CH2), 3.58 (m, 4H, OCH2), 2.57 (dt, J 18.5, 6.6, 2H, NCH2), 2.42 (t, J 4.5, 4H, NCH2), 2.30 (m, 2H, NCH2). δC (75.5 MHz, DMSO) 176.80, 173.22, 148.84, 148.22, 128.35, 127.37, 126.37, 125.68, 109.24, 108.34, 102.03, 66.22, 56.48, 55.33, 41.67. m/z (HR-ESI) Calc. for C17H19N3O4S, [M+H]+: 362.1169. Found 362.1183. HPLC purity >90 %, 6.23 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-(((2,2-dimethyl-1,3-dioxolan-4-yl)methyl)imino)thiazolidin-2-one 7l
Yellow crystals (0.025 g, 38.1 %). Mp 195–198°C. δH (300.13 MHz, DMSO) 7.80 (s, 1H, CH), 7.07 (d, J 11.8, 3H, ArH), 6.12 (s, 2H, CH2), 4.33 (t, J 5.7, 1H, OCH), 4.04 (t, J 8.4, 1H, OCH), 3.75 (t, J 8.4, 1H, OCH), 3.65 (t, J 5.1, 2H, NCH2), 1.37 (s, 3H, CH3), 1.26 (s, 3H, CH3). δC (75.5 MHz, DMSO) 176.86, 173.60, 148.89, 148.21, 128.29, 127.82, 126.04, 125.74, 109.22, 108.87, 108.33, 102.02, 73.35, 66.56, 47.32, 26.86, 25.36. m/z (HR-ESI) Calc. for C17H18N2O5S, [M+H]+: 363.1009. Found 363.1021. HPLC purity >95 %, 9.47 min.
4-(((Z)-((Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-2-oxothiazolidin-4-ylidene)amino)methyl) cyclohexanecarboxylic acid 7m
Yellow crystals (0.082 g, 100 %). Mp 249°C (decomp.). δH (300.13 MHz, DMSO) 9.28 (br s, 1H, NH), 7.77 (s, 1H, CH), 7.07 (d, J 10.4, 3H, ArH), 6.13 (s, 2H,CH2), 4.32 (s, 1H, COCH), 3.45 (dd, J 6.9, 2.3, 2H NCH2), 2.15 (t, J 11.7, 1H,CHCH2), 1.86 (dd, J 36.8, 13.5, 4H, CH2), 1.66 (s, 1H, CH2), 1.30 (dd, J 24.9, 10.6, 2H, CH2), 1.02 (m, 1H, CH2). δC (75.5 MHz, DMSO) 190.51, 176.60, 173.26, 149.61, 149.10, 128.36, 127.16, 126.32, 125.46, 109.06, 108.24, 101.85, 50.52, 42.31, 36.33, 29.37, 28.16. m/z (HR-ESI) Calc. for C19H20N2O5S, [M+H]+: 389.1166. Found 389.1181. HPLC purity 95 %, 9.04 min.
(R)-Methyl 2-((Z)-((Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-2-oxothiazolidin-4-ylidene)amino)-3-(1H-indol-2-yl)propanoate 7n
Brown solid (0.040 g, 49.8 %). Mp94°C(decomp.). m/z (ESI, 20 V) 357.0 (100 %, M+•). HPLC purity 95 %, 10.58 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-((2-((6-nitropyridin-3-yl)amino)ethyl)imino) thiazolidin-2-one 7o
Yellow powder (0.057 g, 76.9 %). Mp 248°C (decomp.). δH (300.13 MHz, DMSO) 9.40 (br s, 1H, NH), 8.92 (d, J 2.7, 1H, ArH), 8.30 (s, 1H, ArH), 8.13 (d, J 9.7, 1H, ArH), 7.69 (s, 1H, CH), 7.06 (m, 3H, ArH), 6.59 (d, J 9.4, 1H, ArH), 6.13 (s, 2H, CH2), 3.69 (s, 4H, NCH2). m/z (HR-ESI) Calc. for C18H15N5O5S, [M+H]+•: 414.0867. Found 414.0887. HPLC purity 90 %, 9.64 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-((3,4-dimethoxybenzyl)imino)thiazolidin-2-one 7p
Yellow powder (0.062 g, 86.9 %). Mp 268°C (decomp.). δH (300.13 MHz, DMSO) 9.64 (br s, 1H, NH), 7.80 (s, 1H, CH), 7.07 (s, 2H, ArH), 7.03 (d, J 8.6, 2H, ArH), 6.93 (m, 2H, ArH), 6.11 (s, 2H, CH2), 4.63 (s, 2H, CH2), 3.76 (s, 3H, CH3), 3.74 (s, 3H, CH3). m/z (HR-ESI) Calc. for C20H18N2O5S, [M−H]−: 397.0864. Found 397.0873. HPLC purity 90 %, 9.75 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-((4-methoxyphenethyl)imino)thiazolidin-2-one 7q
Yellow powder (0.039 g, 57.4 %). Mp 244°C (decomp.). δH (300.13 MHz, DMSO) 9.30 (br s, 1H, NH), 7.71 (s, 1H, CH), 7.17 (d, J 8.6, 2H, ArH), 7.05 (m, 3H, ArH), 6.87 (d, J 8.6, 2H, ArH), 6.12 (s, 2H, CH2), 3.72 (s, 3H, CH3), 3.66 (t, J 7.5, 2H, NCH2), 2.88 (t, J 7.4, 2H, CH2). m/z (HR-ESI) Calc. for C20H18N2O4S, [M+H]+•: 383.1060. Found 383.1050. HPLC purity >99 %, 10.60 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-((furan-2-ylmethyl)imino)thiazolidin-2-one 7r
Yellow powder (0.030 g, 51.0 %). Mp 215–217°C. δH (300.13 MHz, DMSO) 9.68 (br s, 1H, NH), 7.81 (s, 1H, CH), 7.66 (s, 1H, ArH), 7.06 (d, J 13.4, 3H, ArH), 6.45 (dd, J 5.3, 3.7, 2H, ArH), 6.13 (s, 2H, CH2), 4.72 (s, 2H, NCH2). m/z (HR-ESI) Calc. for C16H12N2O4S, [M+H]+•: 329.0591. Found 329.0606. HPLC purity >99 %, 9.80 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-(cyclohexylimino)thiazolidin-2-one 7s
Yellow crystals (0.025 g, 41.6 %). Mp 212–214°C. δH (300.13 MHz, DMSO) 8.96 (br s, 1H, NH), 7.80 (s, 1H, CH), 7.09 (d, J 10.6, 2H, ArH), 7.05 (s, 1H, ArH), 6.13 (s, 2H, CH2), 3.89 (s, 1H, NCH), 1.87 (dd, J 51.5, 10.7, 2H, CH2), 1.71 (dd, J 40.8, 12.2, 2H, CH2), 1.36 (m, 4H, CH2), 1.18 (m, 2H, CH2). m/z (HR-ESI) Calc. for C17H18N2O3S, [M+H]+•: 331.1111. Found 331.1095. HPLC purity >99 %, 10.37 min.
(4Z,5Z)-4-((2-(1H-Indol-3-yl)ethyl)imino)-5-(benzo[d][1,3]dioxol-5-ylmethylene)thiazolidin-2-one 7t
Yellow powder (0.039 g, 56.0 %). Mp 248–251°C. δH (300.13 MHz, DMSO) 10.85 (br s, 1H, NH), 10.75 (s, 1H, NH), 7.71 (s, 1H, CH), 7.59 (d, J 7.4, 1H, ArH), 7.51 (d, J 7.9, 1H, ArH), 7.34 (t, J 8.2, 1H, ArH), 7.21 (s, 1H, ArH), 7.05 (dq, J 14.8, 7.4, 5H, ArH), 6.13 (s, 2H, CH2), 3.77 (t, J 7.4, 2H, NCH2), 3.07 (t, J 7.4, 2H, CH2). m/z (HR-ESI) Calc. for C21H17N3O3S, [M−H]−: 390.0918. Found 390.0937. HPLC purity >99 %, 10.00 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-((pyridin-2-ylmethyl)imino)thiazolidin-2-one 7u
Yellow crystals (0.021 g, 34.8 %). Mp 223–225°C. δH (300.13 MHz, DMSO) 9.86 (br s, 1H, NH), 8.56 (d, J 4.4, 1H, ArH), 7.88 (s, 1H, CH), 7.81 (td, J 7.7, 1.7, 1H, ArH), 7.41 (d, J 7.7, 1H, ArH), 7.33 (m, 1H, ArH), 7.08 (d, J 11.5, 2H, ArH), 6.14 (s, 2H, CH2), 4.81 (s, 2H, NCH2). m/z (HR-ESI) Calc. for C17H13N3O3S, [M+H]+: 340.0750. Found 340.0736. HPLC purity >95 %, 6.45 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-(phenethylimino)thiazolidin-2-one 7v
Yellow powder (0.026 g, 40.6 %). Mp 226–228°C. δH (300.13 MHz, DMSO) 7.72 (s, 1H, CH), 7.25 (m, 5H, ArH), 7.06 (d, J 13.6, 3H, ArH), 6.13 (s, 2H, CH2), 3.72 (t, J 7.5, 2H, NCH2), 2.96 (t, J 7.5, 2H, CH2). m/z (HR-ESI) Calc. for C19H16N2O3S, [M+H]+: 353.0954. Found 353.0967. HPLC purity >99 %, 11.12 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-((3-hydroxypropyl)imino)thiazolidin-2-one 7w
Yellow crystals (0.010 g, 22.8 %). Mp 210°C (decomp.). δH (300.13 MHz, DMSO) 9.30 (br s, 1H, NH), 7.74 (s, 1H, CH), 7.06 (m, 3H, ArH), 6.12 (s, 2H, CH2), 4.57 (s, 1H, OH), 3.51 (m, 4H, CH2), 1.77 (m, 2H, CH2CH2). m/z (ESI, 20V) 307.6 (40 %, M+•). m/z (HR-ESI) Calc. for C14H14N2O4S, [M+H]+: 307.0747. Found 307.0755. HPLC purity >95 %, 7.65 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-(methylimino)thiazolidin-2-one 7x
Yellow powder (0.042 g, 74.1 %). Mp 285°C (decomp.). δH (300.13 MHz, DMSO) 9.35 (br s, 1H, NH), 7.68 (s, 1H, CH), 7.05 (m, 3H, ArH), 6.12 (s, 2H, CH2), 3.03 (d, J 4.4, 3H, NCH3). m/z (ESI, 20 V) 263.3 (100 %, M+•). m/z (HR-ESI) Calc. for C12H10N2O3S, [M−H]−: 261.0339. Found 261.0346. HPLC purity >99 %, 8.25 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-(ethylimino)thiazolidin-2-one 7y
Yellow powder (0.035 g, 58.9 %). Mp 230°C (decomp.). δH (300.13 MHz, DMSO) 9.40 (br s, 1H, NH), 7.78 (s, 1H, CH), 7.06 (d, J 13.0, 3H, ArH), 6.12 (s, 2H, CH2), 3.49 (m, 2H, NCH2), 1.21 (t, J 7.2, 3H, CH2CH3). m/z (ESI, 20 V) 277.3 (100 %, M+•).m/z (HR-ESI) Calc. for C13H12N2O3S, [M+H]+: 277.0641. Found 277.0648. HPLC purity >99 %, 8.97 min.
(4Z,5Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-4-((2-hydroxypropyl)imino)thiazolidin-2-one 7z
Yellow crystals (0.020 g, 33.8 %). Mp 228–230°C (decomp.). δH (300.13 MHz, DMSO) 9.35 (br s, 1H, NH), 7.82 (d, J 4.8, 1H, CH), 7.06 (dd, J 13.8, 4.4, 3H, ArH), 6.13 (d, J 4.4, 2H, CH2), 4.97 (s, 1H, OH), 3.91 (s, 1H, NCH), 3.42 (m, 2H, NCH2), 1.10 (t, J 5.2, 3H, CHCH3). m/z (ESI) 307.3 [M+H]+ m/z (HR-ESI) Calc. for C14H14N2O4S, [M+H]+: 307.0747. Found 307.0750. HPLC purity >99 %, 7.80 min.
General Procedure for the Preparation 4-Amino Acid Substituted Benzodioxol Thiazolidin-2-ones
Fmoc-L-Ala-Wang resin (0.13 mg: loading 0.83 mmol g−1) was suspended in DMF (2.0 mL) for 10 min, and then excess solvent was removed by filtration. The resin was treated with 20% piperidine (2.0 mL) for 5 min. Excess reagent was removed by filtration. This process was repeated, and then L-Ala deprotected beads were thoroughly washed with DMF. Qualitative deprotection was confirmed using the ninhydrin test. L-Ala deprotected beads were taken up in DMF (5.0 mL), added to 5-benzo[1,3]dioxol-5-ylmethylene-4-methylsulfanyl-thiazol-2- one 5 (0.060 g, 0.20 mmol), and the mixture heated to 80°C for 19–23 h. The reaction mixture was cooled, the resin isolated by filtration, washed thoroughly with DMF, MeOH, and ether and then dried. The final product was cleaved from the resin in 95% TFA, 5% H2O (3.0 mL) over 1.5 h, filtered, then the supernatant diluted with acetonitrile/H2O (50 : 50, 50 mL), and placed on a freezedrier overnight. Crystallization from ethanol afforded the title compound as a brown powder unless otherwise stated. The following 4-amino acid substituted benzodioxol thiazolidin- 2-ones 7aa–7ab were prepared in this manner.
(S)-2-((Z)-((Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-2-oxothiazolidin-4-ylidene)amino)propanoic Acid 7aa
A solution of (Z)-5-(benzo[d][1,3]dioxol-5-ylmethylene)-4-(methylthio)thiazol-2(5H)-one (5) (0.060 g) in DMF reacted with Wang-L-Ala-Fmoc (130 mg: loading 0.83 mmol g−1) and after workup afforded a brown powder (0.018 g, 51.0 %), mp 233°C (decomp.). m/z (ESI, 20 V) 321.3 (100%, M+•). m/z (HR-ESI) Calc. for C14H12N2O5S, [M+H]+: 321.0540. Found 321.0533. HPLC: purity >95 %, 8.20 min.
(S)-2-((Z)-((Z)-5-(Benzo[d][1,3]dioxol-5-ylmethylene)-2-oxothiazolidin-4-ylidene)amino)-3-(1H-indol-2-yl)propanoic Acid 7ab
A solution of (Z)-5-(benzo[d][1,3]dioxol-5-ylmethylene)-4-(methylthio)thiazol-2(5H)-one (5) (0.050 g) in DMF reacted with Wang-L-Trp(tBu)-Fmoc (180 mg: loading 0.61 mmol g−1) and after workup afforded a brown powder (0.025 g, 51.0 %). m/z (HR-ESI) Calc. for C22H17N3O5S, [M+H]+•: 436.0967. Found 436.0962. HPLC: purity >98%, 9.80 min.
Acknowledgements
J-A.P. is a recipient of an Australian Postgraduate Award (APA) Scholarship. This work was funded through the National Institutes of Health grants CA43460 and CA62924, the Virginia and D.K. Ludwig Fund for Cancer Research (USA), the Cancer Council Victoria no. 436708 and a National Health and Medical Research Council grant no. 545943 (Australia).
References
- 1.Grischuk AP, Komaritsa ID, Baranov SN. Khimiya Geterotsiklicheskikh Soedinenii. 1966;2:706. [Google Scholar]
- 2.Komaritsa ID, Grischuk AP. Khimiya Geterotsiklicheskikh Soedinenii. 1968;4:706. [Google Scholar]
- 3.Plevachuk NE, Komaritsa ID. Khimiya Geterotsiklicheskikh Soedinenii. 1970;6:159. [Google Scholar]
- 4.Kim H-J, Choo H, Cho YS, No KT, Pae AN. Bioorg. Med. Chem. 2008;16:636. doi: 10.1016/j.bmc.2007.10.047. [DOI] [PubMed] [Google Scholar]
- 5.Calderón F, Barros D, Bueno JM, Coteron JM, Fernandez E, Gamo FJ, Lavandera JL, Leon ML, Macdonald SJF, Mallo A, Manzano P, Porras E, Fiandor JM, Castro J. ACS Med. Chem. Lett. 2011;2:741. doi: 10.1021/ml200135p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinson J-A, Schmidt-Kittler O, Zhu J, Jennings IG, Kinzler KW, Vogelstein B, Chalmers DK, Thompson PE. ChemMedChem. 2011;6:514. doi: 10.1002/cmdc.201000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camps M, Rueckle T, Ji H, Ardissone V, Rintelen F, Shaw J, Ferrandi C, Chabert C, Gillieron C, Francon B, Martin T, Cirillo D, Perrin D, Leroy D, Vitte PA, Hirsch E, Wymann MP, Cirillo R, Schwarz MK, Rommel C. Nat. Med. 2005;11:936. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- 8.Baell JB, Holloway GA. J. Med. Chem. 2010;53:2719. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 9.Enchev V, Chorbadjiev S, Jordanov B. Chem. Heterocycl. Compd. 2002;38:1110. [Google Scholar]
- 10.Irvine MW, Patrick GL, Kewney J, Hastings SF, MacKenzie SJ. Bioorg. Med. Chem. Lett. 2008;18:2032. doi: 10.1016/j.bmcl.2008.01.117. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez P, Polli JE. J. Pharmacol. Exp. Ther. 2008;326:296. doi: 10.1124/jpet.107.135863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burton W, Budde WL, Cheng CC. J. Med. Chem. 1970;13:1009. doi: 10.1021/jm00299a061. [DOI] [PubMed] [Google Scholar]