

Abstract
Proper control of hepatic glucose production is central to whole body glucose homeostasis and its disruption plays a major role in diabetes. Here we demonstrate that, although established as an intracellular lipid chaperone, aP2 is in fact actively secreted from adipocytes to regulate liver glucose metabolism. Fasting and lipolysisrelated signals regulate secretion of aP2 from adipocytes, and circulating aP2 levels are markedly elevated in mouse and human obesity. Recombinant aP2 stimulates glucose production and gluconeogenic activity in primary hepatocytes in vitro and in lean mice in vivo. In contrast, neutralization of secreted aP2 reduces glucose production and corrects the diabetic phenotype of obese mice. Hyperinsulinemiceuglycemic and pancreatic clamp studies demonstrated actions of aP2 in liver upon aP2 administration or neutralization. We conclude that aP2 is a novel adipokine linking adipocytes to hepatic glucose production and that neutralizing secreted aP2 may represent an effective therapeutic strategy for diabetes.
INTRODUCTION
Adipose tissue is the most effective site for energy and nutrient storage as well as release, depending on the energy demands of the organism. This highly conserved function of adipose tissue ensures safe storage at times of plenty and is an integral part of survival during limited access to nutrients. In addition, adipose tissue is also an important endocrine organ responsible for systemic metabolic regulation (Rosen and Spiegelman, 2006). The metabolic effects of adipose tissue are mediated by a variety of hormones, inflammatory cytokines, adipokines, and lipokines, which play a critical role in systemic metabolic homeostasis, and alterations in the endocrine output of adipose tissue link obesity to an array of metabolic disorders (Hotamisligil, 2006; Waki and Tontonoz, 2007). Thus, in both physiological and pathological contexts, adipose tissue is a key organ where nutrient and endogenous signaling molecules interact and integrate; ultimately resulting in systemic regulation or disruption of metabolic homeostasis.
While obesity-induced hormonal changes in adipose tissue are extensively studied, the endocrine function of this tissue during fasting and in regulation of glucose production is poorly understood. In times of nutrient deprivation, the body launches a complex hormonal response to maintain glucose supply to vital organs (Cahill, 2006; Lin and Accili, 2011; Unger and Cherrington, 2012). This response, among other adaptations, results in the breakdown of glycogen and lipid stores to generate substrates and energy, and stimulates glucose production from the liver (Cahill, 2006; Lin and Accili, 2011; Unger and Cherrington, 2012). A key mediator signaling some of these fasting functions, including glucose production, is the pancreatic hormone glucagon, produced by alpha cells of the islets of Langerhans, which counteracts the action of insulin (Cahill, 2006; Lin and Accili, 2011; Unger and Cherrington, 2012). Adipose tissue lipolysis contributes the majority of fatty acids in the serum, which are taken up and oxidized in muscle and activate glucose production in liver; Until now however, no hormonal input emanating from this tissue has been identified that impacts hepatic glucose production and other changes in systemic glucose metabolism associated with fasting or related responses in the long term (Boden, 2003; Chu et al., 2002; Everett-Grueter et al., 2006; Lam et al., 2003).
Interestingly, it is established that hepatic glucose production is dysregulated in obesity and represents a key process leading to development of diabetes. There is an ongoing debate in the field as to whether this response is a consequence of insulin resistance or glucagon sensitivity in the liver, is secondary to an inability of insulin to suppress lipolysis effectively (Lin and Accili, 2011; Unger and Cherrington, 2012), or is due to other yet unknown mechanisms. Whether it is expansion of adipose tissue, dysregulated lipolysis, or both that contributes to hepatic glucose output, the mechanism by which this process is signaled between adipose tissue and liver, and the hormonal mediator(s) carrying out this function also remain unidentified.
In recent years, cytosolic adipose tissue fatty acid binding proteins (FABPs) have emerged as critical molecules integrating intracellular lipid signals under metabolic stress conditions (Furuhashi and Hotamisligil, 2008; Hertzel and Bernlohr, 2000). Adipocytes express one major and one minor isoform of FABP called aP2 and mal1 (FABP4 and FABP5), respectively. Mice deficient in these lipid chaperones exhibit marked protection against a multitude of metabolic abnormalities associated with obesity (Furuhashi et al., 2008; Furuhashi et al., 2007; Maeda et al., 2005; Makowski et al., 2001). Previously, we demonstrated that FABP-deficiency in adipose tissue results in the production of C16:1n7-palmitoleate, and it is through this mechanism that adipose lipid chaperones regulate liver lipogenesis and muscle glucose utilization (Cao et al., 2008). While a major phenotype of these animals also relates to hepatic glucose production, neither we nor others have been able to identify the hormonal mechanism by which hepatic glucose production is regulated by adipose tissue lipid chaperones. Since we have ruled out a role for leptin or adiponectin in this function (Cao et al., 2006), these observations indicate the presence of an additional signal that communicates between adipose tissue and liver to regulate glucose metabolism, including responses during lipolysis.
Here we demonstrate that aP2 is secreted from adipocytes in response to fasting and fasting-related signals and regulates hepatic glucose production. Hence, despite the established dogma linking its biology strictly to its cytosolic activities, aP2 carries out a critical metabolic function by acting as a novel adipokine in the adipo-hepatic axis, which could be an attractive target for therapeutic intervention in diabetes.
RESULTS
aP2 is secreted from adipocytes in vitro
Since its identification, aP2 has been considered a cytosolic protein. However, it was recently identified by a proteomics screen in the supernatant of differentiated 3T3-L1 adipocytes (data not shown) and has been detected in human serum (Xu et al., 2006). These intriguing observations prompted us to examine the possibility of regulated secretion of this protein from adipocytes. Examination of aP2 levels in conditioned medium and cell lysates collected from wild type (WT) and FABP-deficient (both aP2 and mal1) adipocytes revealed the clear presence of this protein in the conditioned medium of WT cells (Fig. 1a). Adiponectin (ACDC) secretion was also readily detectable in adipocytes from both genotypes. In contrast, other highly expressed cytosolic proteins were undetectable under the same conditions, supporting the possibility of active aP2 secretion from adipocytes (Fig. 1a). However, as one of the most abundant cytosolic proteins in adipocytes, the presence of aP2 in conditioned medium could still have been due to non-specific release resulting from cell death and/or lysis. To examine the nature of aP2’s exit from cells, we transfected cells with a Flag-tagged green fluorescent protein (GFP) along with similarly tagged aP2 and AKT and carefully titrated the amounts of each plasmid to ensure that all proteins were expressed at similar levels inside cells. We then probed both conditioned medium and cell lysate with the anti-Flag antibody to eliminate any variation that might be introduced by differing sensitivities among antibodies. In these experiments, aP2 was readily detectable in the conditioned medium while GFP and AKT were undetectable under the same conditions (Fig. 1b). These data indicated that aP2 is secreted and that its presence in conditioned medium was not due to non-specific cell lysis or death. We also expressed Flag-tagged aP2 in aP2−/− adipocytes under the control of CMV promoter and examined its secretion under basal and stimulated conditions (Fig 1.c–d). In this setting, aP2 secretion was detected in the conditioned media by western blot analysis following immunoprecipitation with an anti-Flag antibody. To understand the kinetics and mode of aP2 secretion further, we metabolically labeled cellular proteins in adipocytes and performed pulse chase analysis of secreted aP2 along with a classically secreted adipocyte protein, adiponectin. In these experiments, aP2 was detectable in conditioned medium at one-hour post-chase and continued to increase during the four-hour chase period in a manner similar to adiponectin (Fig. 1e). Interestingly, while adiponectin secretion was blocked by brefeldin A or monensin, neither of these agents exhibited any inhibitory effect on aP2 secretion, indicating that aP2 is released from adipocytes via a non-classical secretory pathway (Fig. 1e). This finding is consistent with the fact that aP2 lacks a signal peptide sequence.
Figure 1. Secretion of aP2 in vitro from cultured cells.
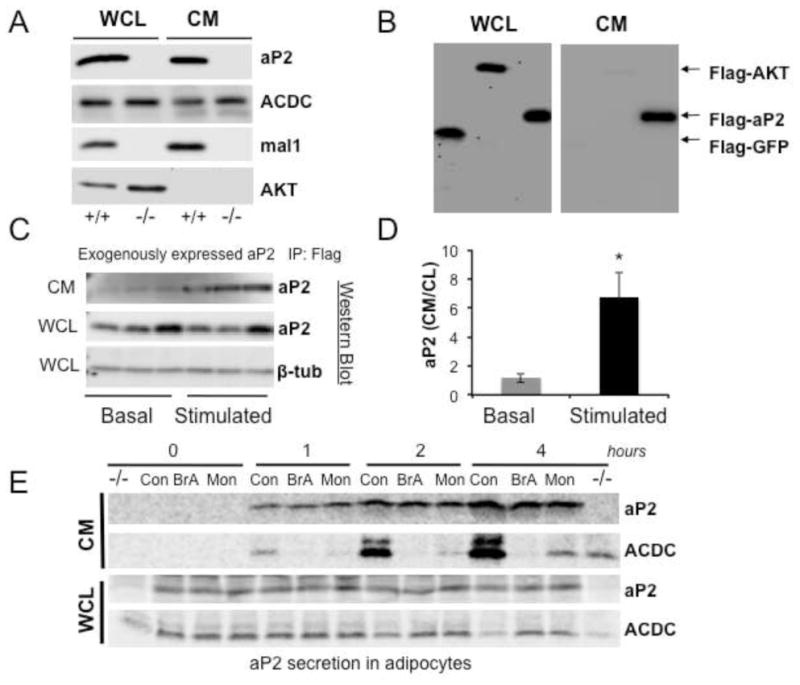
a, aP2 secretion in adipocytes. Whole cell lysate (WCL) and conditioned medium (CM) from differentiated WT (+/+) or AM−/− (−/−) adipocytes lacking both aP2 and mal1 were blotted using anti-aP2, adiponectin (ACDC), mal1, or AKT antibodies. These cell lines were developed in-house. b, aP2 secretion in HEK 293 cells. Whole cell lysates (WCL) or immunoprecipated conditioned medium (CM) from HEK 293 cells transfected with Flag-AKT, Flag-GFP-aP2 or Flag-GFP plasmids were used to detect aP2 by western blotting with an anti-Flag antibody. c, CMV promoter driven aP2 expression and its regulated secretion studied in aP2−/− adipocytes. Flag-aP2 was transfected into aP2−/− cells and its appearance in the CM was examined under basal and forskolin (FSK) stimulated condition (20μM for 1h). CM samples were immunoprecipitated with an anti-Flag antibody and blotted with anti-aP2. Cell lysate was probed with aP2 and β-tubulin (β-tub) antibodies. d, Quantitation of data shown in panel c determined by using ImageJ program. * p < 0.05 in student’s t test. e, Pulse chase analysis of aP2 secretion. Cultured adipocytes were metabolically labeled and then treated with vehicle, brefeldin A (BrA, 10 μg/ml) or monensin (Mon, 5 μM), and samples were taken at indicated time points. Proteins in CM were immunoprecipitated using anti-aP2 or adiponectin (ACDC) antibodies. After electrophoresis, radiolabeled proteins were subjected to autoradiography. An aP2−/− cell lysate was used as negative control. Data are presented as means ± SEM.
aP2 secretion is regulated by nutritional fluctuations and lipolysis
To explore the potential metabolic function of secreted aP2, we first examined aP2 levels in response to metabolic status through nutrient fluctuations and investigated whether aP2 levels change in the fasted and fed states. The circulating aP2 levels in mice fasted for 24 hours were higher compared to levels at the fed state (4-hour feeding following the fast) or during ad libitum feeding (Fig. 2a). Therefore, nutritional status can modulate serum aP2 levels, without any change in the cytosolic pool of the protein in adipose tissue (Fig. 2a), which suggests that serum aP2 might be part of a systemic program that regulates energy homeostasis. During nutrient deprivation, a primary function of adipose tissue in energy homeostasis is to provide fatty acids via lipolysis for other tissues. Since aP2 levels are increased during fasting, we investigated whether aP2 secretion responds to signals that are activated in this state in vivo.
Figure 2. Nutritional status and lipolysis regulates aP2 secretion.
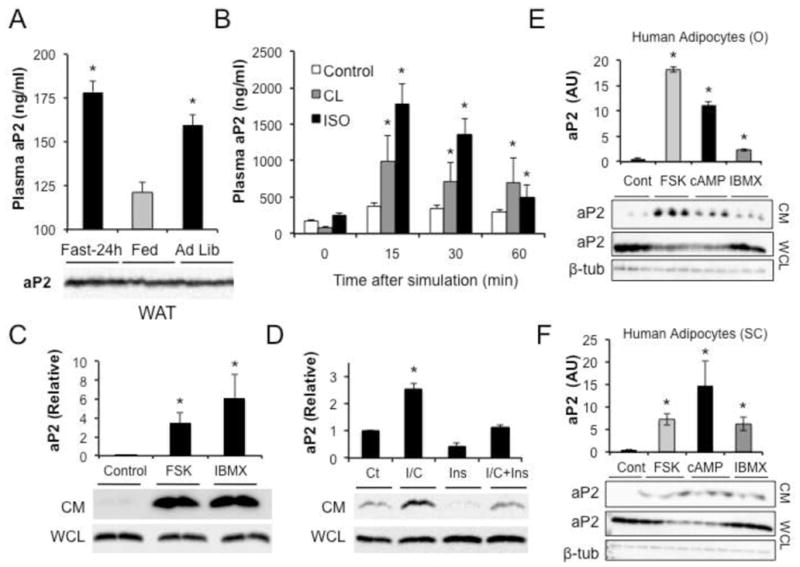
a, Plasma aP2 in mice fasted for 24 hours, refed for 4 hours after a 24-hour fast, or fed ad libitum. Below the graphs are corresponding aP2 western blots in WAT lysates. b, Plasma aP2 levels in mice injected with saline (Control), CL 316243 (CL, 0.1mg/kg) or isoproterenol (ISO, 1mg/kg) to induce lipolysis. At least 6 male mice were used in each experiment. c, aP2 in conditioned medium (CM) or whole cell lysates (WCL) of adipose tissue explants treated with forskolin (FSK, 20μM) or isobutylmethylxanthine (IBMX, 1mM). Below the graphs are corresponding aP2 western blots in CM and WCL. d, aP2 in CM or WCL of adipocytes treated with isobutylmethylxanthine and dibutryl cyclic adenosine monophosphate (IBMX/db-cAMP; I/C, 1mM) and insulin (Ins, 100ng/ml). Below the graphs are corresponding aP2 western blots in CM and WCL. e–f, aP2 in CM or WCL of primary human omental (O) or subcutaneous (SC) adipocytes treated with FSK, db-cAMP, or IBMX. Below the graphs are corresponding aP2 western blots in CM and WCL blotted with anti-aP2 antibody. Beta-tubulin is shown as a loading control. Data are presented as means ± SEM. * p < 0.05 in student’s t test.
We first administered β-adrenergic receptor agonists isoproterenol (pan-β) or CL 316243 (β3-specific) to mice, and verified that each compound activated lipolysis (Fig. S1a). Mice receiving either of the compounds exhibited a rapid increase in plasma aP2 levels compared to vehicle-treated controls (Fig. 2b), indicating that aP2 secretion is affected by lipolytic signals in vivo and that the elevated circulating aP2 in mice during fasting is consistent with increased lipolytic activity in adipose tissue. We also harvested and cultured adipose tissue explants ex vivo, and examined aP2 secretion in response to forskolin (FSK) or isobutylmethylxanthine (IBMX) treatment to study the impact of other fasting-related signals on aP2 secretion. Both of these agents caused a very substantial increase in aP2 secretion from adipose explants (Fig. 2c), confirming that this process is tightly linked to fasting signals and lipolysis ex vivo, as it is in vivo.
Next we examined aP2 secretion in vitro in cultured adipogenic cell lines and examined the impact of IBMX and dibutyryl-cAMP (db-cAMP) treatment in pure populations of adipocytes. Cultured WT adipocytes exposed to IBMX and db-cAMP exhibited a robust increase in aP2 secretion (Fig. 2d). Several other fasting-related signals such beta agonists, branched-chain amino acids, and glycerol also increased aP2 secretion from cultured adipocytes (data not shown). Interestingly, when cells were treated with insulin, both baseline and stimulated aP2 secretion were suppressed (Fig. 2d). In adipocytes treated with increasing doses of CL 316243 or forskolin, we also examined the presence of lactate dehydrogenase (LDH) activity in the conditioned medium. LDH levels did not correlate with appearance of aP2 during the course of treatment with these agents, thus supporting regulated secretion rather than release by death (Fig S1b–c).
In addition to mouse systems, we also examined regulation of aP2 secretion in human primary cultured adipocytes (Fig. 2e–f). In cultured omental (Fig. 2e) and subcutaneous (Fig. 2f) adipocytes, treatment with forskolin, IBMX, or db-cAMP increased aP2 secretion from adipocytes and also stimulated lipolysis (Fig. S2a–d). Taken together, these results demonstrate that fasting-related signals and agents that stimulate lipolysis strongly induce aP2 secretion in both mouse and human adipocytes.
Regulation of aP2 secretion in vivo
Fasting and lipolysis related secretion of aP2 prompted us to examine circulating aP2 levels in physiological and pathological states in vivo in WT and FABP-deficient mouse models. In WT as well as mal1−/− (M−/−, FABP5−/−) mice, aP2 was present at levels ranging from 100 to 300 ng/ml in plasma (Fig. 3a). There was no detectable signal in aP2−/− (A−/−) and aP2-mal1−/− (AM−/−) controls, validating the specificity of the assay (Fig. 3a). Plasma aP2 in WT mice is 10 to 20-fold more abundant than leptin (around 12.5 ng/ml), but still significantly (100-fold) lower than adiponectin levels (5–10 μg/ml). To explore the regulation of circulating aP2 in the context of metabolic disease, we compared the plasma profiles of lean and obese (both genetic and diet induced models) mice. Plasma aP2 was markedly (~ 4-fold) increased in both high fat diet (HFD)-fed and leptin-deficient (ob/ob) mice (Fig. 3b). Increased serum aP2 levels have been reported to be associated with obesity in humans (Tso et al., 2007; Xu et al., 2006). We measured circulating aP2 in a large set of samples with a range of body weight (≈ 1800 males and females). As shown in figure 3c, we found that circulating aP2 levels in obese humans were significantly elevated similar to what we observed in obese mice and correlated strongly with body mass index in both males (r=0.41, p<0.001) and females (r=0.46, p<0.001). Adipocytes also produce mal1 (FABP5), a second but minor FABP isoform at this site (Fig. 1a), which was also detected in plasma and elevated in aP2-deficient animals (Fig. S3a). Unlike aP2, plasma mal1 levels were not significantly elevated in obese mice (Fig. S3b) and there was no nutritional regulation of circulating mal1 levels during fasting and feeding (Fig S3c).
Figure 3. Regulation of aP2 secretion in vivo.
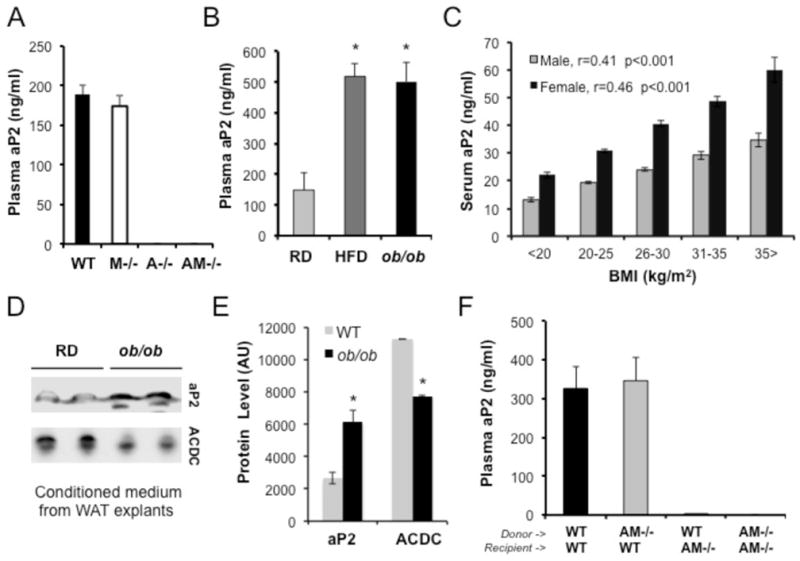
a, Plasma aP2 levels in WT, mal1−/− (M−/−), aP2−/− (A−/−), and aP2-mal1−/− (AM−/−) mice, determined by ELISA. b, Plasma aP2 levels of lean mice (WT regular diet, RD), or mice with dietary (WT high fat diet, HFD) or genetic obesity (ob/ob). c, Serum aP2 levels in human subjects, female (n=910) and male (n=904), as a function of body mass index (BMI). d, aP2 secretion from weight-matched adipose tissue explants of lean and obese mice. Adipose tissue explants were collected from WT mice maintained on regular diet or ob/ob mice and were thoroughly washed with PBS. Fresh medium was added and incubated overnight and collected for western blot analysis using anti-aP2 or adiponectin (ACDC) antibodies. e, Quantitative measurement of the relative secretion of aP2 and adiponectin from adipose tissue explants from WT mice maintained on regular diet or ob/ob mice is graphed. f, Plasma aP2 in mice that have undergone bone marrow transplantation. Bone marrow transplantation was performed between WT and aP2-mal1−/− (AM−/−) mice (as donors and recipients) and plasma aP2 levels were determined by aP2 ELISA in the resulting 4 groups of chimeras. Statistical analysis was performed by student’s t test. Data are presented as means ± SEM. * p < 0.05.
Increased aP2 levels during obesity may be due to elevated aP2 expression, expanded fat mass, increased aP2 secretion, changes in clearance, or a combination of these alterations. Since it is known that obesity does not have a strong impact on overall adipose tissue aP2 expression (Tong et al., 2000), we attempted to determine whether increased volume of fat mass and/or dysregulation of secretion contribute to the elevated circualting aP2 levels in obese mice. We collected fat explants from lean and obese mice (ob/ob) and examined aP2 release ex vivo. In this explant culture, aP2 secretion from an equal mass of adipose tissue from obese mice was still significantly higher than that from lean controls, indicating that obese mice have dysregulated aP2 secretion (Fig. 3d,e). In contrast, adiponectin secretion was significantly reduced in obese explants, verifying the fidelity of this system in capturing the secretory profile of adipose tissue that is seen in obesity (Fig. 3d,e). Hence, increase in plasma aP2 is likely the result of increased secretion per fat mass, which is further augmented with increased overall adiposity or potential alterations in tissue uptake and clearance due to the inflammatory milieu that may occur in obesity.
Obese mice accumulate macrophages in adipose tissue (Weisberg et al., 2003; Xu et al., 2003) and since aP2 is expressed in both adipocytes and macrophages (Furuhashi et al., 2008; Makowski et al., 2001), increased aP2 in blood and in tissue explants could be due to increased aP2 release from macrophages. To determine the cell type responsible for the increased blood aP2 in the context of obesity, we performed bone marrow transplantation between WT and FABP-deficient mice (Fig. 3f). An examination of plasma aP2 in the resulting chimeric mice revealed that bone marrow-derived cells from WT mice could not sustain a detectable level of plasma aP2 in FABP-deficient mice (Fig. 3f). FABP-deficient bone marrow transplants did not have any effect on the circulating aP2 concentrations detected in WT recipients. A similar pattern was also observed for mal1 where the predominant contributor to blood levels was the stromal compartment although a small amount was detected emerging from bone marrow-derived cells (Fig. S3d). Taken together, these results demonstrate that adipocytes, but not hematopoietic cells, are the predominant contributors of circulating FABP in mice, and that aP2 is a nutritionally-regulated potential adipokine with significantly elevated blood levels in obesity.
Circulating aP2 regulates liver glucose production
The tight coupling of aP2 secretion to fasting and lipolytic signals suggests that secreted aP2 might have related metabolic functions. Fasting is coupled to a switch from hepatic glycogenolysis to gluconeogenesis, demanding a higher drive to sustain hepatic glucose production, and is accompanied by decreased glucose utilization (Cahill, 2006; Unger and Cherrington, 2012). Dysregulation of these processes is critical in the development of hyperglycemia and frank diabetes (Lam et al., 2003). As blood aP2 levels are constitutively elevated in the obese, it is possible that aP2 inappropriately signals an aspect of fasting metabolism despite ample nutrient and energy availability in obesity. Since reduced hepatic glucose production is a major feature of genetic FABP-deficiency (Cao et al., 2006), our observations raise the possibility that aP2 may be an adipokine that regulates systemic glucose metabolism in vivo.
To address this possibility we first tested the effects of recombinant aP2 treatment on glucose production in primary isolated hepatocytes. In these cells isolated from overnight fasted animals, treatment with glucagon (3μM), the prototype hormone regulating liver glucose production, resulted in increased glucose production (Fig. 4a). Interestingly, a similar pattern of an effect was also observed upon aP2 treatment where glucose production was increased by 30% (Fig. 4a). In these hepatocytes, treatment with aP2 also resulted in increased expression of two gluconeogenic enzymes, phosphoenolpyruvate carboxykinase 1, Pck1 (Fig. 4b), and glucose-6-phosphatase, G6pc (Fig. S4a). Treatment with recombinant aP2 also significantly (84%) increased the enzymatic activity of Pck1 in primary hepatocytes (Fig. 4c). A mutant aP2 with alterations in its lipid-binding domain (Erbay et al., 2009) lacked the ability to induce Pck1 expression in hepatocytes (Fig. 4d). There was also no change in gluconeogenic gene expression in primary hepatocytes upon treatment with recombinant mal1 (Fig. S4b). Hence, there was selectivity in the actions of aP2 on hepatic gluconeogenic targets.
Figure 4. Regulation of glucose homeostasis by recombinant soluble aP2.
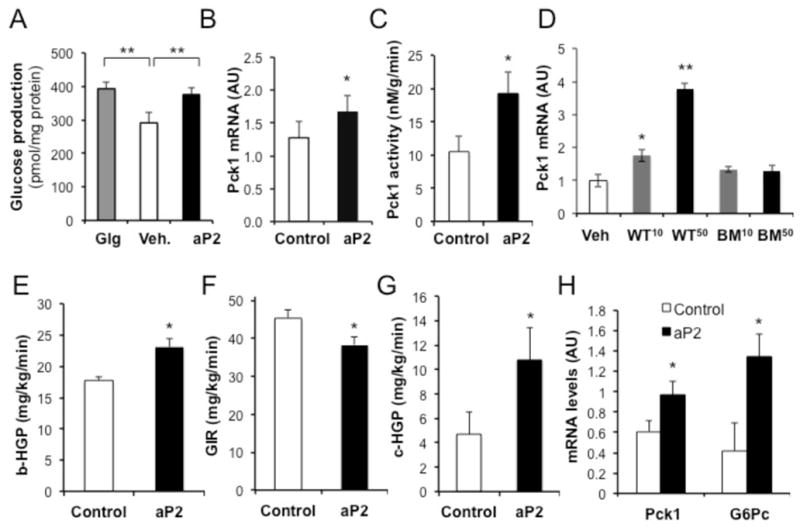
a, Glucose production from primary mouse hepatocytes treated with glucagon (3 μM) and aP2 (10 μg/ml) compared to vehicle (Veh). b, Expression of phosphoenolpyruvate carboxykinase 1 (Pck1) in primary mouse hepatocytes treated with aP2 (10 μg/ml). c, Enzymatic activity of Pck1 in primary mouse hepatocytes treated with aP2 (50 μg/ml). d, Pck1 mRNA expression in primary hepatocytes treated with 10 (WT10 or BM10) or 50 (WT50 or BM50) μg/ml recombinant WT or lipid binding mutant (BM) protein. e, Basal hepatic glucose production rates (b-HGP) in WT mice (5h fasted) after 3 hour infusion with control or recombinant aP2 protein prior to hyperinsulinemic-euglycemic clamp studies with aP2 infusion. f, Glucose infusion rates in the same animals, g, Hepatic glucose production during hyperinsulinemic-euglycemic clamp period (c-HGP). h, Expression of gluconeogenic genes Pck1 and G6pc in liver samples from mice infused with control or recombinant aP2 protein. Statistical analysis was performed by student’s t test. Data are presented as means ± SEM. * p < 0.05 and ** p < 0.01.
If secreted aP2 also functions, at least in part, to regulate hepatic glucose production in vivo, there may be important physiological and pathophysiological implications of such activity and its aberrant regulation in type 2 diabetes. Hence, we experimentally elevated serum aP2 in otherwise metabolically normal mice and examined whole body glucose fluxes using clamp studies (Fig. S4c–d). We produced and purified recombinant protein and first infused aP2 into conscious wild type mice to examine the effects of circulating aP2 on glucose metabolism with a hyperinsulinemic-euglycemic clamp study (Fig. S4e). In this setting, aP2 infusion established a high steady-state plasma aP2 concentration (≈300ng/ml) comparable to levels seen in obese mice (Fig. S4d). During the clamp study, mice receiving aP2 displayed increased basal and clamp hepatic glucose production and required significantly reduced glucose infusion (Fig. 4e,f,g). There was no significant difference in whole body glucose disposal rates (Fig. 4g) under similar extent of hyperinsulinemia (Fig S4g). In aP2-treated mice, there were significant increases in the expression of gluconeogenic enzymes Pck1 and G6pc in liver (Fig. 4h).
We also determined that administration of a single dose of recombinant aP2 into lean mice led to increased levels of plasma aP2 that lasted for several hours (Fig. S5a). We then injected recombinant aP2 twice daily to ensure that mice maintained elevated aP2 in circulation. Administration of recombinant aP2 in this regimen for two weeks did not alter the body weight or serum free fatty acid levels of mice during this period (Fig. S5b–c). These lean healthy animals, however, exhibited mild glucose intolerance after receiving recombinant aP2 for two weeks, as determined by a glucose tolerance test (Fig. S5d). Taken together, these observations indicated that secreted aP2 regulates hepatic glucose production in vivo and that raising its levels, alone, could impair glucose metabolism, even in the absence of any dietary contribution and alterations in body weight.
We next performed pancreatic clamp studies (Fig. S6a–c) to address the direct ability of aP2 to target liver glucose metabolism in vivo. For this, we used aP2-deficient genetic background mice and reconstituted circulating aP2 by infusing recombinant protein. We were able to achieve blood aP2 levels comparable to obese animals (Fig. 5a) and proceeded with pancreatic clamp experiments. Infusion of aP2 did not alter plasma levels of mal1, adiponectin, and glucagon levels (Fig. 5b–d). In this setting we also showed increased hepatic glucose production (Fig. 5e, Fig. S6d) and decreased glucose infusion rates upon aP2 administration (Fig. 5f), but no significant effect on glucose disposal (Fig. 5g). In line with previous cellular and in vivo experiments, we also observed increased expression of gluconeogenic enzymes, Pck1 and G6pc, in the liver samples (Fig. S6e,f), although the former did not reach significance (p=0.06). Pck1 activity in the liver tissue was significantly (85%) increased in aP2-treated animals (Fig. 5h). There were no differences in hepatic lipid and glycogen content, lipogenic (Fasn and Scd1) and inflammatory (IL1β, TNFα, and IL6) gene expression upon aP2 treatment (Fig. S7a–h). Hence, these experiments demonstrate that aP2 can act on liver to stimulate glucose in WT mice as well as upon reconstitution in aP2-deficient animals, but do not rule out potential aP2 action on peripheral sites.
Figure 5. Effect of aP2 on hepatic glucose production in pancreatic clamp studies.
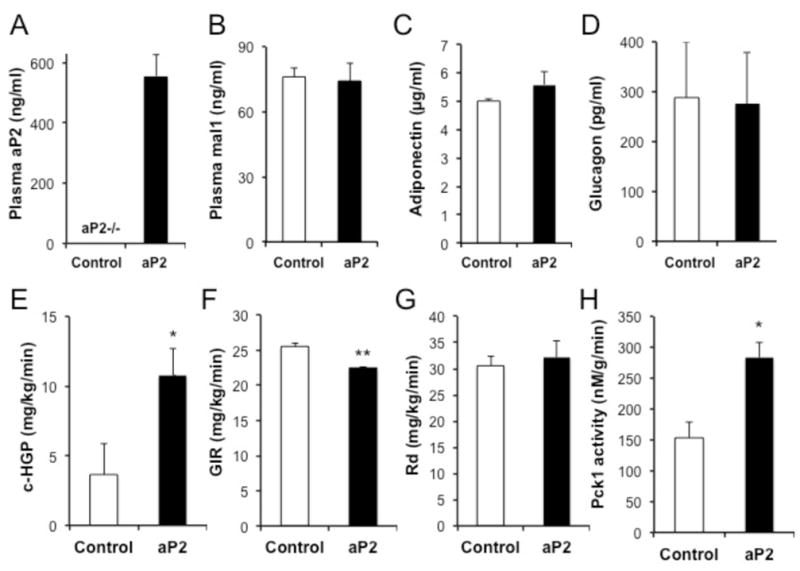
a, Reconstitution of serum aP2 in the aP2−/− mice at levels comparable to obese mice were achieved by recombinant aP2 infusion at a dose of 8μg/kg/min, in lean, 10-week-old, male mice. Plasma mal1 (b), adiponectin (c), and glucagon (d) levels in aP2−/− mice infused with either control or recombinant aP2 protein. e, clamp-hepatic glucose production (c-HGP), f, glucose infusion rates (GIR) and g, rate of whole body glucose disposal (RD) in aP2−/− mice that were infused with recombinant aP2 during the pancreatic clamp study. h, Phosphoenolpyruvate carboxykinase 1 (Pck1) activity in liver samples harvested from aP2−/− mice following infusion with recombinant aP2 protein. Statistical analysis was performed by student’s t test. Data are presented as means ± SEM. * p < 0.05, ** p < 0.01, n=8 in aP2 infusion and n=6 in control group.
Serum aP2 critically regulates hepatic glucose metabolism in mice
Stimulation of aP2 secretion upon lipolysis and the marked increase in its serum levels in obesity raises the possibility that circulating aP2 may be a candidate hormone linking adipose tissue to hepatic glucose production and lead to its dysregulation in obesity. To investigate this hypothesis, we developed a neutralizing antibody to interfere with circulating aP2. We verified that this antibody specifically detected aP2 with high sensitivity and specificity (Fig. S5e) and then injected into mice with dietary obesity for two weeks and examined metabolic outcomes. There was a reduction in plasma aP2 levels in antibody-treated mice (Fig. 6a), which occurred without any alteration in aP2 protein levels in the adipose tissue (Fig. 6b). We also verified that this reduction in blood aP2 was not due to interference with our assay system, as short-term administration of the antibody did not alter aP2 levels in lean mice, and measurements in antibody-spiked plasma samples did not interfere with the assay (data not shown). Antibody administration did not alter the body weight (Fig. 6c) but caused a significant decrease in blood glucose levels within two weeks of treatment compared to controls treated with a pre-immune IgG (Fig. 6d). No difference was detected in blood free fatty acid levels between groups (Fig. 6e), In a glucose tolerance test, mice receiving the aP2 antibody exhibited significantly improved glucose disposal curves compared to control animals (Fig. 6f). The changes in glucose disposal curves evaluated by the area under the curve also exhibited significant differences (Fig. 6g). Taken together, these results demonstrated significantly improved whole body glucose metabolism upon treatment with the anti-aP2 antibody.
Figure 6. Improved glucose homeostasis in obese mice after aP2 neutralization.
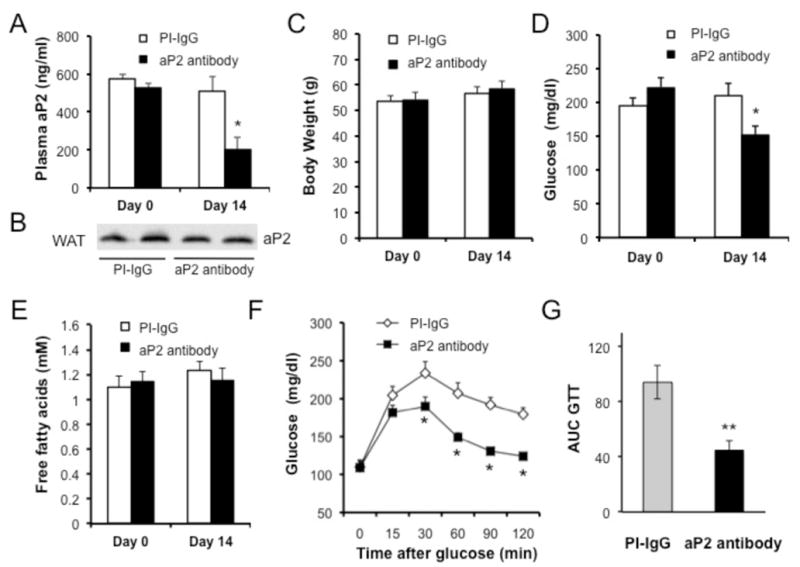
a, Plasma aP2 levels in mice with dietary obesity before and after administration of control pre-immune (PI-IgG) or an anti-aP2 antibody for two weeks, determined by aP2 ELISA. b, aP2 protein expression in total protein extracts of white adipose tissue (WAT) from obese mice after treatment with PI-IgG or anti-aP2 antibody for two weeks. c, Body weight and d, glucose levels in obese mice treated with control PI-IgG or anti-aP2 antibody for two weeks. Body weight measurements and blood glucose levels were determined after 6 hours of daytime food withdrawal. e, Free fatty acid levels in mice before and after antibody treatment f, Glucose tolerance test (GTT) in obese mice after aP2 neutralization for two weeks (1g/kg glucose). g, Graph depicting the area under the curve (AUC) calculations for GTT shown in panel f. Statistical analysis was performed by student’s t test. Data are presented as means ± SEM. * p<0.05, ** p<0.01.
We then asked whether the effects of aP2-neutralization on glucose metabolism were also related to its effects on hepatic glucose metabolism. Therefore, we examined total body glucose fluxes and tissue-specific effects by performing hyperinsulinemic-euglycemic clamp studies in mice treated with aP2 antibody. Neutralization of aP2 in obese mice resulted in significantly decreased basal and clamp hepatic glucose production (Fig. 7a,b), indicating that the liver is the primary target of circulating aP2 in regulating glucose metabolism. During the clamp studies, obese mice injected with the antibody required significantly increased glucose infusion rates (Fig. 7c), but exhibited no changes in their rate of overall glucose disposal compared to controls (Fig. 7d). These results indicated that the elevated glucose infusion rate in these animals was driven mainly by decreased hepatic glucose production, which is also supported by decreased gluconeogenic gene expression (Pck1 and G6pc) in the liver tissues of these animals (Fig. 7e). These data are in line with what has been observed in whole body FABP-deficient mice with genetic or dietary obesity (Cao et al., 2006) and consistent with the effects obtained with soluble aP2 in this study. Collectively, the effects of aP2 on hepatic glucose metabolism appear to be primarily mediated by the secreted form of this protein (Fig. 7f).
Figure 7. aP2 neutralization affects hepatic glucose production.
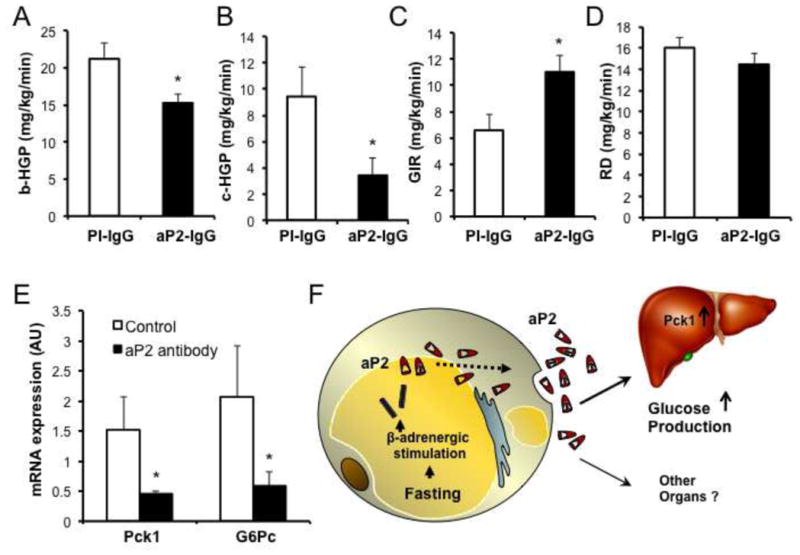
a, Basal hepatic glucose production (b-HGP) in WT, high fat diet-fed, obese mice during hyperinsulinemic-euglycemic clamps following aP2 neutralization. b, Hepatic glucose production during the clamp period (c-HGP) in obese mice after aP2 neutralization. c, Glucose infusion rate (GIR) in obese mice following aP2 neutralization. d, Rate of whole body glucose disposal (RD) during the clamp period in obese mice following aP2 neutralization. e, Expression of gluconeogenic genes phosphoenolpyruvate carboxykinase 1 (Pck1) and glucose-6-phosphatase (G6pc) following neutralization of aP2. At least 10 male mice were used in each experiment. Statistical analysis was performed by student’s t test. Data are presented as means ± SEM. * p < 0.05. f, Model of aP2 action following secretion from adipocytes. aP2 is a fasting-regulated hormone responsive to β-adrenergic stimuli and lipolytic signals, and acts on liver to stimulate glucose production. Other potential target organs and cells remain unknown.
DISCUSSION
The present study provides evidence that aP2 is an adipokine that is regulated by nutritional status and obesity. In mouse and human adipocytes, aP2 secretion is activated by fasting-related signals, including lipolytic agents. In mice, serum aP2 is entirely derived from adipocytes, with a marked increase in dietary or genetic obesity models. Importantly, serum aP2 levels are also strongly and positively correlated with obesity in humans. Depletion of serum aP2 in obese mice suppresses the elevated hepatic glucose production, while the converse—increasing serum aP2 in lean mice—leads to enhance hepatic glucose production. These results indicate that secreted aP2 may be an important component of the adipo-hepatic communication system regulating liver glucose production (Fig. 7f).
Serum free fatty acids represent a key energy source during fasting. It is also recognized that elevated lipolysis and increased serum fatty acid levels are linked to the dysregulation of systemic glucose homeostasis and are one of the critical underlying causes of obesity-induced metabolic pathologies (Boden, 2003). Excess fatty acids cause insulin resistance in muscle and liver by reducing glucose utilization and attenuating insulin-mediated suppression of glucose production, respectively. Utilizing well-controlled hormonal conditions and clamp studies, fatty acids have also been shown to increase liver glucose production (Chu et al., 2002). This effect has been attributed to the activation of gluconeogenesis pathways by fatty acids (Boden, 2003; Lam et al., 2003), but an effect on insulin action has not been ruled out. Furthermore, numerous mouse models and conditions have uncoupled liver glucose production from increased serum fatty acids (Everett-Grueter et al., 2006; Fery et al., 1996; Savage et al., 2007) both in health and in the presence of diabetes. These observations indicate the potential involvement of an unknown factor(s) required to stimulate hepatic gluconeogenesis, especially during fasting conditions or obesity where there is insulin resistance and hence poor control of lipolysis, somewhat resembling the fasting condition in the presence of plenty (Fu et al., 2012). Our study identifies aP2 as a candidate adipocyte hormone mediating this important endocrine function.
The secretion of aP2 from adipocytes occurs at baseline conditions and responds to several signals associated with fasting condition. It would be further revealing to explore the mechanisms by which these signals stimulate aP2 secretion from adipocytes. Since aP2 lacks a signal peptide and its secretion from adipocytes is resistant to blocking of the classic secretory pathway, it is likely that its secretion in these cells occurs through a non-classical, alternative mechanism. This may represent a novel mechanism employed by adipocytes to support their secretory output and would also be an important area to explore in the future and may offer further insights into the biology of aP2 as an adipokine. Additionally in obesity, adipose tissue exhibits signs of inflammation and cell death, which may contribute to the presence of high levels of aP2 in circulation. Regardless, the apparent role of aP2 as a required serum component for dysregulated liver glucose production argues that obesity-induced hyper-aP2-emia might contribute to the elevated hepatic glucose production, which is the hallmark of hyperglycemia, in subjects with type 2 diabetes (Olefsky and Nolan, 1995). This would be consistent with the fact that during the transition from insulin resistance to hyperglycemia, fatty acid concentrations do not change significantly and hence may not explain, alone, the alterations in liver glucose metabolism. In fact, emerging data have strongly linked serum aP2 levels with metabolic disease risk in humans (Cabre et al.; Furuhashi et al., 2011; Kralisch and Fasshauer, 2013; Peeters et al., 2011; Xu et al., 2006; Yoo et al., 2011) and even suggested that circulating aP2’s relation to metabolic risk is significantly stronger than fasting free fatty acids Karakas et al., 2009.
In our study, we detected direct actions of aP2 in liver cells to regulate glucose production. However, in vitro systems present limitations to study glucose production faithfully. Therefore, it is still possible that regulation of hepatic glucose production by aP2 may also involve indirect mechanisms on other hormonal pathways such as glucagon on liver in vivo. Understanding of the potential cell surface signaling mechanisms and whether aP2 engages similar downstream mediators such as cAMP production and PKA activation, and transcription factors such as PGC1α and HNF4 that result in the regulation of gluconeogenic targets should shed light into these possibilities Lin and Accili, 2011. We have produced strong evidence for a direct action of aP2 on liver using pancreatic clamp studies in aP2-deficient mice reconstituted with recombinant aP2. However, we cannot rule out the involvement of other signals and hormones in collaboration with aP2 to regulate this or other aspects of glucose metabolism systemically to contribute to development of diabetes. Future studies should address this interesting possibility. Regardless, our findings support an exciting possibility that neutralizing serum aP2 may overcome the hurdles in exploiting aP2 as a therapeutic target by small molecules and represent an efficient approach to treat diabetes and possibly other metabolic diseases.
EXPERIMENTAL PROCEDURES
Animals
Mice with homozygous null mutations in aP2 and mal1 were backcrossed more than 12 generations into a C57BL/6J genetic background as previously described (Maeda et al., 2005). The genetic backgrounds of mice are also verified by congenic genotyping of 288 loci markers for the C57Bl/6J background using an ABI3130 genetic analyzer. Mice were maintained on regular chow diet (RD, PicoLab 5058 Lab Diet) or placed on high-fat diet (Research Diets, Inc., D12492i) at 4 weeks of age for 20 weeks to induce dietary obesity. Leptin-deficient (ob/ob) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Animals used were 24 to 28 weeks of age for lean and dietary models and 16 weeks of age for the ob/ob model, unless otherwise stated in the text. Regardless of the model, all mice used in experiments were males and maintained on a 12-hour light and dark cycle. In each animal experiment at least 10 mice were used, unless otherwise stated in the text. Glucose tolerance tests, hyperinsulinemic-euglycemic clamp, and pancreatic clamp experiments were performed as described previously (Furuhashi et al., 2007; Maeda et al., 2005; Rossetti et al., 1997). The Harvard Medical Area Standing Committee on Animals approved all studies.
Bone marrow transplantation
Six–week-old recipient mice were irradiated with two 5 Gy doses (total 10 Gy) from a cesium source separated by a 4-hour interval in order to minimize radiation toxicity. Bone marrow was collected by flushing the femurs and tibias from sex-matched donor mice (6–8 weeks of age) with Gibco RPMI 1640 medium (Invitrogen, Carlsbad, CA), as described Furuhashi et al., 2008. Four hours after the second irradiation, 5 × 106 bone marrow cells in 0.2 ml medium were injected intravenously. Starting one week before and 4 weeks following bone marrow transplantation, 100 mg/l neomycin and 10 mg/l polymyxin B sulfate were added to the acidified drinking water.
Metabolic labeling and pulse chase experiments
Cultured adipocytes were plated onto six-well plates and incubated with DMEM containing 0.2% BSA for 16 h. The cells were washed with PBS twice and incubated with methionine- and cysteine-free DMEM without serum for 1 hour to deplete the intracellular pools. Thereafter, the cells were incubated with 1 ml of methionine- and cysteine-free DMEM without serum containing 100 μCi/ml of L-[35S]-methionine and L[35S]-cysteine (GE Healthcare) for 4 hours. Then, the labeling medium was replaced with 1.2 ml of DMEM (serum-free) with vehicle, brefeldin A (10 μg/ml) or monensin (5 μM), and chased up to 4 hours. To prepare cell lysates, the cells were washed with PBS twice, and resuspended in Buffer A (10 mM Tris-HCl pH 7.5, 150mM NaCl, 5mM EDTA, 1% Nonidet P-40 and proteinase inhibitor cocktail). After centrifugation to remove undissolved cellular debris, the cell lysates were subjected to immunoprecipitation. A total of 500 μl of cell lysate was mixed with 500μl of Buffer A without EDTA and NP-40 and precleared by protein G sepharose (GE Healthcare) for 30 minutes and immunoprecipitated with either adiponectin antiserum (kindly provided by Dr. Philipp Scherer, UT Southwestern Medical Center) or aP2 antibody overnight followed by incubation with 40 μl of protein G sepharose for 2 hours. Forty five μl of 10x Buffer B (1x Buffer B: 10mM Tris-HCl pH 7.5, 150mM NaCl, 1mM EDTA, 1% NP-40) was added to the aliquots of the medium from metabolically labeled cells (250 μl) and DMEM (200 μl), and immunoprecipitation was performed as described above. Immune complexes were washed three times with Buffer B, and subjected to SDS-PAGE followed by autoradiography.
Primary culture of mouse hepatocytes and glucose production assays
Hepatocytes were isolated from overnight fasted mice by the collagenase perfusion method, as described previously (Sekiya et al., 2007). Cells were resuspended in Williams-E medium containing 5% FBS and seeded on collagen-coated dishes at 4.5 × 104 cells/cm2. After 4 h, cells were washed and cultured in fresh medium. For glucose production, cells were serum starved with 0.2% BSA overnight. Thereafter, the medium was replaced with phenol- and glucose-free DMEM, supplemented with 2mM propionic acid, and the cells were incubated for 2 hours with vehicle, recombinant aP2 (10 μg/ml) or glucagon (3 μM). The glucose concentration was determined using Amplex Red Glucose/Glucose Oxidase Assay kit (Invitrogen). The cells were lysed with 0.1N NaOH overnight, and the protein concentration was determined to normalize the values for glucose production.
Production, purification, and administration of recombinant aP2 and aP2 antibody
Recombinant aP2 or control Gus protein with 6x His tag was produced in E. coli and purified with B-PER 6xHis Spin Purification Kit (Pierce Biotechnology, Inc) followed by endotoxin removal with a commercial system (Lonza Inc.). One hundred μg of control or aP2 protein was intraperitoneally injected into WT mice maintained on regular chow diet twice a day for two weeks. The rabbit polyclonal antibody against mouse aP2 was produced using recombinant full-length aP2 protein and the antibody was purified from the serum of the final bleed using the NAb™ Spin system (Pierce Biotechnology, Inc). Pre-immune serum was purified similarly and used as control. Purified antibody was diluted in saline to 1μg/μl and injected at a dose of 5 mg/kg into mice on high-fat diet for 14 days. The aP2 protein used in hyperinsulinemic and pancreatic clamp studies was produced and purified by the UCB pharmaceutical company.
Hyperinsulinemic-euglycemic or pancreatic clamp studies with aP2 infusion
Four days prior to experiments, mice were anesthetized and the right jugular vein was catheterized with a PE-10 polyethylene tube (inside and outside diameters, 0.28 mm and 0.61 mm, respectively; Becton Dickinson, Franklin Lakes, NJ) filled with heparin solution (100 USP U/ml). The distal end of the catheter was tunneled under the skin, exteriorized in the intrascapular area, heat-sealed and then placed in a pocket under the skin until the day of the experiment when it was re-exteriorized. Hyperinsulinemic-euglycemic and pancreatic clamps were performed by modification of reported procedures (Furuhashi et al., 2008; Rossetti et al., 1997). Steady state tracer analysis was used for calculations and glucose specific activity was verified for steady state. Prior to pancreatic clamp experiments, we validated the protocol and determined that glucagon levels were in the physiological range (77 pg/ml) during the clamp period.
Data quantitation and statistical analysis
The western blot quantitations were made using ImageJ, a Java-based image processing program developed at the National Institutes of Health that gives pixel values to the corresponding image, thus enabling presentation of the data sets with statistics. The images generated by BioRad VersaDoc system were analyzed using ImageJ. The background noise signal was subtracted and the values obtained were corrected to the cellular aP2 in all the quantitations. Data are presented as mean±SEM. Statistical significance was determined by student’s t test and * denotes significance at p<0.05, **denotes significance at p<0.01.
Supplementary Material
Highlights.
Lipid chaperone aP2 is a novel adipokine with elevated serum levels in obesity
Recombinant aP2 stimulates hepatic glucose production, in vitro and in vivo
Antibody-mediated neutralization of serum aP2 reverses diabetes in obese mice
Serum aP2 could be a promising drug target for the treatment of diabetes
Acknowledgments
This work was supported in part by a National Institutes of Health (NIH) grant DK064360 (to GSH). HC was supported by fellowships from the NIH Roadmap (DK71507-04) and the American Diabetes Association. ME was supported by the Roadmap grant R90 DK071507 from NIH. MS was supported by a fellowship from the Manpei Suzuki Diabetes Foundation. MF was supported by fellowships from Japan Society for the Promotion of Science and American Diabetes Association. We thank Michael Alcala for technical help, Eric Rimm for providing human samples, Fahri Saatcioglu for scientific insights and discussion, Megan Washack for editorial assistance, and Ana Paula Arruda for the graphical abstract. The technology described in this manuscript is licensed to the biopharmaceutical company UCB, Union Chimique Belge.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Boden G. Effects of free fatty acids on gluconeogenesis and glycogenolysis. Life Sci. 2003;72:977–988. doi: 10.1016/s0024-3205(02)02350-0. [DOI] [PubMed] [Google Scholar]
- Cabre A, Babio N, Lazaro I, Bullo M, Garcia-Arellano A, Masana L, Salas-Salvado J. FABP4 predicts atherogenic dyslipidemia development. The PREDIMED study. Atherosclerosis. 2012;222:229–234. doi: 10.1016/j.atherosclerosis.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Cahill GF., Jr Fuel metabolism in starvation. Annu Rev Nutr. 2006;26:1–22. doi: 10.1146/annurev.nutr.26.061505.111258. [DOI] [PubMed] [Google Scholar]
- Cao H, Gerhold K, Mayers JR, Wiest MM, Watkins SM, Hotamisligil GS. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell. 2008;134:933–944. doi: 10.1016/j.cell.2008.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Maeda K, Gorgun CZ, Kim HJ, Park SY, Shulman GI, Kim JK, Hotamisligil GS. Regulation of metabolic responses by adipocyte/macrophage Fatty Acid-binding proteins in leptin-deficient mice. Diabetes. 2006;55:1915–1922. doi: 10.2337/db05-1496. [DOI] [PubMed] [Google Scholar]
- Cassidy A, Chiuve SE, Manson JE, Rexrode KM, Girman CJ, Rimm EB. Potential role for plasma placental growth factor in predicting coronary heart disease risk in women. Arterioscler Thromb Vasc Biol. 2009;29:134–139. doi: 10.1161/ATVBAHA.108.171066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CA, Sherck SM, Igawa K, Sindelar DK, Neal DW, Emshwiller M, Cherrington AD. Effects of free fatty acids on hepatic glycogenolysis and gluconeogenesis in conscious dogs. Am J Physiol Endocrinol Metab. 2002;282:E402–411. doi: 10.1152/ajpendo.00136.2001. [DOI] [PubMed] [Google Scholar]
- Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, Fazio S, Wiest MM, Watkins SM, Linton MF, Hotamisligil GS. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med. 2009;15:1383–1391. doi: 10.1038/nm.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett-Grueter C, Edgerton DS, Donahue EP, Vaughan S, Chu CA, Sindelar DK, Cherrington AD. The effect of an acute elevation of NEFA concentrations on glucagon-stimulated hepatic glucose output. Am J Physiol Endocrinol Metab. 2006;291:E449–459. doi: 10.1152/ajpendo.00043.2006. [DOI] [PubMed] [Google Scholar]
- Fery F, Plat L, Melot C, Balasse EO. Role of fat-derived substrates in the regulation of gluconeogenesis during fasting. Am J Physiol. 1996;270:E822–830. doi: 10.1152/ajpendo.1996.270.5.E822. [DOI] [PubMed] [Google Scholar]
- Fu S, Fan J, Blanco J, Gimenez-Cassina A, Danial NN, Watkins SM, Hotamisligil GS. Polysome profiling in liver identifies dynamic regulation of endoplasmic reticulum translatome by obesity and fasting. PLoS genetics. 2012;8:e1002902. doi: 10.1371/journal.pgen.1002902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi M, Fucho R, Gorgun CZ, Tuncman G, Cao H, Hotamisligil GS. Adipocyte/macrophage fatty acid-binding proteins contribute to metabolic deterioration through actions in both macrophages and adipocytes in mice. J Clin Invest. 2008;118:2640–2650. doi: 10.1172/JCI34750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi M, Hotamisligil GS. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat Rev Drug Discov. 2008;7:489–503. doi: 10.1038/nrd2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi M, Ishimura S, Ota H, Hayashi M, Nishitani T, Tanaka M, Yoshida H, Shimamoto K, Hotamisligil GS, Miura T. Serum fatty acid-binding protein 4 is a predictor of cardiovascular events in end-stage renal disease. PLoS One. 2011;6:e27356. doi: 10.1371/journal.pone.0027356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi M, Tuncman G, Gorgun CZ, Makowski L, Atsumi G, Vaillancourt E, Kono K, Babaev VR, Fazio S, Linton MF, Sulsky R, Robl JA, Parker RA, Hotamisligil GS. Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature. 2007;447:959–965. doi: 10.1038/nature05844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannucci E, Liu Y, Hollis BW, Rimm EB. 25-hydroxyvitamin D and risk of myocardial infarction in men: a prospective study. Arch Intern Med. 2008;168:1174–1180. doi: 10.1001/archinte.168.11.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertzel AV, Bernlohr DA. The mammalian fatty acid-binding protein multigene family: molecular and genetic insights into function. Trends Endocrinol Metab. 2000;11:175–180. doi: 10.1016/s1043-2760(00)00257-5. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Karakas SE, Almario RU, Kim K. Serum fatty acid binding protein 4, free fatty acids, and metabolic risk markers. Metabolism. 2009;58:1002–1007. doi: 10.1016/j.metabol.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralisch S, Fasshauer M. Adipocyte fatty acid binding protein: a novel adipokine involved in the pathogenesis of metabolic and vascular disease? Diabetologia. 2013;56:10–21. doi: 10.1007/s00125-012-2737-4. [DOI] [PubMed] [Google Scholar]
- Lam TK, Carpentier A, Lewis GF, van de Werve G, Fantus IG, Giacca A. Mechanisms of the free fatty acid-induced increase in hepatic glucose production. Am J Physiol Endocrinol Metab. 2003;284:E863–873. doi: 10.1152/ajpendo.00033.2003. [DOI] [PubMed] [Google Scholar]
- Lin HV, Accili D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 2011;14:9–19. doi: 10.1016/j.cmet.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Cao H, Kono K, Gorgun CZ, Furuhashi M, Uysal KT, Cao Q, Atsumi G, Malone H, Krishnan B, Minokoshi Y, Kahn BB, Parker RA, Hotamisligil GS. Adipocyte/macrophage fatty acid binding proteins control integrated metabolic responses in obesity and diabetes. Cell Metab. 2005;1:107–119. doi: 10.1016/j.cmet.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Makowski L, Boord JB, Maeda K, Babaev VR, Uysal KT, Morgan MA, Parker RA, Suttles J, Fazio S, Hotamisligil GS, Linton MF. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat Med. 2001;7:699–705. doi: 10.1038/89076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olefsky JM, Nolan JJ. Insulin resistance and non-insulin-dependent diabetes mellitus: cellular and molecular mechanisms. Am J Clin Nutr. 1995;61:980S–986S. doi: 10.1093/ajcn/61.4.980S. [DOI] [PubMed] [Google Scholar]
- Peeters W, de Kleijn DP, Vink A, van de Weg S, Schoneveld AH, Sze SK, van der Spek PJ, de Vries JP, Moll FL, Pasterkamp G. Adipocyte fatty acid binding protein in atherosclerotic plaques is associated with local vulnerability and is predictive for the occurrence of adverse cardiovascular events. Eur Heart J. 2011;32:1758–1768. doi: 10.1093/eurheartj/ehq387. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. 2006;444:847–853. doi: 10.1038/nature05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetti L, Chen W, Hu M, Hawkins M, Barzilai N, Efrat S. Abnormal regulation of HGP by hyperglycemia in mice with a disrupted glucokinase allele. Am J Physiol. 1997;273:E743–750. doi: 10.1152/ajpendo.1997.273.4.E743. [DOI] [PubMed] [Google Scholar]
- Savage DB, Petersen KF, Shulman GI. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiological reviews. 2007;87:507–520. doi: 10.1152/physrev.00024.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya M, Yahagi N, Matsuzaka T, Takeuchi Y, Nakagawa Y, Takahashi H, Okazaki H, Iizuka Y, Ohashi K, Gotoda T, Ishibashi S, Nagai R, Yamazaki T, Kadowaki T, Yamada N, Osuga J, Shimano H. SREBP-1-independent regulation of lipogenic gene expression in adipocytes. J Lipid Res. 2007;48:1581–1591. doi: 10.1194/jlr.M700033-JLR200. [DOI] [PubMed] [Google Scholar]
- Tong Q, Dalgin G, Xu H, Ting CN, Leiden JM, Hotamisligil GS. Function of GATA transcription factors in preadipocyte-adipocyte transition. Science. 2000;290:134–138. doi: 10.1126/science.290.5489.134. [DOI] [PubMed] [Google Scholar]
- Tso AW, Xu A, Sham PC, Wat NM, Wang Y, Fong CH, Cheung BM, Janus ED, Lam KS. Serum adipocyte fatty acid binding protein as a new biomarker predicting the development of type 2 diabetes: a 10-year prospective study in a Chinese cohort. Diabetes Care. 2007;30:2667–2672. doi: 10.2337/dc07-0413. [DOI] [PubMed] [Google Scholar]
- Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122:4–12. doi: 10.1172/JCI60016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waki H, Tontonoz P. Endocrine functions of adipose tissue. Annu Rev Pathol. 2007;2:31–56. doi: 10.1146/annurev.pathol.2.010506.091859. [DOI] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu A, Wang Y, Xu JY, Stejskal D, Tam S, Zhang J, Wat NM, Wong WK, Lam KS. Adipocyte fatty acid-binding protein is a plasma biomarker closely associated with obesity and metabolic syndrome. Clin Chem. 2006;52:405–413. doi: 10.1373/clinchem.2005.062463. [DOI] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo HJ, Kim S, Park MS, Choi HY, Yang SJ, Seo JA, Kim SG, Kim NH, Baik SH, Choi DS, Choi KM. Serum adipocyte fatty acid-binding protein is associated independently with vascular inflammation: analysis with (18)F-fluorodeoxyglucose positron emission tomography. J Clin Endocrinol Metab. 2011;96:E488–492. doi: 10.1210/jc.2010-1473. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.