

Abstract
The redox-sensitive transcription factor NFκB mediates the expression of genes involved in inflammation and cell survival. Thioredoxin reductase-1 (TR1) and its substrate thioredoxin-1 (Trx1) act together to reduce oxidized cysteine residues within the DNA binding domain of NFκB and promote maximal DNA binding activity in vitro. It is not clear, however, if NFκB is regulated via this mechanism within living cells. The purpose of the present study was to determine the mechanism of NFκB modulation by TR1 in cells stimulated with the inflammatory cytokine tumor necrosis factor-α (TNF). In both control cells and in cells depleted of TR1 activity through chemical inhibition or siRNA knock down, TNF stimulation resulted in degradation of the cytoplasmic NFκB inhibitor IκB-α and translocation of NFκB to the nucleus. Similarly, the DNA binding activity and redox state of NFκB were unaffected by TR1 depletion. In contrast, NFκB-mediated gene expression was markedly inhibited in cells lacking TR1 activity, suggesting that the transactivation potential of NFκB is sensitive to changes in TR1 activity. Consistent with this concept, phosphorylation of the transactivation domain of NFκB was inhibited in the presence of curcumin. Surprisingly, another TR1 inhibitor, 1-chloro-2,4-dinitrobenzene, had no effect, and siRNA knock down of TR1 actually increased phosphorylation at this site. These results demonstrate that TR1 activity controls the transactivation potential of NFκB, and that more than one mechanism may mediate this effect.
Keywords: Thioredoxin, thioredoxin reductase, transcription, redox, oxidation, NFκB, DNA binding
INTRODUCTION
Nuclear factor-kappaB (NFκB) is a transcription factor that controls the expression of genes involved in apoptosis, cell survival and inflammation [1]. NFκB is constitutively expressed but maintained in an inactive cytoplasmic complex through its association with IκB proteins [2]. A variety of stimuli, exemplified by the inflammatory cytokine tumor necrosis factor-α (TNF), activate the canonical pathway whereby a phosphorylation cascade is initiated that results in the phosphorylation, ubiquitination and degradation of the negative regulator IκB-α, allowing translocation of liberated NFκB into the nucleus where it binds specific enhancer sites within target genes. Nuclear translocation of NFκB alone does not result in maximal NFκB transcriptional activity. Other layers of regulation, including reversible oxidation, phosphorylation and acetylation of the transcription factor, are also important in modulating NFκB activity (for a review see [3–6]). The redox state of conserved cysteine residues within the DNA binding domain of NFκB heterodimers, corresponding to Cys62 of the p50 subunit and Cys38 of the p65 subunit, is thought to be a key determinant of the DNA binding activity of NFκB. In vitro DNA binding assays have shown that oxidation of Cys62 to a sulfenic acid prevents NFκB from binding to DNA, and that reduction to a thiol restores binding [7].
A number of lines of evidence point to the thioredoxin system, consisting of thioredoxin-1 (Trx1) and the NADPH-dependent enzyme thioredoxin reductase-1 (TR1), as being responsible for maintaining Cys62 of p50 in the thiol form. Trx1 is a small redox active protein that receives reducing equivalents from TR1 and transfers them to its substrates. In vitro, Trx1 can reduce p50 directly [7–8] or indirectly through the redox chaperone protein Ref-1 [9–10]. In cells, NFκB activity is enhanced upon over-expression of a nuclear-targeted Trx1 fusion protein, whereas a redox-inactive mutant of Trx1 has no effect [11]. These data are consistent with a model of redox regulation of NFκB transcriptional activity in which the reduced form of Trx1 reduces NFκB (either directly or through Ref-1), allowing DNA binding and transcription of target genes to proceed. Because Trx1 is reduced by TR1, this model predicts that inhibition of TR1 activity would block the flow of reducing equivalents from NADPH to NFκB, resulting in the accumulation of oxidized NFκB and decreased expression of NFκB target genes. Indeed, the TR inhibitors mercury, aurothiomalate, sulforaphane, aurothioglucose and BBSKE inhibit NFκB-mediated gene expression [12–17], but their effects on the redox state of NFκB have not been investigated.
Recently we reported that inhibition of TR1 activity by siRNA-mediated knock down did not result in accumulation of oxidized Trx1 [18], raising the question of whether downstream targets of Trx1 like NFκB became oxidized under these conditions. In light of the apparent disconnect between TR1 activity and Trx1 redox state, we re-examined the mechanism by which TR1 inhibition affects NFκB activity. We used HeLa cells in the majority of the experiments because the thioredoxin system and compartmentalized redox regulation of NFκB have been well-characterized in this cell line [11, 18–22]. In addition, we used RAW264.7 cells to assess whether the mechanisms at work in HeLa cells were also operating in other cell lines and in response to different activating stimuli. The data demonstrate that depletion of TR1 activity through chemical inhibition or siRNA-mediated knock down inhibited NFκB at the level of the nucleus, as would be predicted from the model described above. However, the inhibition was not dependent on the accumulation of oxidized Trx1, nor was it mediated by oxidation of cysteines within either the p50 or p65 subunits of NFκB. Together, these findings demonstrate that TR1 controls the transactivation potential of NFκB independently from effects on DNA binding activity mediated by Trx1.
MATERIALS AND METHODS
Cell culture
HeLa cells (American Tissue Culture Collection, Manassas, VA) are an established epithelial cell line isolated from a human cervical adenocarcinoma. The original donor was a 30 year old African-American woman. RAW264.7 cells are a murine monocyte/macrophage cell line isolated from a tumor induced by Abelson murine leukemia virus in BALB/c mice. The cells were incubated at 37°C under humidified atmosphere with 5% CO2 in Dulbecco’s modification of Eagle’s Medium (DMEM) containing 4.5g/L glucose, L-glutamine, and sodium pyruvate and supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA) and penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO). For all experiments, cells were plated at a density of 12,000 cells/cm2 and were confluent at the time the experiment was conducted.
TR1 knock down
Small interfering RNA was used to knock down TR1. 24 hours after plating, cells were transfected with siRNA against TR1 (siTR1), Trx1 (siTrx1) or control non-targeting siRNA (siNT) using Dharmafect transfection reagent (all reagents Dharmacon Inc., Lafayette, CO). Preliminary experiments used additional controls that were mock-transfected or untransfected, and these controls were similar to siNT transfected cells. Optimal knock down at the protein level occurred 48 hours after transfection.
Measurement of TR activity
Thioredoxin reductase activity was measured by the end-point insulin reduction assay for total TR enzymatic activity in biological samples with spectrophotometric detection [23]. Reagents and purified rat liver TR1 were from Sigma. Recombinant Trx1 was obtained from American Diagnostica (Stamford, CT). Because the insulin endpoint assay for TR activity measures the combined activities of both TR1 and TR2, the results of this assay are referred to as total TR activity.
Measurement of NFκB-dependent reporter gene
Cells were transfected with PathDetect NFκB-luciferase reporter plasmid (Stratagene, La Jolla, CA) using Fugene HD transfection reagent (Roche, Indianapolis, IN). The κB element in this reporter (5′-GGGGACTTTCC-3′) is identical to one of the 3 κB elements in the promoter for the IκB-α gene. To control for differences in transfection efficiency, cells were co-transfected with a plasmid encoding renilla luciferase under the control of the thymidine kinase promoter. Firefly luciferase and renilla luciferase activities were measured with the Dual-Luciferase Reporter Assay System (Promega, Madison, WI), and firefly luciferase activity was normalized by dividing by renilla luciferase activity.
Real time-PCR analysis of gene expression
Gene expression of the NFκB -dependent gene IκB-α was analyzed by real time-PCR. Cells were lysed and mRNA was extracted using an mRNA purification kit (Ambion, Austin, TX) then converted to cDNA using reverse transcriptase (Applied Biosystems, Foster City, CA). TaqMan gene expression assays were used to probe IκB-α mRNA levels on the ABI Prism platform (Applied Biosystems, Foster City, CA). β-actin levels were used to normalize IκB-α levels using the comparative CT method, and calibrator cDNA was used to normalize results between experiments.
Western blot analysis
Cellular proteins were prepared as either whole cell lysates [24] or as cytosolic and nuclear fractions [25]. Equal amounts of protein, as measured by the BioRadDC protein assay kit using γ-globulin as a protein standard, were separated by SDS-PAGE. Separated proteins were transferred to nitrocellulose membranes, blocked and probed with primary antibody. Trx1 antibody was from American Diagnostica (Stamford, CT) and phospho-Ser536-p65 antibody was from Cell Signaling (Danvers, MA). All other primary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Secondary antibodies were conjugated with Alexafluor-680 (Invitrogen, Carlsbad, CA) or IRDye800 (Rockland Immunochemicals, Gilbertsville, PA), and fluorescence was detected using the Odyssey imaging system (Li-Cor Biotechnology, Lincoln, NE). Densitometry analysis was performed with the Li-Cor imaging software.
Electrophoretic mobility shift analysis (EMSA)
Nuclear fractions were prepared and analyzed as described in [25]. Equal amounts of nuclear protein were incubated with an oligonucleotide probe containing the NFκB consensus binding site from the luciferase reporter described above (5′-GGGGACTTTCC-3′) that had been labeled with γ-32P-ATP by T4 polynucleotide kinase (Invitrogen, Carlsbad, CA). Unbound probe was separated from protein-bound probe by native polyacrylamide electrophoresis. To identify the subunit composition of the protein bound to the probe, supershifts were performed by incubating the protein-probe mixture with antibodies specific for p50 or p65 (Santa Cruz Biotechnology, Santa Cruz, CA) prior to separation. Shifted and supershifted bands were visualized by autoradiography.
Trx1 redox western blot analysis
Oxidized and reduced forms of Trx1 were separated and detected using the previously described thioredoxin redox western blot [26]. Briefly, cells were lysed in 6 M guanidine-HCl, 50 mM Tris-HCl, 3 mM EDTA and 0.5% Triton-X-100 in the presence of 50 mM iodoacetic acid (IAA) (all reagents Sigma) and incubated for 30 minutes at 37°C, resulting in carboxymethylation of reduced, but not oxidized, cysteines. Proteins were separated by native PAGE, and Trx1 was detected by standard western blot procedures, as described above. Densitometry was performed using LI-COR imaging software and compiled from 3 independent experiments. The redox state of Trx1 was calculated by dividing the sum of the 1 disulfide and 2 disulfide forms by the sum of all forms (oxidized and reduced) and expressed as percent oxidized.
A modified thioredoxin redox western blot technique was used to separate and identify the redox states of Trx1 in subcellular fractions following knockdown of TR1 [27]. Cells were fractionated by the method of Dignam as described in [28], but DTT was omitted and 50 mM IAA was included in the lysis buffer. After centrifugation, the cytosolic (supernatant) and nuclear fractions (pellet) were further carboxymethylated in denaturing guanidinium buffer as described above for the redox western blot method [27]. Equal volumes of lysate from each treatment group were analyzed by western blot.
Measurement of p50 and p65 redox states
The redox states of p50 and p65 were measured using the method developed for use with bacterial Trx and mitochondrial Trx2 [29–30]. Cellular proteins were precipitated in cold 10% trichloroacetic acid (TCA), and pellets were washed with cold acetone. Proteins were dissolved in derivatization buffer containing 0.67 M Tris, pH 8, 2% SDS, 1 mM EDTA, and 15 mM 4-acetamido-4′-maleimidylstilbene-2,2′-disolfonic acid, disodium salt (AMS; Invitrogen, Carlsbad, CA). To prepare fully reduced and oxidized proteins, AMS was omitted from the derivatization buffer and either DTT or H2O2 was added for 30 minutes at 37°C, followed by re-precipitation and solubilization in derivatization buffer containing AMS. After AMS derivatization for 1 hour at 37°C, all samples were again precipitated in TCA, washed in acetone and solubilized in 1X red loading buffer (New England Biolabs, Ipswich, MA) for separation by SDS-PAGE and standard western blotting.
Over-expression of wild type and mutant p65
The plasmid encoding wild type p65 was a kind gift from Katia Vancompernolle. After subcloning the coding region into pcDNA3.1, the cysteine in the DNA binding domain (at position 38) was mutated to alanine by site-directed mutagenesis [26]. HeLa cells were transiently transfected with plasmids encoding wild type and mutant p65 using Fugene HD.
RESULTS
Inhibition of TR1 activity blocks NFκB-mediated gene expression
We used both chemical inhibition and siRNA-mediated knock down of TR1 to study the effects of TR1 on NFκB activity. Treatment of cells with the TR1 inhibitors 1-chloro-2,4-dinitrobenzene (CDNB) and curcumin inhibited total TR activity (the combined activities of TR1 and mitochondrial TR2) by more than 90% within 1 hour of exposure, and this level of inhibition persisted for at least 8 hours (Fig. 1A). TNF-induced expression of an NFκB reporter gene was completely inhibited by either CDNB or curcumin (Fig. 1B). CDNB and curcumin had no effect on basal NFκB activity (not shown). In response to TNF, NFκB-dependent luciferase activity increased 19-fold in vehicle-treated cells, but this induction was blocked in cells treated with CDNB or curcumin. PCR analysis confirmed that CDNB and curcumin blocked transcriptional activation of the luciferase reporter gene (not shown).
Fig. 1. Curcumin and CDNB inhibit TR1 activity and block expression of an NFκB-dependent reporter gene.
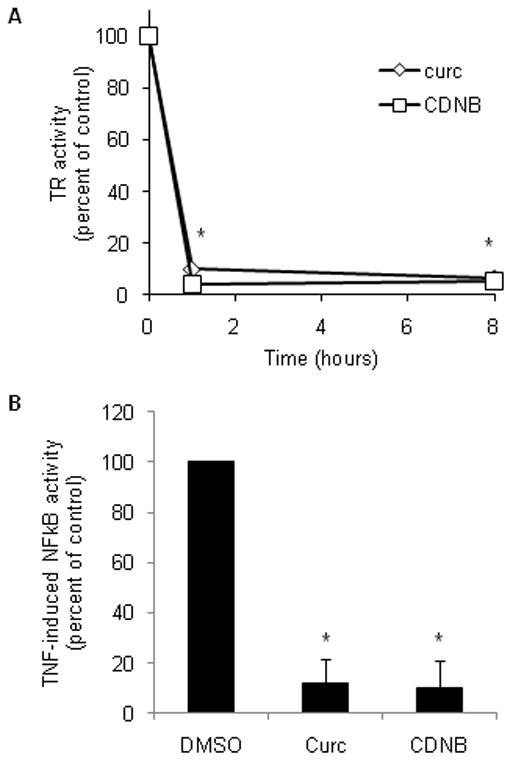
(A) HeLa cells were exposed to vehicle (DMSO), 50 μM curcumin (diamonds) or 30 μM CDNB (squares) for 1 hour or 8 hours, then TR activity (the combined activities of TR1 and TR2) was measured in whole cell lysates. Activity is expressed as the percent of activity in the DMSO-treated control cell lysates from 3 independent experiments. The asterisks indicate the time points at which TR activity was significantly lower (p<0.05) in cells treated with curcumin or CDNB relative to DMSO-treated control cells. (B) The expression of an NFκB reporter gene was measured in HeLa cells treated with chemical inhibitors of TR1 and stimulated with TNF. Cells were co-transfected with plasmids encoding NFκB-dependent luciferase reporter construct and a control reporter construct for 24 hours, and then treated with vehicle (DMSO), 50 μM curcumin or 30 μM CDNB with or without 10 ng/ml TNF for 3 hours. Data are expressed as a percentage of the induction by TNF in vehicle controls. Error bars represent standard deviation for 3 independent experiments, and an asterisk indicates p<0.05.
Knock down of TR1 by transfection with siRNA for 48 hours lowered TR activity (Fig. 2A) and TR1 protein levels (Fig. 2B) by 90%, while leaving Trx1 levels unchanged (Fig. 2B). Under these conditions, TNF-induced expression of the NFκB luciferase reporter was 80% lower than in control cells transfected with non-targeting siRNA (siNT; Fig. 2C). In contrast, knock down of the TR1 substrate Trx1 had no effect on TNF-induced NFκB-mediated gene expression (Fig. 2C). Real time-PCR revealed that mRNA levels of the endogenous NFκB target gene IκB-α were induced 6.6-fold by TNF in siNT control cells, whereas in siTR1 cells IκB-α was induced only 3.4-fold, corresponding to a 48% decrease relative to siNT controls (Fig. 2D). There was no significant difference in basal expression of IκB-α expression between siNT and siTR1 cells. Therefore, both an endogenous NFκB target gene and expression of an NFκB-dependent reporter gene were inhibited in cells depleted of TR1.
Fig. 2. Knock down of TR1, but not Trx1, inhibits NFκB-mediated gene expression.
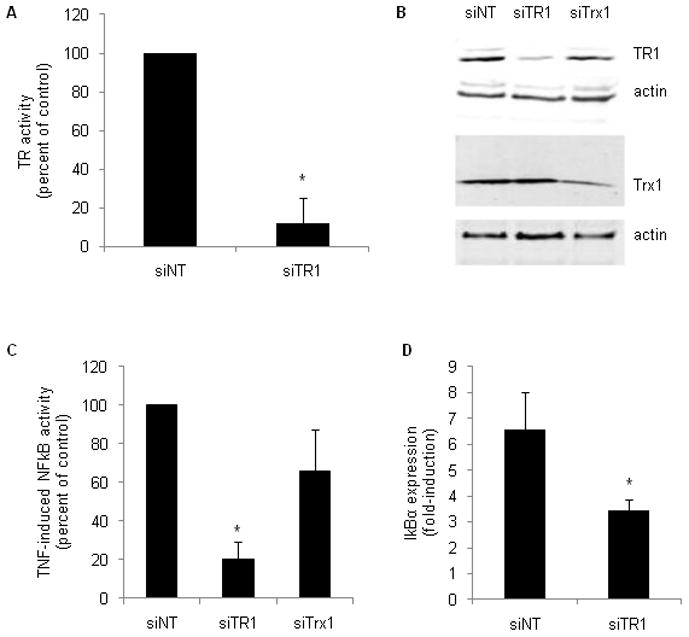
HeLa cells were transfected with non-targeting siRNA (siNT) or siRNA targeting TR1 (siTR1) or Trx1 (siTrx1). TR activity (A) and western blot analysis of TR1 and Trx1 protein levels (B) were performed in whole cell lysates 48 hours after transfection. Blots were re-probed for β-actin to demonstrate equal protein loading. NFκB-mediated gene expression was measured by luciferase reporter and real time-PCR analysis of endogenous gene expression. For the reporter gene assay (C), cells were transfected with the indicated siRNA for 24 hours, followed by an additional 24 hour transfection with the reporter plasmids. 10 ng/ml TNF was added during the last 3 hours of transfection. To measure endogenous gene expression (D), 10 ng/ml TNF was added 45 hours after transfection with siRNA and continued for another 3 hours. RNA was extracted and analyzed by real time PCR as described in Materials and Methods.
TR1 activity does not affect cytoplasmic activation, nuclear translocation or DNA binding activity of NFκB
To determine which step in the activation of NFκB was inhibited in cells lacking TR1 activity, cytosolic and nuclear events in the NFκB signaling pathway were investigated. Degradation of cytoplasmic IκB-α in response to TNF was unaffected by curcumin and only slightly inhibited by CDNB (Fig. 3A), indicating that the compounds were not inhibiting IκB kinase or proteasomal function. Western blots of nuclear fractions showed that translocation of NFκB subunits p50 and p65 into the nucleus in response to TNFα stimulation was also unaffected by treatment with curcumin and, corresponding to the effect on IκB-α, was slightly inhibited by CDNB (Fig. 3B). These data demonstrate that cytoplasmic activation and nuclear translocation of NFκB are largely unaffected by treatment with curcumin or CDNB.
Fig. 3. TR1 inhibition does not affect TNF-induced degradation of cytosolic IκB-α, nuclear translocation of p50 and p65 or DNA binding activity of NFκB.
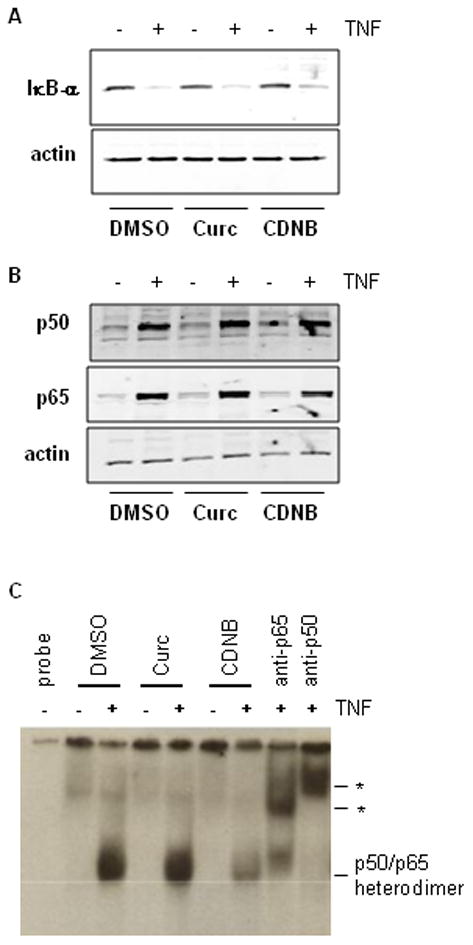
(A) Cytosolic IκB-α and (B) nuclear p50 and p65 were measured by western blot in curcumin- and CDNB-treated cells that were either unstimulated (−) or co-stimulated (+) with TNF for 1 hour. Blots were re-probed for β-actin to demonstrate equal protein loading. (C) DNA binding activity was assessed by EMSA of nuclear extracts from cells treated as in (A) and (B). The position of the major DNA-binding species, the p50/p65 heterodimer is indicated. Asterisks mark the positions of the supershifted bands obtained by incubation with antibodies against p50 and p65 (lanes labeled “anti-p50” and “anti-p65”). Each of the experiments in this figure was repeated at least 3 times with similar results.
To investigate whether curcumin or CDNB inhibited DNA binding activity, an EMSA was conducted on nuclear fractions from cells co-treated with curcumin or CDNB and TNF (Fig. 3C). There was very little NFκB binding to the oligonucleotide probe in unstimulated cells. Treatment with TNF stimulated DNA binding activity. Supershift analysis revealed that the protein-DNA band contained both p50 and p65 subunits of NFκB. Nuclear fractions from curcumin-treated cells showed as much DNA binding of the p50/p65 heterodimer upon TNF stimulation as did vehicle-treated cells, indicating that loss of DNA binding ability does not account for the inhibition of NFκB signaling. CDNB treatment partially inhibited TNF-induced DNA binding activity. This is in agreement with the pattern of IκB degradation and p50/p65 nuclear translocation for samples treated with CDNB, reflecting less total NFκB activation following CDNB treatment as compared to controls and curcumin treated samples. DNA binding activity (Fig. 3C) in nuclear extracts correlated with p50 and p65 levels (Fig. 3B). Therefore, curcumin and CDNB did not inhibit the DNA binding activity of NFκB, demonstrating that these inhibitors were not alkylating the DNA binding domain cysteines because this type of modification is essentially irreversible under the condition of the EMSA.
NFκB is a critical mediator of gene expression during the innate immune response. Stimulation of macrophages with bacterial endotoxin (lipopolysaccharide; LPS) induces the expression of the cytokine TNF, among other inflammatory mediators, via activation of NFκB [31]. When macrophages were exposed to the TR1 inhibitors CDNB and curcumin, there was a dose-dependent decrease in LPS-induced TNF expression (Fig. 4A). Similar to the effect seen in HeLa cells, TR1 inhibition did not affect degradation of cytosolic IκB-α or nuclear translocation of p65 (Fig. 4B). Therefore, modulation of NFκB transactivation potential by TR1 is a mechanism common to at least 2 different physiologically relevant stimuli in at least 2 cell types.
Fig. 4. LPS-stimulated TNF production by RAW264.7 cells is inhibited at the nuclear level by TR1 inhibitors.
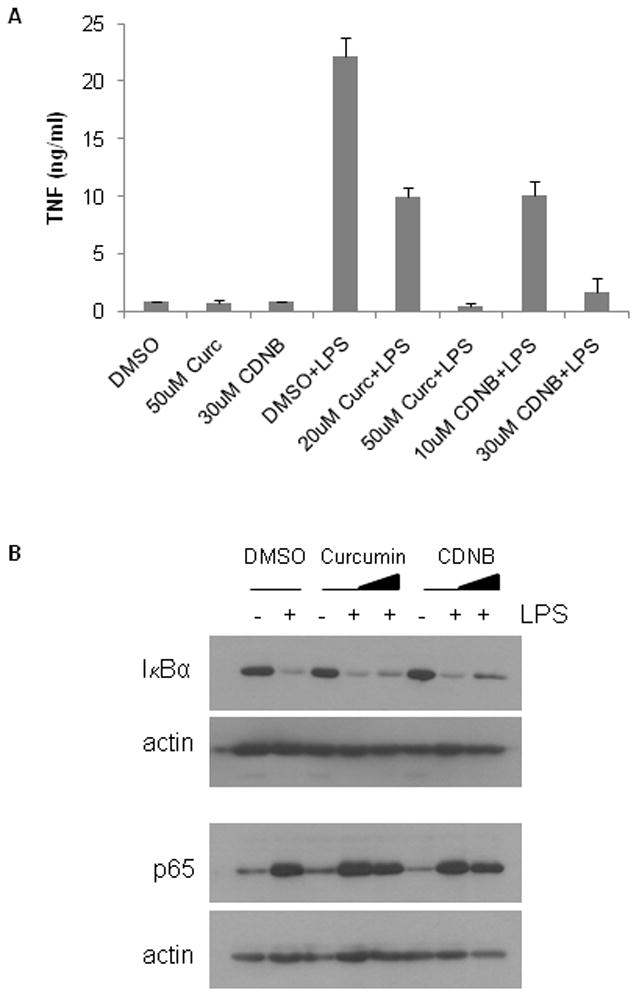
(A) TNF production by RAW264.7 murine macrophages. Cells were incubated with vehicle (DMSO) or the indicated concentrations of CDNB or curcumin (curc) in the absence or presence of 100 ng/ml LPS for 24 hours. TNF released into the conditioned media was measured by ELISA. (B) Cytoplasmic activation and nuclear translocation of NFκB in LPS-stimulated RAW264.7 cells. Cells were incubated with increasing concentrations of curcumin (0, 10 and 50 μM) or CDNB (0, 10 and 30 μM) and either left unstimulated (−) or were stimulated (+) with 100 ng/ml LPS for 15 minutes. Cytoplasmic IκB-α and nuclear p50 and p65 were determined by western blot. Blots were re-probed for β-actin to show equal loading.
As with the chemical inhibitors, TR1 knockdown inhibited NFκB at the nuclear level. Western blots of cytosolic fractions revealed that TNF-induced degradation of IκB-α was unaffected by TR1 knockdown (Fig. 5A), and western blots of the corresponding nuclear fractions showed that the NFκB subunits p50 and p65 translocated to the nucleus in response to TNF in both control and TR1 knock down cells (Fig. 5B). An EMSA confirmed that the subunit composition of nuclear NFκB was not affected in response to TR1 knockdown and that TNF-stimulated NFκB binding in siTR1 cells was no different from the NFκB binding of controls (Fig. 5C).
Fig. 5. Knock down of TR1 does not alter cytoplasmic activation or nuclear DNA binding of NFκB.
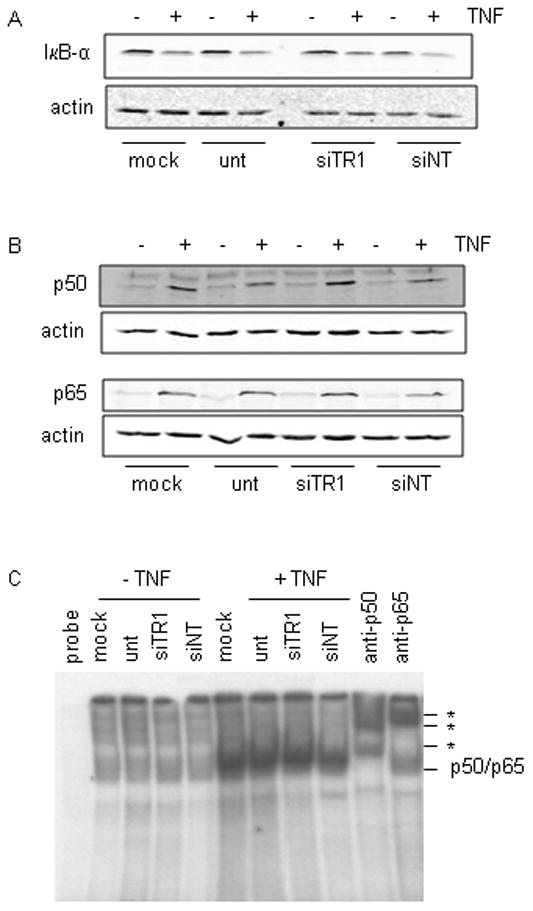
HeLa cells were transfected with siTR1 or transfection controls consisting of non-targeting siRNA (siNT), mock transfection (mock; no siRNA) or untransfected (unt; no siRNA or Dharmafect transfection reagent) for 48 hours, then stimulated with 10 ng/ml TNF for 1 hour where indicated. (A) Western blot of cytoplasmic IκB-α. (B) Western blot of nuclear proteins for p50 and p65 levels. Blots were re-probed for β-actin to demonstrate equal protein loading. (C) Nuclear proteins were analyzed by EMSA as described in the legend to Fig. 3.
Nuclear NFκB inhibition does not correlate with Trx1 oxidation
Trx1 is involved in the transfer of reducing equivalents from TR1 to NFκB [7, 9–10]. If the flow of reducing equivalents from TR1 to NFκB was being blocked by curcumin or CDNB, or limited by TR1 knock down, then increased oxidation of Trx1 would be expected. In order to determine if Trx1 oxidation was associated with the NFκB inhibition observed following treatment with the TR1 inhibitors curcumin and CDNB, the redox state of Trx1 in response to these treatments was investigated using the Trx1 redox western blot technique. Curcumin treatment (Fig. 6A) resulted in time-dependent oxidation of Trx1, with significant formation of the active site disulfide beginning at 2 hours. Maximum oxidation of Trx1 by curcumin was 37% and occurred at 16 hours. Treatment with CDNB oxidized Trx1 more extensively (Fig. 6B), resulting in the formation of both the 1-disulfide and 2-disulfide forms of the protein [26]. Maximum oxidation of Trx1 following CDNB treatment reached a plateau of 75% oxidized at 4 hours which persisted at all subsequent time points.
Fig. 6. Trx1 is oxidized following treatment with curcumin or CDNB, but not following siRNA-mediated knock down of TR1.
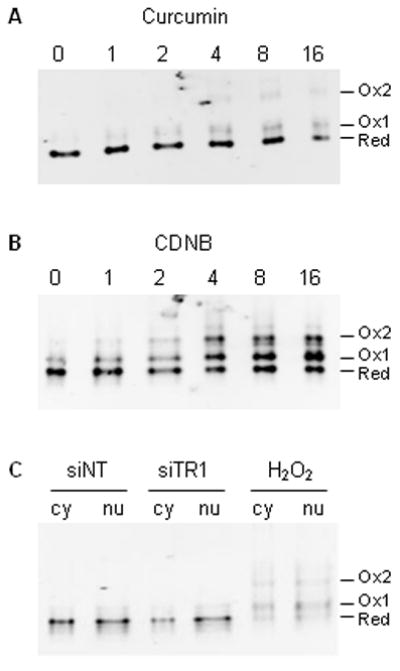
HeLa cells were treated with 50 μM curcumin (A) or 30 μM CDNB (B) for up to 16 hours. Following treatment, the redox state of Trx1 was determined using the Trx1 redox western blot technique. In (C), cells were transfected for 48 hours with non-targeting siRNA (siNT) or siRNA against TR1 (siTR1). A separate culture was treated with 1 mM H2O2 for 2 minutes as a positive control for Trx1 oxidation. The redox state of Trx1 in cytosolic and nuclear fractions was determined as described in Materials and Methods.
We previously demonstrated that knockdown of TR1 does not result in the oxidation of the total cellular pool of Trx1 [18]. It was still possible that nuclear pool of Trx1 was selectively oxidized, and that this pool was more directly involved in regulating NFκB activity. Compartmental redox western blot analysis of Trx1 showed that there was no significant difference in the amount of oxidized Trx1 between the siTR1 cells and the siNT controls in either the cytosolic or nuclear compartments (Fig. 6C). Peroxide-treated positive controls show the expected extensive oxidation of Trx1 in both the cytosolic and nuclear fractions (Fig. 6C).
The redox state of NFκB is preserved in cells with limiting TR1 activity
Oxidation of specific cysteine residues within the NFκB DNA binding domain has been shown to inhibit NFκB activity. To determine whether NFκB oxidation accounted for the loss of NFκB activity in cells exposed to chemical TR1 inhibitors, we measured the redox states of the NFκB subunits p50 and p65. To do this, cellular proteins were derivatized with AMS, a maleimide that reacts with reduced thiols but not oxidized forms of cysteine. Each AMS molecule incorporated adds 0.5 kDa to the mass of the protein. Therefore, more-reduced forms of the protein run more slowly on a denaturing gel than more-oxidized forms, and these different forms can be detected by immunoblotting. When AMS was omitted, both p50 and p65 exhibited the expected electrophoretic mobilities. When proteins in cellular lysates were reduced with DTT then derivatized with AMS the proteins again ran as a single band, but with slower mobilities resulting from the incorporation of AMS into the completely reduced forms (compare lanes 1 and 2 in Fig. 7A). When lysates were oxidized with diamide, hydrogen peroxide or tert-butylhydroperoxide prior to reaction with AMS, again a single band resulted upon western blotting for p50 or p65, but this time with a mobility identical to that of underivatized protein and corresponding to the completely-oxidized form of the protein. When the native redox states were assessed by immediately trapping all cellular thiols with AMS, the data showed that both p50 and p65 existed almost exclusively in their fully-reduced forms. Incubation of cells with curcumin or CDNB had no effect on the redox states of p50 or p65; these proteins continued to be in the fully-reduced forms (Fig. 7A, lanes 7–12).
Fig. 7. Neither p50 nor p65 is oxidized in cells with limiting TR1 activity.
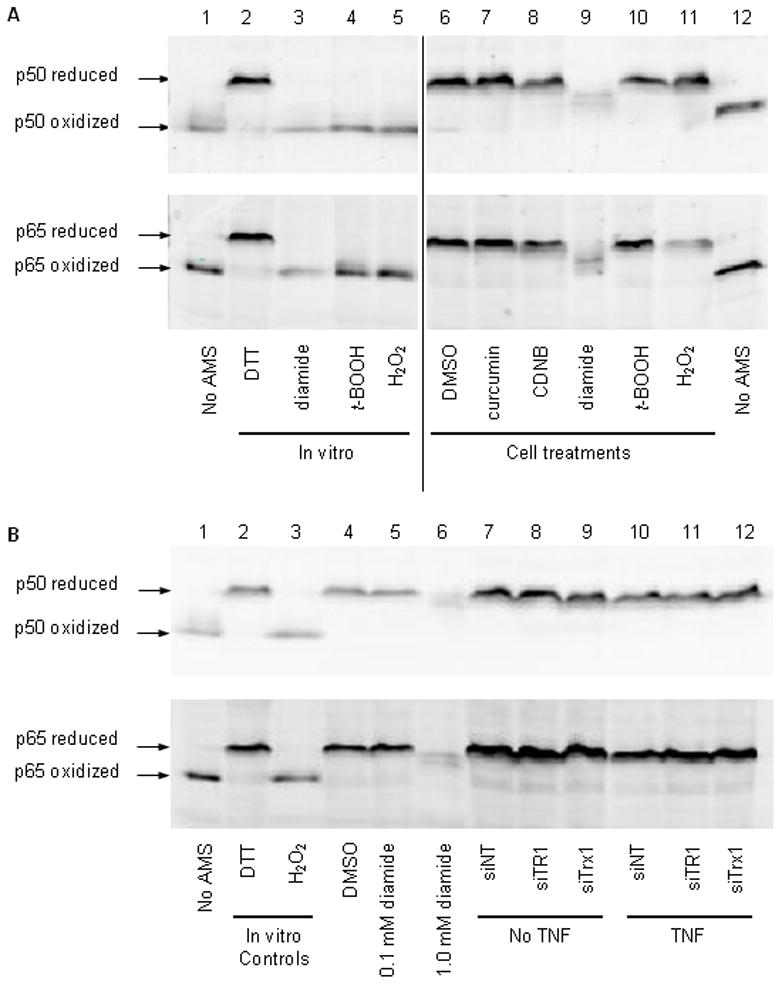
The redox states of p50 and p65 were determined in cells and cell lysates exposed to thiol oxidants and inhibitors of TR1 (A) or in cells depleted of TR1 by siRNA (B). In panel (A), the in vitro conditions refer to treatments made to re-solubilized TCA-precipitated proteins prior to derivatization with AMS, and the cell treatments refer to additions made to the cell cultures prior to lysis and derivatization with AMS. Lanes 1 and 12 contain cellular proteins that were not derivatized with AMS (no AMS). The concentrations used for in vitro and cell treatments were the same: 100 mM DTT, 10 mM diamide, 1 mM tert-butylhydroperoxide (t-BOOH), 10 mM hydrogen peroxide (H2O2), 0.1% DMSO (vehicle control), 50 μM curcumin and 30 μM CDNB. (B) Oxidized and reduced mobility controls (lanes 1 and 2, respectively) were prepared as in (A) by treating lysates in vitro with 100 mM DTT or 10 mM hydrogen peroxide. Positive controls for cellular oxidation of p50 and p65 were produced by treating cells with vehicle control (0.1% DMSO) or increasing concentrations of diamide in vitro for 30 minutes. Lanes 7 through 12 show the effects of different 48 hour siRNA knock downs in the absence (no TNF) or presence (TNF) of 50 ng/ml TNF in the final 30 minutes. Twice as much total protein was loaded in lanes 7–12 as in lanes 1–6. Arrows indicate the positions of fully oxidized and fully reduced forms of p50 and p65.
Next the ability of known thiol oxidants to oxidize NFκB subunits was assessed. Surprisingly, neither hydrogen peroxide nor tert-butylhydroperoxide were able to oxidize p50 or p65 in intact cells, whereas the same concentrations of these oxidants were able to oxidize the denatured proteins in the cell lysates (compare cell treatments to in vitro treatments in Fig. 7A). Of the oxidants tested, only diamide was able to oxidize p50 and p65 in cells. A high concentration of diamide resulted in loss of completely-oxidized p50 and p65 and the appearance of a mixture of partially oxidized forms. These data suggest that at least some of the cysteine residues within p50 and p65 are oxidized in diamide-treated cells. It should be noted that we consistently observed a loss in overall signal intensity upon incubation of either lysates or cells with diamide. This may reflect the formation of disulfide cross-links between NFκB subunits and other cellular proteins under oxidizing conditions.
Similar to the results with CDNB and curcumin, p50 and p65 remained fully reduced in cells depleted of TR1 by siRNA knock down (Fig. 7B). Exposure of cells to the inflammatory cytokine TNF has been reported to result in elevated ROS levels [32] and oxidation of the mitochondrial forms of thioredoxin (Trx2) and thioredoxin reductase (TR2) [33–34]. Therefore, we measured the redox states of p50 and p65 in the absence and presence of TNF stimulation. The results showed that TNF had no effect on the redox state of either p50 or p65, even in TR1 or Trx1 knock down cells (Fig. 7B) or in cells exposed to curcumin or CDNB (not shown).
Transactivation by a redox-insensitive mutant of NFκB is inhibited by TR1 knock down
The data in Fig. 7 strongly suggest that the DNA binding domain cysteines of NFκB are not mediating the effects of TR1 knock down on NFκB activity. To verify this conclusion, cells were transfected with a redox-insensitive mutant of p65 in which the DNA binding domain cysteine was mutated to alanine. In agreement with the observation that p65 is not oxidized in TR1 knock down cells, the activity of mutant p65 was inhibited to the same extent as wild type p65 upon over-expression in siTR1 knock down cells (Fig. 8). Therefore, the inhibitory effect of TR1 knock down is not mediated by changes in the redox state of the DNA binding domain of p65.
Fig. 8. Knockdown of TR1 leads to inhibition of NFκB activity and this inhibition does not involve the DNA binding domain cysteine of p65.
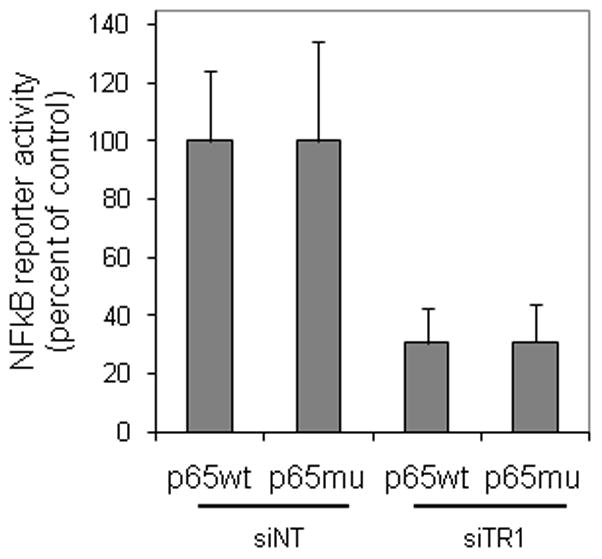
Cells were transfected with siRNA against TR1 or non-targeting controls (siNT) for 24 hours, and then transfected with reporter constructs for NFκB activity and a plasmid encoding mutant or wild type p65. Luciferase activity was measured after an additional 24 hours. Data represent the mean ± SEM of normalized NFκB-dependent luciferase activity from 3 independent experiments expressed as percent of the activity of the enzyme in siNT transfected cells.
Phosphorylation of Ser536 of p65 is differentially sensitive to curcumin, CDNB and siRNA knock down of TR1
A number of posttranslational modifications have been shown to alter the transactivation potential of NFκB (reviewed in [6]). One of the best characterized modifications is phosphorylation of Ser536 within the transactivation domain of p65. The data in Fig. 9 show that stimulation of cells with TNF resulted in transient phosphorylation of Ser536 of p65 which peaked 10 minutes after TNF stimulation and returned almost to basal levels after 1 hour. When TR1 was inhibited with curcumin, both basal and TNF-induced phosphorylation at Ser536 were inhibited. Although p65 translocated into the nucleus in response to TNF (see Fig. 3), it was not properly phosphorylated in the presence of curcumin (Fig. 9A). These results provide an explanation for the observation that NFκB is not transcriptionally active in cells exposed to curcumin even though it has translocated to the nucleus.
Fig. 9. Curcumin, CDNB and siRNA knock down of TR1 have different effects on phosphorylation of Ser536 of p65.
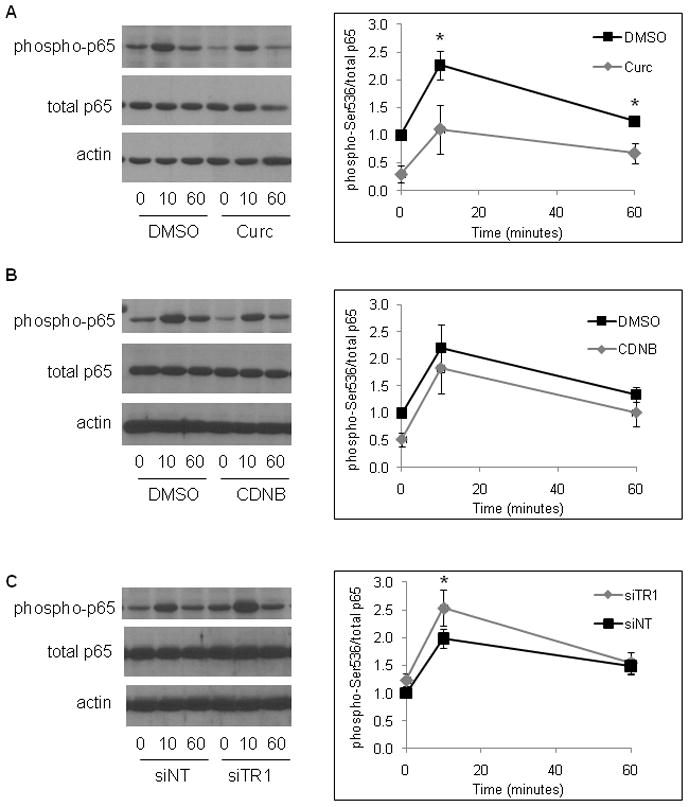
(A) HeLa cells were either untreated (0 minutes) or treated with 10 ng/ml TNF for 10 or 60 minutes in the presence of DMSO (vehicle control) or 50 μM curcumin. Western blots of whole cell lysates were prepared using antibodies specific for phospho-Ser536-p65 (phospho-p65), total p65 and β-actin (actin). The graph on the right shows the intensity of the band corresponding to the phosphorylated form of p65 normalized to the total amount of p65 from 3 independent experiments (mean ± SEM; * indicates p < 0.05). (B) Cells were stimulated with TNF in the absence and presence of 30 μM CDNB. Western blotting was performed as in part (A). Densitometric analysis of 3 independent experiments showed no statistical differences between DMSO (vehicle) and CDNB (right panel). (C) Cells were transfected with siRNA specific for TR1 (siTR1) or non-targeting siRNA (siNT) for 48 hours, then stimulated with TNF and analyzed by western blotting as in (A) and (B). TNF-induced phosphorylation was consistently higher in siTR1-transfected cells; the graph on the right represents the mean ± SEM of 7 independent experiments (* indicates p < 0.05).
CDNB, in contrast to curcumin, had no effect on the phosphorylation state of Ser536 in either unstimulated or TNF-stimulated cells (Fig. 9B), suggesting that curcumin and CDNB inhibit NFκB-mediated transactivation by different mechanisms. In contrast to both curcumin and CDNB, siRNA knock down of TR1 increased the maximal phosphorylation levels seen 10 minutes after TNF stimulation (Fig. 9C).
DISCUSSION
The data presented here show that loss of TR1 inhibits the transcriptional activity of NFκB while leaving DNA binding and all earlier steps in the activation pathway intact. TNF-induced IκB-α degradation, nuclear translocation of NFκB subunits, proper formation of p50/p65 heterodimers and DNA binding activity were not altered by curcumin, CDNB or siRNA knock down of TR1. Inhibition of NFκB-mediated gene expression did not require oxidation or alkylation of NFκB, or even the presence of a DNA binding domain cysteine. These data point to a novel mechanism by which TR1 modulates NFκB, a mechanism that affects transactivation rather than DNA binding activity.
TNF-induced phosphorylation of Ser536 within the transactivation domain of the p65 subunit of NFκB was inhibited by curcumin. Phosphorylation of Ser536 increases the transactivation potential of NFκB [6], and loss of this activating modification explains the inhibitory effect of curcumin. However, neither CDNB nor siRNA knock down of TR1 inhibited phosphorylation at this site, demonstrating that there is likely to be more than one mechanism by which TR1 activity controls the transactivation potential of NFκB. Phosphorylation or acetylation at one of the other sites within p65 that are known to alter transactivation potential may be sensitive to TR1 activity. Alternatively, the signaling pathways that control NFκB activation may be regulated by TR1. For example, inhibitors of phosphatidylcholine-phospholipase C, protein kinase C, phosphatidylinositol 3-kinase and phosphodiesterase block transactivation by NFκB while leaving upstream steps intact [35–37]. It is possible that TR1 influences NFκB activity by regulating the signaling cascades in which these proteins participate. It is known that many of the phosphatases and kinases involved in NFκB activation are redox sensitive [3, 5], but the role of TR1 in regulating these enzymes is unclear. A closer examination of the effect of TR1 on the redox states and activities of enzymes involved in the NFκB pathway may reveal novel TR1 substrates.
The p50/p65 heterodimer is the predominant form of NFκB in most cells, and it is this form that we have shown to be sensitive to regulation by TR1. Supershift analysis showed that p50/p65 was the major DNA binding species in the nuclei of TNF-stimulated HeLa cells, and this was not altered by inhibition of TR1. The sensitivity of p65 to TR1 inhibition was confirmed in the over-expression experiments shown in Fig. 8. Furthermore, we showed that Cys38 was not involved in TR1-mediated regulation of NFκB. Taken together, these results show that transactivation of gene expression by p65, most likely when heterodimerized with p50, is dependent on TR1 activity. It remains to be determined whether other NFκB subunits are regulated by TR1 activity in a similar manner.
The observation that TR1 regulated NFκB activity through a mechanism that was independent of NFκB oxidation was somewhat surprising. Although in vitro experiments have clearly demonstrated that oxidation of specific cysteine residues of NFκB inhibits its ability to bind to DNA [7, 9, 38–39], this particular mechanism was not employed under conditions of limiting TR1 activity. While chemical inhibitors of TR1 completely blocked NFκB activity at the nuclear level, they had no effect on the redox state of p50 or p65, the two NFκB subunits present in the heterodimer within the nucleus. Similarly, the inhibition of nuclear NFκB activity observed following TR1 knockdown occurred in the absence of NFκB oxidation. This is the first study to directly measure the redox state of NFκB subunits in cells as a function of TR1 activity, and the results suggest a novel mechanism of TR1 regulation of nuclear NFκB activity through a mechanism that does not involve direct redox regulation of NFκB.
Two other groups have examined the redox state of NFκB within cells under basal and stimulated conditions. Nishi et al., reported that p50 became more reduced in J-50 cells upon translocation to the nucleus in response to phorbol myristate acetate [40]. In contrast, Hansen et al., found that p50 became more oxidized after HeLa cells were stimulated with hydrogen peroxide [21]. In the present study, the redox state of p50 (and p65) did not change in HeLa cells upon stimulation with TNF. All of the p50 cysteines were labeled with AMS under the denaturing conditions used in our study, indicating that the protein was completely reduced even in unstimulated cells. Under conditions where NFκB is oxidized, such as in cells exposed to oxidants [21] or oxidant-inducing drugs [41], TR1 activity may be important for both the DNA binding activity and transactivation potential of NFκB.
In the present study, the majority of the inhibition of NFκB activity occurred at a point downstream of IκB-α degradation and nuclear translocation of the p50/p65 heterodimer, although CDNB demonstrated some partial inhibition of these early activation steps. Brennan and O’Neill, showed that curcumin and CDNB could bind directly to p50 and inhibit its DNA binding activity when measured by EMSA, but only at concentrations greater than those used in the present study [42]. They also found that curcumin, but not CDNB, could inhibit IKK activity in Jurkat cells, providing a second mechanism by which curcumin could inhibit the NFκB pathway. Inhibition of this cytoplasmic activation step has also been observed in fibroblasts [43], and constitutively active IKK was inhibited by curcumin in human mantle cell lymphoma cells [44]. Therefore, curcumin and CDNB can inhibit NFκB activity through a variety of mechanisms in different cell types, but under the conditions used here, inhibition of transactivation was the predominant mechanism.
Trx1 was oxidized in cells in which TR1 was inhibited with either curcumin or CDNB, but not in cells depleted of TR1 by siRNA. We have previously shown that the redox state of total cellular Trx1 does not change in cells depleted of TR1 [18], and in the current study we extend these findings by demonstrating that distinct subcellular pools of Trx1 were not oxidized under these conditions either. This is an important observation because nuclear Trx1 has been specifically implicated in regulating NFκB activity [11]. In a recent study, activity of an NFκB reporter was inhibited in cells engineered to produce ROS within the nucleus [20]. As in the present study, this inhibition occurred in the absence of nuclear Trx1 oxidation [20], but the redox state of NFκB was not measured. We have also reported that different chemical inhibitors of TR1 can have different effects on the thioredoxin system. Methylated arsenic, like curcumin and CDNB in the current study, resulted in increased Trx1 oxidation in cells, whereas aurothioglucose, like siRNA knock down of TR1, had no effect on Trx1 redox state [18]. The mitochondrial form of TR [45] or another selenoprotein [46] may have been responsible for keeping Trx1 reduced when TR1 was knocked down.
The results presented here identify a novel mechanism by which the thioredoxin system controls gene expression. In contrast to the previously-described regulation of DNA binding activity by Trx1, the regulation of transactivation of NFκB by TR1 is independent of changes in the redox states of either Trx1 or NFκB. This novel mechanism may be particularly relevant under the reducing conditions normally encountered within cells, where NFκB would not be expected to be oxidized.
Acknowledgments
This work was supported by NIH grants ES003819 and ES012260 (WHW), P01 AA017103 (CJM), R01 AA0015970 (CJM), R01 AA018016 (CJM), R01 DK071765 (CJM), R37 AA010762 (CJM), R01 AA018869 (CJM), P30 AA019360 (CJM), RC2AA019385 (CJM), and the Department of Veterans Affairs (CJM).
The abbreviations used are
- AMS
4-acetamido-4′-maleimidylstilbene-2,2′-disolfonic acid, disodium salt
- CDNB
1-chloro-2,4-dinitrobenzene
- DMEM
Dulbecco’s modification of Eagle’s medium
- DMSO
dimethylsulfoxide
- DTT
dithiothreitol
- IAA
iodoacetic acid
- LPS
lipopolysaccharide
- SDS
sodium dodecylsulfate
- t-BOOH
tert-butyhydroperoxide
- TNF
tumor necrosis factor-α
- Trx1
thioredoxin-1
- TR1
thioredoxin reductase-1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 2.Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11:372–377. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- 3.Flohe L, Brigelius-Flohe R, Saliou C, Traber MG, Packer L. Redox regulation of NF-kappa B activation. Free Radic Biol Med. 1997;22:1115–1126. doi: 10.1016/s0891-5849(96)00501-1. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 5.Pantano C, Reynaert NL, van der Vliet A, Janssen-Heininger YM. Redox-sensitive kinases of the nuclear factor-kappaB signaling pathway. Antioxid Redox Signal. 2006;8:1791–1806. doi: 10.1089/ars.2006.8.1791. [DOI] [PubMed] [Google Scholar]
- 6.Neumann M, Naumann M. Beyond IkappaBs: alternative regulation of NF-kappaB activity. Faseb J. 2007;21:2642–2654. doi: 10.1096/fj.06-7615rev. [DOI] [PubMed] [Google Scholar]
- 7.Matthews JR, Wakasugi N, Virelizier JL, Yodoi J, Hay RT. Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res. 1992;20:3821–3830. doi: 10.1093/nar/20.15.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qin J, Clore GM, Kennedy WM, Huth JR, Gronenborn AM. Solution structure of human thioredoxin in a mixed disulfide intermediate complex with its target peptide from the transcription factor NF kappa B. Structure. 1995;3:289–297. doi: 10.1016/s0969-2126(01)00159-9. [DOI] [PubMed] [Google Scholar]
- 9.Mitomo K, Nakayama K, Fujimoto K, Sun X, Seki S, Yamamoto K. Two different cellular redox systems regulate the DNA-binding activity of the p50 subunit of NF-kappa B in vitro. Gene. 1994;145:197–203. doi: 10.1016/0378-1119(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 10.Ando K, Hirao S, Kabe Y, Ogura Y, Sato I, Yamaguchi Y, Wada T, Handa H. A new APE1/Ref-1-dependent pathway leading to reduction of NF-kappaB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008;36:4327–4336. doi: 10.1093/nar/gkn416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirota K, Murata M, Sachi Y, Nakamura H, Takeuchi J, Mori K, Yodoi J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J Biol Chem. 1999;274:27891–27897. doi: 10.1074/jbc.274.39.27891. [DOI] [PubMed] [Google Scholar]
- 12.Dieguez-Acuna FJ, Ellis ME, Kushleika J, Woods JS. Mercuric ion attenuates nuclear factor-kappaB activation and DNA binding in normal rat kidney epithelial cells: implications for mercury-induced nephrotoxicity. Toxicol Appl Pharmacol. 2001;173:176–187. doi: 10.1006/taap.2001.9195. [DOI] [PubMed] [Google Scholar]
- 13.Carvalho CM, Chew EH, Hashemy SI, Lu J. Holmgren AInhibition of the human thioredoxin system. A molecular mechanism of mercury toxicity. J Biol Chem. 2008;283:11913–11923. doi: 10.1074/jbc.M710133200. [DOI] [PubMed] [Google Scholar]
- 14.Sakurai A, Yuasa K, Shoji Y, Himeno S, Tsujimoto M, Kunimoto M, Imura N, Hara S. Overexpression of thioredoxin reductase 1 regulates NF-kappa B activation. J Cell Physiol. 2004;198:22–30. doi: 10.1002/jcp.10377. [DOI] [PubMed] [Google Scholar]
- 15.Heiss E, Gerhauser C. Time-dependent modulation of thioredoxin reductase activity might contribute to sulforaphane-mediated inhibition of NF-kappaB binding to DNA. Antioxid Redox Signal. 2005;7:1601–1611. doi: 10.1089/ars.2005.7.1601. [DOI] [PubMed] [Google Scholar]
- 16.Ueno H, Kajihara H, Nakamura H, Yodoi J, Nakamuro K. Contribution of Thioredoxin Reductase to T-Cell Mitogenesis and NF-kB DNA-Binding Promoted by Selenite. Antioxid Redox Signal. 2007;9:115–121. doi: 10.1089/ars.2007.9.115. [DOI] [PubMed] [Google Scholar]
- 17.Lan L, Zhao F, Wang Y, Zeng H. The mechanism of apoptosis induced by a novel thioredoxin reductase inhibitor in A549 cells: possible involvement of nuclear factor-kappaB-dependent pathway. Eur J Pharmacol. 2007;555:83–92. doi: 10.1016/j.ejphar.2006.10.037. [DOI] [PubMed] [Google Scholar]
- 18.Watson WH, Heilman JM, Hughes LL, Spielberger JC. Thioredoxin reductase-1 knock down does not result in thioredoxin-1 oxidation. Biochem Biophys Res Commun. 2008;368:832–836. doi: 10.1016/j.bbrc.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smart DK, Ortiz KL, Mattson D, Bradbury CM, Bisht KS, Sieck LK, Brechbiel MW, Gius D. Thioredoxin reductase as a potential molecular target for anticancer agents that induce oxidative stress. Cancer Res. 2004;64:6716–6724. doi: 10.1158/0008-5472.CAN-03-3990. [DOI] [PubMed] [Google Scholar]
- 20.Halvey PJ, Hansen JM, Johnson JM, Go YM, Samali A, Jones DP. Selective oxidative stress in cell nuclei by nuclear-targeted D-amino acid oxidase. Antioxid Redox Signal. 2007;9:807–816. doi: 10.1089/ars.2007.1526. [DOI] [PubMed] [Google Scholar]
- 21.Hansen JM, Moriarty-Craige S, Jones DP. Nuclear and cytoplasmic peroxiredoxin-1 differentially regulate NF-kappaB activities. Free Radic Biol Med. 2007;43:282–288. doi: 10.1016/j.freeradbiomed.2007.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu J, Yoshida Y, Yamashita U. DNA-binding activity of NF-kappaB and phosphorylation of p65 are induced by N-acetylcysteine through phosphatidylinositol (PI) 3-kinase. Mol Immunol. 2008;45:3984–3989. doi: 10.1016/j.molimm.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 23.Arner ES, Holmgren A. Current Protocols in Toxicology. New York: John Wiley and Sons; 2005. Measurement of thioredoxin and thioredoxin reductase; pp. 7.4.1–7.4.14. [DOI] [PubMed] [Google Scholar]
- 24.Fang J, Lu J, Holmgren A. Thioredoxin reductase is irreversibly modified by curcumin: a novel molecular mechanism for its anticancer activity. J Biol Chem. 2005;280:25284–25290. doi: 10.1074/jbc.M414645200. [DOI] [PubMed] [Google Scholar]
- 25.Janssen YM, Sen CK. Nuclear factor kappa B activity in response to oxidants and antioxidants. Methods Enzymol. 1999;300:363–374. doi: 10.1016/s0076-6879(99)00141-x. [DOI] [PubMed] [Google Scholar]
- 26.Watson WH, Pohl J, Montfort WR, Stuchlik O, Reed MS, Powis G, Jones DP. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J Biol Chem. 2003;278:33408–33415. doi: 10.1074/jbc.M211107200. [DOI] [PubMed] [Google Scholar]
- 27.Watson WH, Jones DP. Oxidation of nuclear thioredoxin during oxidative stress. FEBS Lett. 2003;543:144–147. doi: 10.1016/s0014-5793(03)00430-7. [DOI] [PubMed] [Google Scholar]
- 28.Mercier PA, Winegarden NA, Westwood JT. Human heat shock factor 1 is predominantly a nuclear protein before and after heat stress. J Cell Sci. 1999;112(Pt 16):2765–2774. doi: 10.1242/jcs.112.16.2765. [DOI] [PubMed] [Google Scholar]
- 29.Ritz D, Beckwith J. Redox state of cytoplasmic thioredoxin. Methods Enzymol. 2002;347:360–370. doi: 10.1016/s0076-6879(02)47036-x. [DOI] [PubMed] [Google Scholar]
- 30.Halvey PJ, Watson WH, Hansen JM, Go YM, Samali A, Jones DP. Compartmental oxidation of thiol-disulphide redox couples during epidermal growth factor signalling. Biochem J. 2005;386:215–219. doi: 10.1042/BJ20041829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yi AK, Yoon JG, Hong SC, Redford TW, Krieg AM. Lipopolysaccharide and CpG DNA synergize for tumor necrosis factor-alpha production through activation of NF-kappaB. Int Immunol. 2001;13:1391–1404. doi: 10.1093/intimm/13.11.1391. [DOI] [PubMed] [Google Scholar]
- 32.Higuchi Y, Otsu K, Nishida K, Hirotani S, Nakayama H, Yamaguchi O, Matsumura Y, Ueno H, Tada M, Hori M. Involvement of reactive oxygen species-mediated NF-kappa B activation in TNF-alpha-induced cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2002;34:233–240. doi: 10.1006/jmcc.2001.1505. [DOI] [PubMed] [Google Scholar]
- 33.Hansen JM, Zhang H, Jones DP. Mitochondrial thioredoxin-2 has a key role in determining tumor necrosis factor-alpha-induced reactive oxygen species generation, NF-kappaB activation, and apoptosis. Toxicol Sci. 2006;91:643–650. doi: 10.1093/toxsci/kfj175. [DOI] [PubMed] [Google Scholar]
- 34.Kim JR, Lee SM, Cho SH, Kim JH, Kim BH, Kwon J, Choi CY, Kim YD, Lee SR. Oxidation of thioredoxin reductase in HeLa cells stimulated with tumor necrosis factor-alpha. FEBS Lett. 2004;567:189–196. doi: 10.1016/j.febslet.2004.04.055. [DOI] [PubMed] [Google Scholar]
- 35.Bergmann M, Hart L, Lindsay M, Barnes PJ, Newton R. IkappaBalpha degradation and nuclear factor-kappaB DNA binding are insufficient for interleukin-1beta and tumor necrosis factor-alpha-induced kappaB-dependent transcription. Requirement for an additional activation pathway. J Biol Chem. 1998;273:6607–6610. doi: 10.1074/jbc.273.12.6607. [DOI] [PubMed] [Google Scholar]
- 36.Sizemore N, Leung S, Stark GR. Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the NF-kappaB p65/RelA subunit. Mol Cell Biol. 1999;19:4798–4805. doi: 10.1128/mcb.19.7.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gobejishvili L, Barve S, Joshi-Barve S, McClain C. Enhanced PDE4B expression augments LPS-inducible TNF expression in ethanol-primed monocytes: relevance to alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2008;295:G718–724. doi: 10.1152/ajpgi.90232.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harikumar KB, Kunnumakkara AB, Ahn KS, Anand P, Krishnan S, Guha S, Aggarwal BB. Modification of the cysteine residues in IkappaBalpha kinase and NF-kappaB (p65) by xanthohumol leads to suppression of NF-kappaB-regulated gene products and potentiation of apoptosis in leukemia cells. Blood. 2009;113:2003–2013. doi: 10.1182/blood-2008-04-151944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toledano MB, Leonard WJ. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc Natl Acad Sci U S A. 1991;88:4328–4332. doi: 10.1073/pnas.88.10.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishi T, Shimizu N, Hiramoto M, Sato I, Yamaguchi Y, Hasegawa M, Aizawa S, Tanaka H, Kataoka K, Watanabe H, Handa H. Spatial redox regulation of a critical cysteine residue of NF-kappa B in vivo. J Biol Chem. 2002;277:44548–44556. doi: 10.1074/jbc.M202970200. [DOI] [PubMed] [Google Scholar]
- 41.Jing Y, Yang J, Wang Y, Li H, Chen Y, Hu Q, Shi G, Tang X, Yi J. Alteration of subcellular redox equilibrium and the consequent oxidative modification of nuclear factor kappaB are critical for anticancer cytotoxicity by emodin, a reactive oxygen species-producing agent. Free Radic Biol Med. 2006;40:2183–2197. doi: 10.1016/j.freeradbiomed.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 42.Brennan P, O’Neill LA. Inhibition of nuclear factor kappaB by direct modification in whole cells--mechanism of action of nordihydroguaiaritic acid, curcumin and thiol modifiers. Biochem Pharmacol. 1998;55:965–973. doi: 10.1016/s0006-2952(97)00535-2. [DOI] [PubMed] [Google Scholar]
- 43.Renard P, Delaive E, Van Steenbrugge M, Remacle J, Raes M. Is the effect of interleukin-1 on glutathione oxidation in cultured human fibroblasts involved in nuclear factor-kappaB activation? Antioxid Redox Signal. 2001;3:329–340. doi: 10.1089/152308601300185269. [DOI] [PubMed] [Google Scholar]
- 44.Shishodia S, Amin HM, Lai R, Aggarwal BB. Curcumin (diferuloylmethane) inhibits constitutive NF-kappaB activation, induces G1/S arrest, suppresses proliferation, and induces apoptosis in mantle cell lymphoma. Biochem Pharmacol. 2005;70:700–713. doi: 10.1016/j.bcp.2005.04.043. [DOI] [PubMed] [Google Scholar]
- 45.Turanov AA, Su D, Gladyshev VN. Characterization of alternative cytosolic forms and cellular targets of mouse mitochondrial thioredoxin reductase. J Biol Chem. 2006;281:22953–22963. doi: 10.1074/jbc.M604326200. [DOI] [PubMed] [Google Scholar]
- 46.Turanov AA, Kehr S, Marino SM, Yoo MH, Carlson BA, Hatfield DL, Gladyshev VN. Mammalian thioredoxin reductase 1: roles in redox homoeostasis and characterization of cellular targets. Biochem J. 2010;430:285–293. doi: 10.1042/BJ20091378. [DOI] [PMC free article] [PubMed] [Google Scholar]