

Abstract
Commensal flora can promote both immunity to pathogens and mucosal inflammation. How commensal driven inflammation is regulated in the context of infection remains poorly understood. Here, we show that during acute mucosal infection, Ly6Chi inflammatory monocytes acquire a tissue specific regulatory phenotype associated with production of the lipid mediator prostaglandin E2 (PGE2). Notably, in response to commensals, Ly6Chi monocytes can directly inhibit neutrophil activation in a PGE2-dependent manner. Further, in the absence of inflammatory monocytes, mice develop severe neutrophil-mediated pathology that can be controlled by PGE2 analog treatment. Complementing these findings, inhibition of PGE2 led to enhanced neutrophil activation and host mortality. These data demonstrate a previously unappreciated dual action of inflammatory monocytes in controlling pathogen expansion while limiting commensal mediated damage to the gut. Collectively, our results place inflammatory monocyte derived PGE2 at the center of a commensal driven regulatory loop required to control host-commensal dialogue during inflammation.
Mammalian barrier surfaces house complex commensal communities whose combined membership outnumbers host somatic cells1. Recent studies have highlighted the fundamental role of the commensal flora in the control of tissue development and metabolism2. Additionally, responsiveness to various pathogensrelies on the establishment of a dynamic equilibrium underpinned by the stimulatory-capacity of the flora. In this context, we and others, demonstrated that defined bacteria, or bacteria-derived products, can have an adjuvant effect in promoting mucosal immune responses to oral infections and vaccination3,4. Some of this control relies on the capacity of the flora to activate antigen presenting cell function, modulate IgA production5,6 or stimulate release of a broad spectrum of anti-microbial factors from epithelial cells7. However, this stimulatory property of the flora can be a double-edged sword. Indeed commensals share in common with pathogens, the expression of a large number of pathogen-associated molecular patterns (PAMPs) with strong inflammatory potential2. Mucosal tissues, and in particular the gastrointestinal (GI) tract, are primary sites of infection and in the context of inflammation, immune reactivity towards the microbiota represents considerable risk to the host8. Indeed, commensals have been linked to pathology in a number of mucosal infections4,9 and a variety of inflammatory disorders including Crohn’s disease10.
In the GI tract, under steady state conditions, complementary regulatory elements are in place to promote tolerance and to control inflammatory responses to the microbiota11,12 These include CD103+ dendritic cells (DCs) that are able to induce regulatory T(reg) cells13,14 and tissue resident CD11c+CX3CR1hi macrophages that constitutively produce the immunoregulatory cytokine IL-1012. However, upon pathogen invasion, inflammatory responses must rapidly develop within the mucosal environment and these responses can be associated with significant impairments to this endogenous regulatory network15-17. Additionally, during acute inflammation, Ly6Chi inflammatory monocytes and neutrophils become dominant recruited cell populations18,19. Monocytes and monocyte-derived cell types, such as TNF-α/iNOS producing (TIP)-DC, are critical for microbial clearance in a number of mucosal and systemic infection models, including Citrobacter rodentium20, Herpes simplex virus21, Aspergillus Fumigatus22, Toxoplasma gondii23 and Listeria monocytogenes24. Neutrophils not only play an important role in microbial clearance but are also responsible for collateral damage to tissue as a result of their capacity to release reactive oxygen species (ROS), superoxides, and a number of destructive proteases, alongside inflammatory cytokines25. Whether specific mechanisms exist to regulate the pathogenic potential of neutrophils during mucosal inflammation is unknown. Moreover, how, under inflammatory settings, the delicate balance between the protective and inflammatory role of the flora is controlled, remains an unanswered question.
Toxoplasma gondii is a well-established model used to investigate the balance between pathogen control and collateral damage26. Oral infection of certain strains of mice leads to Th1-mediated intestinal immunopathology characterized by ileitis in which severity depends upon the inflammatory potential of commensal bacteria9. However, despite this severe inflammation, T. gondii infected mice can survive the infectious challenge. We utilized this well-established model of acute mucosal infection to uncover dominant regulatory mechanisms of commensal driven pathology at mucosal sites.
Here, we found that, in response to commensal-derived signals, Ly6Chi inflammatory monocytes acquire a regulatory capacity during acute infection and specifically control neutrophil activation. Further, our work uncovers monocyte-derived PGE2 as a major mediator of immune regulation in the gastrointestinal (GI) tract via its capacity to directly limit activation of neutrophils. Together, our work describes a novel regulatory loop in which commensals limit their own pathologic potential by imposing a regulatory phenotype on Ly6Chi inflammatory monocytes. Such features allow these cells to efficiently control parasite expansion while simultaneously limiting collateral damage.
Results
Collapse of regulatory network and recruitment of inflammatory monocytes during mucosal T. gondii infection
To uncover the factors controlling commensal driven pathology at mucosal sites, we first investigated the dynamics of steady-state regulatory versus recruited inflammatory cells during acute gastrointestinal (GI) inflammation. To address this, we employed a model of T. gondii infection in which C57BL/6 mice were infected orally with 10 cysts of ME-49 (C1 clone)15. Parasite burden in the gut peaks at day 8 post-infection (p.i.), by which time animals develop an acute inflammatory response that is associated with rapid weight loss (Fig. 1a). In this setting, >80% of animals survive, eventually regaining weight by day 18 p.i. At the acute stage of infection, dramatic alterations to the steady-state regulatory network of the small intestine were apparent. Notably, the frequency and absolute number of lamina propria (Lp) CD11b+CD103+ DC, that contribute to Treg-induction13,14, were significantly reduced (Fig. 1b,c). Consistent with this observation, and as we have previously described15, Foxp3+ Treg collapsed at the peak of the inflammatory response (Supplementary Fig. 1a). Furthermore, by day 6 post-infection CD11c+CX3CR1hi macrophages had downregulated their capacity to make IL-10 (Supplementary Fig. 1b).
Figure 1. Collapse of regulatory network and recruitment of inflammatory monocytes during mucosal T. gondii infection.
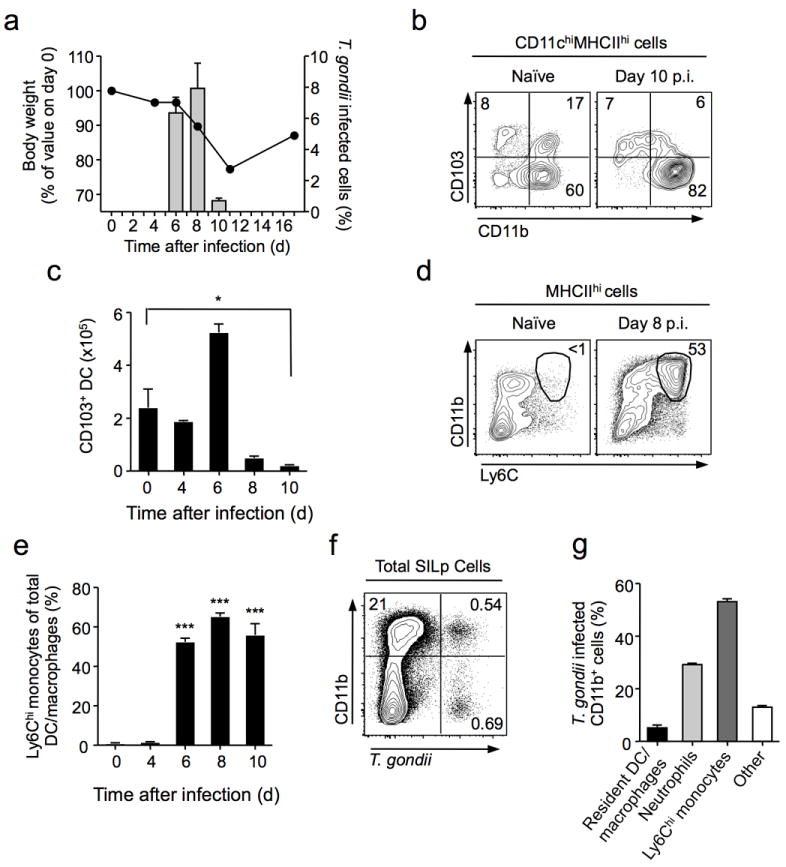
C57BL/6 mice were orally infected with 10 bradyzoite cysts of the red-fluorescent protein (RFP) expressing ME-49 clone, C1. (a) Body weight was monitored over the course of infection (Line). T. gondii-RFP burden was measured by flow-cytometry within the total live cell population isolated from small intestine lamina propria (SILp) (Bars). (b) Frequencies of CD11b+CD103+ dendritic cells (DC) were assessed by flow cytometry in naive (left panel) and day 10 infected (right panel) SILp. Plots are gated on live CD11c+MHCIIhi cells. Numbers in quadrants represent percentage of cells in gate. (c) Absolute numbers of CD11b+CD103+ DC isolated from the SILp at indicated time pointsafter infection. (d) Flow cytometric assessment of recruited inflammatory monocytes (Ly6ChiCD11bhi) in SILp of naïve or day 8 infected mice. Cells are gated on live MHCIIhi cells. Numbers adjacent to gates represent percentage of cells in gate. (e) The frequency of Ly6Chi inflammatory monocytes within the total SILp DC and macrophage population was assessed by flow cytometry over the course of infection. Statistical comparisons calculated against day 0 values. (f) T. gondii (C1-RFP) infected cells within CD11bhi (DC/Macrophages/Mononcytes/Neutrophils) and CD11blo (lymphocytes) populations were assessed by flow cytometry at day 8 post-infection. (g) Distribution of T. gondii infected cell populations within the CD11bhiRFP+ gate. Bar graph summarizes the average frequencies of cells. Data are representative of two independent experiments (n=3), results shown as mean ± SEM. Statistical comparisons were performed using the Student’s t test (*P<0.05, ***P<0.001).
In concert with these impairments to the regulatory network, Ly6ChiMHCIIhi inflammatory monocytes accumulated in the Lp over the course of infection, typically composing over 50% of total mucosal CD11b+ cells by day 8 p.i. (Fig. 1d). At this time point, Ly6Chi monocytes represented the dominant mononuclear phagocyte (MP) population in the small intestine Lp (SILp) (Fig. 1e, Supplementary Fig. 2a,b) and are the most infected cells in the tissue (Fig. 1f,g). Thus, mucosal T. gondii infection results in a global collapse of the steady-state regulatory network concomitant with a dramatic recruitment of Ly6Chi Mo.
Ly6Chi inflammatory monocytes adopt pro-inflammatory and regulatory features at mucosal sites
Previous work described parasite killing as the primary role for Ly6Chi monocytes during T. gondii infection23. However, given the prevalence of this cell-type in the SILp, along with the defect in regulatory populations during acute inflammation, we postulated that Ly6Chi monocytes may take on a more complex function. To explore this possibility, SILp Ly6Chi Mo, were sorted from the inflamed gut, and compared by microarray analysis to their blood precursors from infected mice (Ly6Chi classical monocytes)27(Supplementary Fig. 2c,d). Consistent with their anti-parasitic function, factors with a pro-inflammatory role were upregulated, including iNOS28 and IL-629 at both the gene and protein level (Fig. 2a,b,c). Moreover, IL-1α, and IL-1β, cytokines that play an effector role in multiple infectious settings were also enhanced (Fig. 2a,b,c). Ly6Chi monocytes also upregulated expression of various chemokines including, CXCL2, CCL3, and CCL4 (Fig. 2a,b), that are involved in the recruitment of lymphocytes, neutrophils, and monocytes to sites of inflammation. Additionally, although not enhanced at the transcriptional level, TNF-α protein, was significantly augmented (Fig. 2d).
Figure 2. Ly6Chi inflammatory monocytes have dual features in the SILp.
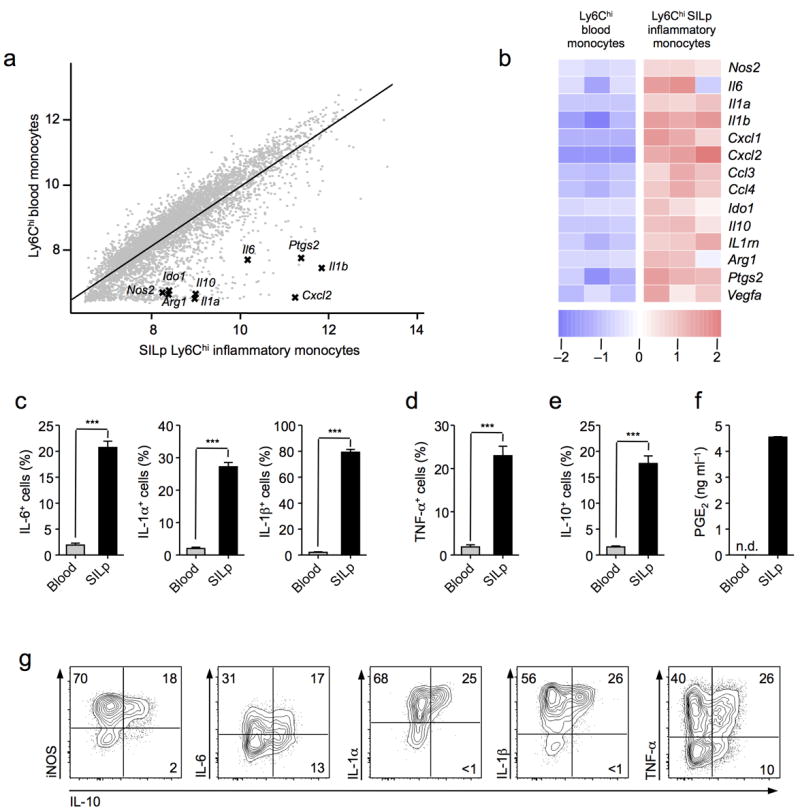
(a) Comparison of microarray expression values in FACS sorted Ly6Chi inflammatory monocytes (CD115+CD11b+), and Ly6Chi small intestine lamina propria (SILp) monocytes (MHCIIhiCD11b+). Both populations were purified by FACS from T. gondii infected C57BL/6 mice at day 8 post infection.(b) Heat map representation of key pro-inflammatory and regulatory genes identified from the profiling in (a). Each column represents an individual biological replicate. (c,d,e) Intracellular cytokine staining for IL-6, IL-1α, IL-1β, TNF-α, and IL-10, in blood Ly6Chi monocytes or SILp Ly6Chi monocytes in whole tissue suspensions cultured for 3 hrs in the presence of brefeldin A. Cells were gated on the live Ly6ChiCD11bhiMHCIIhi population. (f) Enzyme immunoassay (EIA) for PGE2 in supernatants from FACS sorted Ly6Chi blood Mo, and Ly6Chi SILp monocytes in whole tissue suspensions after18 hrs culture. (g) Intracellular cytokine staining for IL-10 in conjunction with iNOS, IL-6, IL-1α, IL-1β, and TNF-α in SILp Ly6Chi monocytes cultured for 3 hr in the presence of brefeldin A. (a,b) Data are from one experiment with three biological replicates, each consisting of 1×105 Ly6Chi blood monocytes, or Ly6ChiSILp monocytes (3–4 mice per replicate). (c-g) Data are representative of three independent experiments with three mice per group, results shown as mean ± SEM.Statistical comparisons were performed using the Student’s t test (***P<0.001).
Concomitant with the induction of inflammatory mediators, numerous genes ascribed to have immunoregulatory potential were also increased in SILp Ly6Chi monocytes (Fig. 2a,b,e). These included IL-1030 and Arginase-131, that both play a suppressive role in T. gondii infection, as well as indoleamine 2,3-dioxygenase (IDO)32. One of the most upregulated genes was Ptgs2 encoding for cyclooxygenase (COX)-2, a key enzyme in prostaglandin synthesis33 (Fig.2a,b). The production of the lipid mediator prostaglandin E2 (PGE2) was confirmed by culturing cell sorted Ly6Chi SILp monocytes from T. gondii infected mice (Fig. 2f). Notably, prostaglandin E2 (PGE2) can have suppressive effects on innate cell function34. The co-expression of immunoregulatory and inflammatory features by mucosal Ly6Chi monocytes was unlikely a feature of a unique subset within this population as intracellular cytokine staining revealed Ly6Chi monocytes co-producing inflammatory and regulatory cytokines (Fig. 2g). Thus, as a consequence of their exposure to the inflamed mucosal milieu, Ly6Chi monocytes adopt a mixed phenotype, acquiring both inflammatory and regulatory features.
Regulatory phenotype of Ly6Chi inflammatory monocytes is induced by commensal stimuli
We next investigated if such expression of dual features was specific to Ly6Chi monocytes in the small intestine lamina propria (SILp) milieu. Although T. gondii invades through the gut, it establishes a systemic infection, driving recruitment of inflammatory cells to multiple sites, including the spleen (Supplementary Fig. 3a). We found that while Ly6Chi monocytes from all infected tissues produced TNF-α to a similar extent (Fig. 3a,d), IL-10 and PGE2 were selectively expressed by Ly6Chi monocytes isolated from the gut (Fig. 3b,e,f).
Figure 3. Ly6Chi inflammatory monocytes acquire regulatory features in response to commensal stimuli.
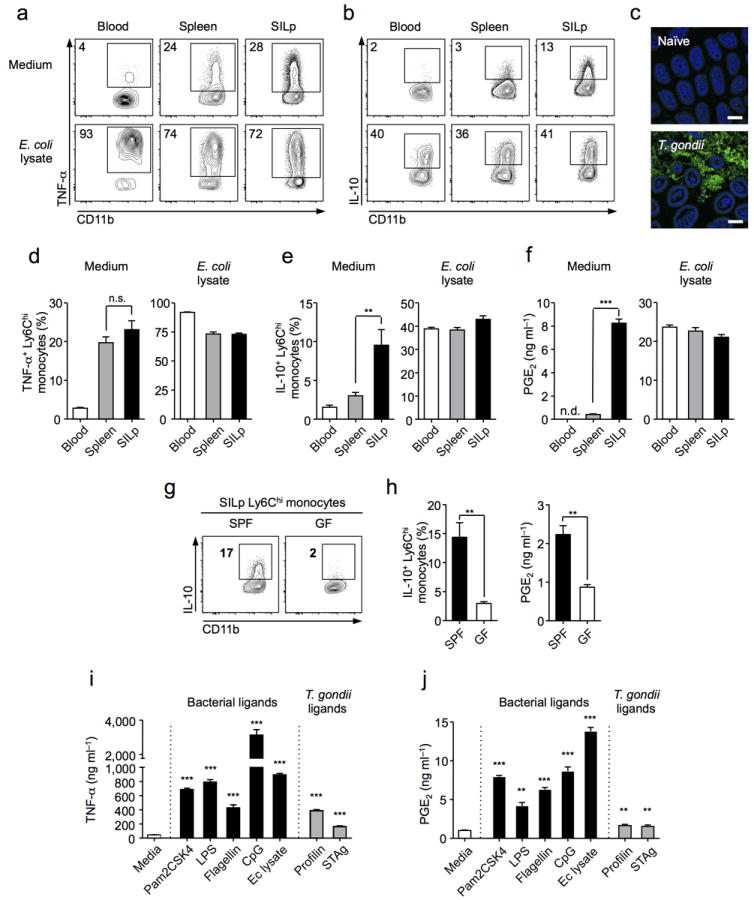
(a,b) At day 8 post-infection cells were isolated from the blood, spleen, and small intestine lamina propria (SILp), and cultured with brefeldin A alone (top panels), or with E. coli lysate (5 μg/ml) in the presence of brefeldin A (bottom panels), for intracellular cytokine staining. Plots gated on Ly6ChiCD11bhiMHCIIhi monocytes. (c) Ileum of naïve or T. gondii infected mice were sectioned and samples hybridized for FISH with a fluorescent probe specific to eubacterial 16S sequences and counter-stained with DAPI (Scale bar 50 μm). (d,e) Histograms represent mean percentage of cytokine positive cells from individual animals (n=3). (f) On day 8 p.i. Ly6Chi monocytes were sorted by FACS and cultured for 18 hrs alone or with E. coli lysate. PGE2 was measured in triplicate supernatants by EIA. (g) Cell suspensions from the SILp of T. gondii-infected specific-pathogen free (SPF) or germ free (GF) mice were intracellular cytokine stained for IL-10. Plots gated on Ly6ChiCD11bhiMHCIIhi monocytes (n=5). (h) SILp Ly6Chi monocytes were sorted and cultured for 18 hrs. PGE2 was measured in triplicate supernatants by EIA. (i) On day 8 p.i. Ly6Chi monocytes were sorted from the SILp and cultured for 18 hrs, alone, or with bacterial ligands (Black bars), or T. gondii ligands (Grey bars). TNF-α was measured in triplicate supernatants by ELISA. (j) Cells were sorted and stimulated as in (i). PGE2 was measured in triplicate supernatants by EIA. Statistical comparisons are calculated against medium alone (i,j). Data are representative of three independent experiments, results are mean ± SEM. Statistical comparisons performed using the Student’s t test (**P<0.01, ***P<0.001, n.s. non significant).
A defining feature of mucosal sites is their juxtaposition with an abundant and diverse commensal flora35. In particular, during T. gondii infection, the gastrointestinal immune system becomes highly exposed to microbial derived products resulting from tissue damage and bacterial translocation dominated by γ-proteobacteria such as Escherichia coli8 (Fig. 3c, data not shown). To assess if IL-10 and PGE2 production could arise as a consequence of exposure to commensals, germ free (GF) mice, that are raised and maintained devoid of live microbes, were infected with T.gondii. At day 9 post-infection (p.i.) the ability of small intestine Ly6Chi monocytes to produce IL-10 and PGE2 was significantly decreased in GF compared to specific pathogen free (SPF) control mice (Fig. 3g,h). To further address the capacity of microbial products to trigger regulatory factors,Ly6Chi monocytes from the SILp and spleen were stimulated with a lysate derived from a commensal strain of E. coli isolated from T. gondii infected mice. In response to microbial stimulation, splenic Ly6Chi monocytes upregulated their production of IL-10 and PGE2, as well as TNF-α, to a similar extent as SILp Ly6Chi Mo, resulting in a normalization of the phenotype between these two populations (Fig. 3a,b,d,e,f). Thus, acquisition of regulatory features is not limited to a subset of Ly6Chi monocytes but the result of commensal driven activation. More specifically, Ly6Chi monocytes produced TNF-α, IL-10, and PGE2, in response to a broad range of bacterial-derived ligands, including those recognized by TLR-2, -4, -5, and -9 (Fig. 3i,j, Supplementary Fig. 3b). Ly6Chi monocytes also responded to soluble T. gondii antigen (STAg), and the T. gondii-derived ligand profilin in a pro-inflammatory manner that was associated with limited production of IL-10, or PGE2 (Fig. 3i,j, Supplementary Fig. 3b). Together these data demonstrate that Ly6Chi Mo, acquire the ability to produce IL-10 and PGE2 at inflamed mucosa and that this response is promoted by commensal exposure.
Ly6Chi inflammatory monocytes prevent neutrophil-mediated pathology in the small intestine
The unusual phenotype of Ly6Chi monocytes during T. gondii infection suggests that these cells may exert regulatory functions at mucosal sites. To address this possibility, we utilized Ccr2 deficient mice that have a defect in monocyte exit from the bone marrow and, hence, tissue accumulation36. Prior to infection, the numbers and frequencies and activation levels of DC subsets, macrophages, neutrophils and CD4+ T cells were identical in the GI tract of Ccr2 deficient mice compared to wild-type controls (data not shown and Supplementary Fig. 4). As expected, by day 8 p.i., pronounced differences were apparent in the ratio of Ly6ChiMHCIIhi monocytes to Ly6G+ neutrophils in the gut (Fig. 4a). Further, T. gondii infection in Ccr2 deficient mice was characterized by more severe mucosal pathology than WT mice controls, evidenced by dramatic destruction of the crypt architecture and influx of inflammatory cells (Fig. 4b, Supplementary Fig. 5a). As previously described for WT mice, in Ccr2 deficient animals, T.gondii mediated pathology is dependent upon neutrophils37, commensals9 and TNF-α47 (Supplementary Fig. 5). Although, previous reports suggested that increased pathology in Ccr2−/− mice was associated with uncontrolled parasite expansion, we found dramatic changes in neutrophil activation prior to detectable changes in parasite burden in the gut and other sites (Fig. 4c,d, Supplementary Fig. 6a,b,c). Increased activation of neutrophils was characterized by a greater frequency of neutrophils expressing TNF-α IL-6, IL-1α, and IL-1β at a higher intensity in Ccr2 deficient mice compared to WT animals (Fig. 4d,e,f,g). Notably, production of reactive oxygen species (ROS), a major driver of immunopathology38, was also significantly increased in neutrophils from Ccr2−/− compared to WT mice (Fig. 4e,h). These changes in neutrophil activation were not associated with changes in absolute numbers of neutrophils, frequencies of DC or resident macrophage subsets, Th1 responses or aberrant increases in parasitic load (Supplementary Fig. 6). Altogether these data support the idea that in addition to their previously described role in parasite killing, Ly6Chi monocytes may also limit neutrophil mediated mucosal immunopathology.
Figure 4. Impaired Ly6Chi inflammatory monocyte recruitment leads to enhanced neutrophil mediated pathology.
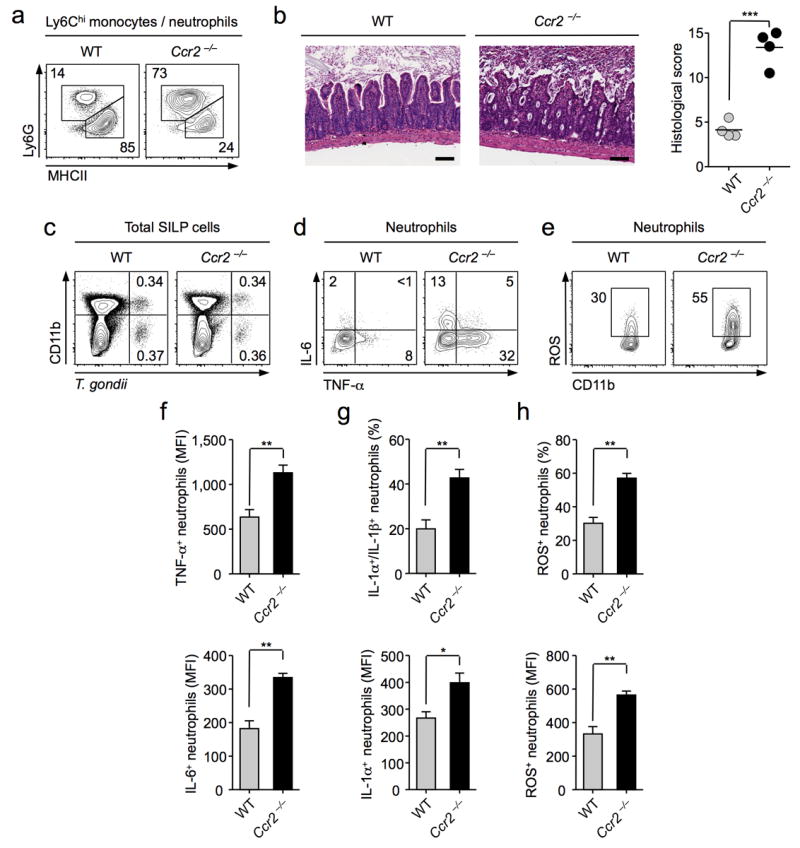
C57BL/6 or Ccr2−/− mice were infected orally with 10 cysts of T. gondii C1. (a) Assessment of neutrophil to inflammatory monocyte ratios by flow-cytometry. Cells were first gated on Ly6Chi/int CD11bhi cells, and populations identified by staining Ly6G and MHCII. Numbers represent the percentage of cells in each gate. (b) Representative H&E stained section of small intestine from day 8 infected wild-type (WT) and Ccr2−/− animals (Scale bar 100 μm) with graph depicting histological scoring. (n=4) (c) Representative plots of T. gondii infected haematopoietic cells at day 8 post-infection assessed by flow-cytometry. (d) SILp tissue suspensions were treated with BFA for 3 hrs. Cells were stained for neutrophil markers (Ly6G+CD11bhi) and TNF-α and IL-6 examined by intracellular cytokine staining. Numbers represent the percentage of cells in each quadrant. (e) Single cell suspensions from SILp were incubated with Dihydrorhodamine (DHR) 123 and cells were stained for surface neutrophil markers and reactive oxygen species (ROS) production assessed by flow-cytometry. (f) Bar graph summarizes the average mean fluorescence intensity (MFI) of TNF-α and IL-6 in neutrophils. (n=3). (g) As in (d), IL-1α and IL-1β were assessed by intracellular cytokine staining. Bar graphs summarize mean percentage of cells co-expressing IL-1α, and IL-1β, and MFI of IL-1α ± SEM. (n=3). (h) Bar graphs summarize the mean percentage of ROS+ cells, and MFI of ROS, ± SEM. (n=3). Data are representative of three independent experiments. Statistical comparisons were performed using the Student’s t test (*P<0.05, **P<0.01, ***P<0.001).
Ly6Chi inflammatory monocytes directly regulate neutrophil activation
Neutrophils and TNF-α are major drivers of mucosal pathology, including during T. gondii infection37,39. Consistent with a pathogenic role for neutrophils in response to commensals they produced a robust TNF-α response to E. coli lysate but failed to express PGE2 or to respond to T. gondii products (Supplementary Fig. 7, Supplementary Fig. 8b). As such, we hypothesized that neutrophils could represent a direct target for the regulatory effect of Ly6Chi Mo. In support of this, monocytes and neutrophils, that both express lysozyme, co-localize in discrete foci along the infected gut (Fig. 5a,b,c). We first assessed whether, in vitro, factors secreted by monocytes were capable of controlling commensal-driven neutrophil activation. To this end, cell sorted, Ly6Chi monocytes were cultured overnight in the presence of E. coli lysate and their supernatant collected. Notably, exposure of purified neutrophils to Ly6Chi monocytes but not neutrophil supernatant significantly reduced their ability to produce TNF-α to activating stimuli (Fig. 5d,e). Although the factors regulating neutrophils have been poorly explored, some reports have suggested a role for IL-1040, TGF-β41, IL-2742, adenosine43, and PGE244. We next assessed a potential role for these factors in mediating the suppressive effects of Ly6Chi Mo, by using blocking antibodies or inhibitors targeting their activity in vitro. Using this approach, we excluded a role for TGF-β, adenosine and IL-27 in the aforementioned suppressive effect (Fig. 5f). Although, antibody inhibition of IL-10R-signaling had a small but significant effect on TNF-α production by neutrophils, the effect observed could not account for the overall inhibition obtained with Ly6Chi monocyte supernatant (Fig. 5f). Strikingly, pre-incubation of neutrophils with inhibitors to PGE2 receptors (EP2 and EP445) completely abrogated the suppressive effect of Ly6Chi monocyte supernatant on neutrophils (Fig. 5g). Further, treatment of Ly6Chi monocytes with indomethacin that inhibits COX-1/2, the rate limiting enzymes in PGE2 synthesis, reversed the suppressive phenotype observed with Ly6Chi monocyte supernatant (Fig. 5h). Complementing these findings, at the acute stage of T.gondii infection, Ly6Chi monocytes that represent the dominant cell type in the inflamed gut are also the major source of PGE2 in the lamina propria (Fig. 1e, Fig. 5i) and treatment of neutrophils with increasing concentrations of purified PGE2 potently suppressed their capacity to produce TNF-α or ROS (Fig.5j). Of relevance to human settings, CD14+ monocytes isolated from human peripheral blood mononuclear cells (PBMC) but not neutrophils also produced large amounts of PGE2 in response to commensal stimuli. Moreover, PGE2 suppresses human neutrophil activation in vitro (Supplementary Fig. 8). Together, these data demonstrate that PGE2 is a potent suppressor of neutrophil activation in vitro and that activated Ly6Chi monocytes can directly inhibit commensal-driven neutrophil activation in a PGE2-dependent manner.
Figure 5. Ly6Chi inflammatory monocytes regulate neutrophil activation via production of PGE2.
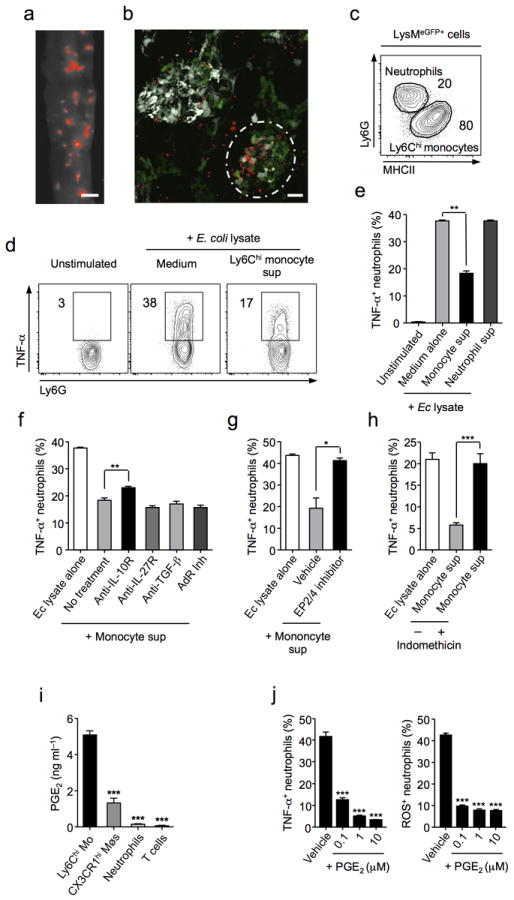
(a) Parasite localization visualized using RFP-expressing T. gondii in whole tissue mounts of jejunal sections from day 8 infection (Scale bar 2 mm). (b) Fixed jejunal tissue from day 8 infected CD11cYFPxLysMeGFP mice was whole mounted and sites of T. gondii invasion analysed by confocal microscopy (CD11c, white; LysM, Green; T. gondii, Red) (Scale bar 20 μm). Circle denotes foci of high parasite density. (c) Frequency of neutrophils (Ly6G+) and monocytes (MHCIIhi) within LysMeGFP population from T. gondii infected SILp. (d) Bone marrow neutrophils (Ly6G+CD11b+) were sorted by FACS and stimulated by E. coli lysate with Ly6Chi monocyte supernatant or control medium. Production of TNF-α was assessed by intracellular cytokine staining. (e) TNF-α production by neutrophils stimulated with E. coli lysate and treated with control medium, monocyte supernatant, or neutrophil supernatant. (f) Neutrophils were stimulated as in (d) and treated with Anti-IL-10R, Anti-IL-27R, Anti-TGF-β, or pan adenosine receptor (AdR) inhibitors in combination with monocyte supernatant and TNF-α production assessed. (g) Neutrophils were stimulated as in (d) and pre-treated with either inhibitors to the PGE2-receptors EP2 and EP4 or vehicle. (h) Neutrophils were stimulated as in (d) and treated with monocyte supernatants that were generated in the presence or absence of indomethacin and TNF-α production assessed. (i) SILp Ly6Chi monocytes, CX3CR1hi resident macrophages, neutrophils and T cells were sorted by FACS and cultured at equal concentration for 18 hrs. PGE2 was measured in triplicate supernatants by EIA. (j) Neutrophils were stimulated as in (d) and treated with increasing concentrations of purified PGE2. TNF-α production was assessed by flow cytometry. Additionally, neutrophils were treated with formyl-peptide, fMLP in the presence of increasing concentrations of purified PGE2. ROS production was assessed by flow cytometry. Histograms represent TNF-α production and ROS production ± SEM. Data are representative of three independent experiments, results are mean ± SEM. Statistical comparisons performed using the Student’s t test (*P<0.05, **P<0.01, ***P<0.001).
PGE2 and Ly6Chi monocytes regulate neutrophil activation in vivo
We next investigated the role of PGE2 and Ly6Chi inflammatory monocytes in the regulation of neutrophil activation and gut immunopathology during acute inflammation. As previously reported, inflammatory monocytes poorly accumulate in tissues following transfer (data not shown)19,21. Thus, to address the capacity of Ly6Chi monocytes to suppress neutrophil activation in vivo, we took advantage of an alternative model of T. gondii infection. To this end, we infected Ccr2 deficient mice intraperitoneally (i.p.) with parasite cysts46. To mimic the dominant gut commensal signal, E.coli lysate was injected i.p, 4 days post infection (Supplementary Fig. 9a). Notably, i.p. injection of pre-activated Ly6Chi monocytes dramatically suppressed the capacity of neutrophils to produce TNF-α in response to commensals (Figure 6 a,b). To link this suppression to the capacity of Ly6Chi Mos to produce PGE2, we pre-incubated these cells with either indomethacin (COX1 and COX2 inhibitor) or celecoxib to specifically inhibit COX2 activity47. In both cases, the capacity of Ly6Chi monocytes to suppress neutrophil activation in vivo was abolished (Fig. 6a,b). These results demonstrate that in vivo the capacity of Ly6Chi monocytes to suppress neutrophil activation depends upon the capacity of these cells to produce PGE2 in response to commensal stimuli.
Figure 6. PGE2 modulates neutrophil activation in vivo.
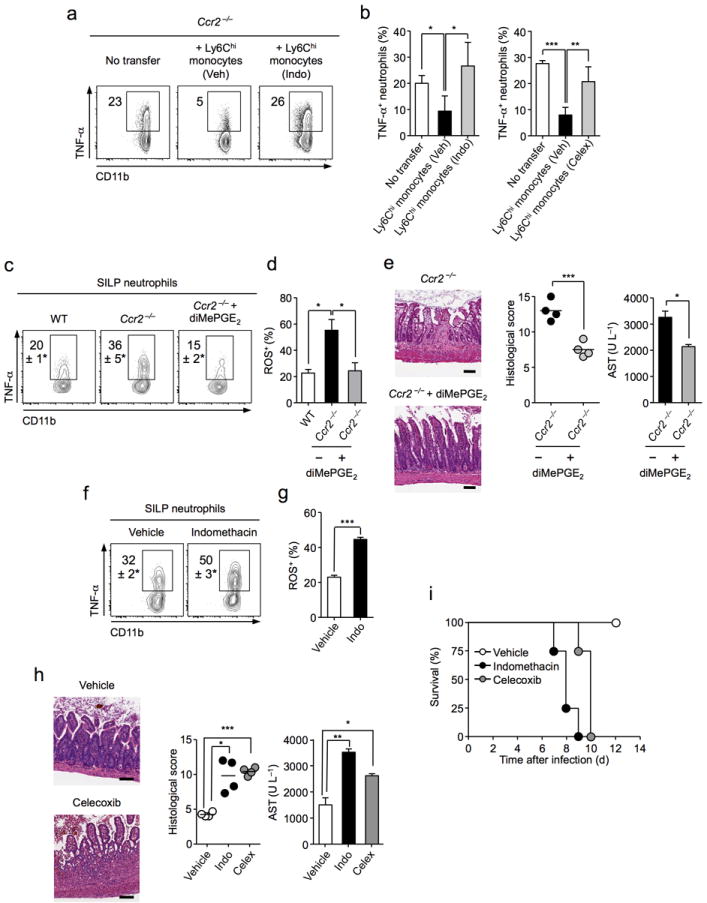
(a-b) C57BL/6 mice were intraperitoneally (i.p.) inoculated with 20 T. gondii cysts. Monocytes, sorted from bone marrow of orally infected mice, were stimulated with E. coli lysate in the presence of indomethicin (Indo), celecoxib (Celex), or vehicle (Veh). Treated monocytes were transferred i.p. at 4 days p.i. and mice subsequently administered E. coli lysate i.p. (a) Cells were isolated by peritoneal lavage and cultured in the presence of BFA and stained for neutrophil markers (Ly6G+CD11bhi) and TNF-α examined by intracellular cytokine staining. (b) Histograms represent TNF-α production ± SEM of two separate experiments. (c-e) Wild-type (WT), or Ccr2−/− mice were orally infected with T. gondii. Ccr2−/− mice were treated with 16,16-dimethyl (diMe) PGE2 or vehicle from days 6-8 post infection. (c) At day 8 p.i. cells were isolated from the small intestine lamina propria (SILp) and neutrophil production of TNF-α assessed. Numbers represent percentage of cells in each gate ± SEM. (d) Histogram represents the percentage of ROS producing SILp neutrophils assessed by flow cytometry ± SEM. (e) Representative H&E staininig of small intestine from infected Ccr2−/− mice treated with diMePGE2 or vehicle (Scale bar 100 μm). Graph depicts histological scoring. Amounts of aspartate aminotransferase (AST) were assessed in sera of animals (n=4). (f-i) WT mice were orally infected with T. gondii. On days 6–8 p.i. mice were treated intraperitoneally with indomethacin or vehicle. (f) Representative plot of TNF-α production gated on neutrophils from vehicle or indomethacin treatment. (g) SILp neutrophil production of ROS was assessed by flow-cytometry. Histograms represent mean production of ROS ± SEM (n=3). (h) Representative H&E stained section of small intestine from vehicle or celecoxib (Celex) treated groups (Scale bar 100 μm). Graph depicts histological scoring. Amounts of AST were assessed in sera of animals (n=4). (i) Survival curve of animals treated with indomethicin or celecoxib on days 6–8 post infection. Data are representative of at least two independent experiments. Data presented as mean ± SEM (n=3). Statistical comparisons performed using the Student’s t test (*P<0.05, **P<0.01, ***P<0.001).
We further explored the capacity of a long-lived analog of PGE2 (16,16-dimethyl PGE2 (diMe-PGE2)48 to regulate neutrophil activity and pathology during T. gondii infection. Significantly, short-term diMePGE2 treatment of wild-type mice, from day 6 to day 8 p.i., had no effect on parasite burden but led to decreased tissue pathology and reduced efflux of inflammatory cells into the lumen (Supplementary Fig. 9b,g). We next assessed if PGE2 could compensate for a lack of Ly6Chi monocytes in Ccr2−/− mice. Strikingly, treatment of Ccr2 deficient mice with diMe-PGE2 significantly reduced the capacity of neutrophils to express TNF-α and reactive oxygen species and reduced mucosal pathology (Fig. 6 c,d,e). These changes in neutrophil activation were not associated with alterations in their absolute number, levels of parasite infection, or changes in effector T cell polarization (Supplementary Fig. 9c,d,e).
Drugs limiting COX activity can have detrimental effects on the GI tract49. We next explored the possibility that these adverse reactions could be associated with aberrant neutrophil activation in the inflamed mucosa. To experimentally test this possibility, we blocked PGE2 production during infection by treating mice with the non-steroidal anti-inflammatory drug (NSAID) indomethicin or the COX-2-specific inhibitor celecoxib. In support of a regulatory role for PGE2, NSAID treatment led to augmented TNF-α and ROS production by neutrophils and enhanced mucosal and liver pathology (Fig. 6f,g,h). Notably, inhibition of prostaglandin synthesis by indomethacin, or the COX-2-specific inhibitor celecoxib, was associated with increased mortality following T. gondii infection (Fig. 6i). Increased mortality was not associated with changes in parasite numbers (Supplementary Fig. 9f,g).
Altogether, these data demonstrate that during mucosal inflammation PGE2 and in particular Ly6Chi Mo-derived PGE2 represents a powerful regulatory arm, capable of limiting lethal neutrophil mediated immunopathology and that impairment to this regulatory pathway can lead to severe pathological consequences (Supplementary Fig. 10).
Discussion
Our work uncovers a novel regulatory loop at mucosal sites in which commensals trigger a program involved in limiting their own pathogenic potential. In particular we show that (i) recruited inflammatory monocytes take on tissue-specific regulatory features in response to commensal-stimuli, (ii) inflammatory monocytes control neutrophil pathogenic potential, and (iii) neutrophil activation is specifically inhibited by PGE2 derived from Ly6Chi inflammatory monocytes at mucosal sites.
Maintenance of mucosal integrity in the face of acute inflammation represents a formidable task for the immune system. At steady state, specialized gastrointestinal DC and resident macrophages help to maintain tolerance to the flora and food antigens50. During pathogen encounter, this regulated environment must adapt to control microbial expansion. In line with this, we found that during acute infection with T. gondii, the steady-state regulatory network is significantly remodelled. Specifically, CD103+ DCs that are associated with Treg cell generation13,14 become depleted from the inflamed tissue, and resident macrophages produce limited IL-10. These results complement our previous observation that Treg cells collapse during mucosal infection15. However, despite this breakdown of regulation, the host can survive implying that alternative mechanisms, specialized in the control of recruited cells, arise during inflammation. More particularly our data clearly establishes that recruited Ly6Chi inflammatory Mo, represent an integral regulatory component at inflamed mucosal sites.
Ly6Chi monocytes are proposed to play a dominant role in the control of the invading pathogen in various settings18,21,22. During T. gondii infection, lack of Ly6Chi monocytes led to enhanced mortality resulting from uncontrolled parasite replication23. Following chemical induced injury in the gut, these cells can contribute to the pathological process by promoting inflammation19. In the present study, we found that Ly6Chi monocytes, and not resident cells, play a major regulatory role in the inflamed gut during acute infection. Indeed, mice deficient in Ly6Chi monocytes exhibited augmented ileal pathology compared to their wild-type counterparts prior to detectable changes in parasite burden. Although, our work does not dispute a role for Ly6Chi monocytes in parasite killing it adds another layer of complexity to their function by revealing a critical role in regulating mucosal inflammation.
Commensal flora modulate various aspects of immune responses and can enhance protection against pathogens. However, the flora is also a major contributor to inflammatory disorders including Crohn’s disease2. In particular, following T. gondii infection, increased bacterial density and contact with the epithelium can directly contribute to neutrophil-mediated tissue damage9. Supporting this, we found that neutrophils strongly responded to commensal but not T. gondii-derived ligands in a pro-inflammatory manner. Such a response highlights the dominant role for neutrophils in controlling bacterial translocation25,51. On the other hand, if unregulated neutrophil responses can lead to severe pathologic consequences25. Despite the central role of neutrophils in driving mucosal pathology the mechanisms modulating the delicate balance between their role in commensal control versus tissue damage are poorly understood.
One striking observation in this model, is that regulation of commensal induced pathology is triggered by commensals themselves in an auto-regulatory loop. Indeed, we found that Ly6Chi monocytes produce various regulatory factors including IL-10 and the lipid mediator PGE2 specifically in the gut mucosa and in response to commensals both in vivo and in vitro. Although the capacity of resident CX3CR1hi macrophages to produce IL-10 is compromised during infection we found that these cells remain able to produce a small amount of PGE2, suggesting that part of the effect of this mediator could arise from other cell types. Nevertheless, in the inflamed mucosa, Ly6Chi monocytes represent the dominant cell type and the major source of this lipid mediator. Similarly high levels of PGE2 were found to be elicited from human CD14+ blood monocytes in response to commensal stimuli. Acquisition of regulatory features by highly activated effector cells has been well documented in effector T cells including during intraperitoneal T.gondii infection where highly-polarized Th1 cells co-produce IL-10 in tandem with IFN-γ52,53. Paralleling this, Ly6Chi monocytes entering the GI tract, respond to commensal ligands by expressing the additional layer of control required to sustain tissue integrity during inflammation. As previously described for activated T cells52,53, such features endow them with the dual capacity to control parasite burden while limiting collateral damage to tissue.
A number of studies have reported that Ly6Chi monocytes can differentiate into CD11c+ TNF-α/iNOS-producing (TIP)-DC in the spleen in response to inflammation24. Indeed, we found that during T. gondii infection, TIP-DC were highly represented in the spleen. On the other hand, the acquisition of regulatory factors by Ly6Chi monocytes has been reported in a number of settings. In particular, myeloid-derived suppressor cells (MDSC), a common feature of cancer54 and sepsis55, are derived from this population. Nevertheless, a role for MDSC or regulatory monocytes has not been reported in the inflamed gut and PGE2 has not been associated with their suppressive capacity. Our present data are to the best of our knowledge the first demonstration of a direct role for inflammatory monocytes in controlling neutrophil function and gut pathology.
Multiple layers of regulation are required to limit mucosal damage during acute inflammation2. A number of studies have focused on the role of PGE2 in the resolution of inflammation, and in chronic inflammatory conditions, but its mechanisms of regulation during an acute response are less well understood56. Our work proposes that via its capacity to target neutrophils at the acute stage of the immune response, the lipid mediator PGE2 represents a major arm of this regulatory network. Although in this study PGE2 was identified as a dominant mechanism of neutrophil suppression, the capacity of Ly6Chi monocytes to produce IL-10, arginase and indoleamine 2,3-dioxygenase (IDO) strongly suggest that these cells can exert additional regulatory functions in the gut. Further, during mucosal inflammation, the effect of PGE2 is likely to extend beyond neutrophils as this molecule can control a large array of cell populations. Indeed, PGE2 has been linked to the maintenance of the gut epithelial layer and has pleiotropic effects on the immune system, including modulation of DC activation, suppression of NK cell function, and alterations to effector T cell polarization34.
Of high relevance to clinical settings, genome-wide association studies (GWAS) linked both TNF-α and pathways associated with the production of ROS to the etiology of IBD57. Crohn’s disease-associated alleles also correlate with expression levels of PGE2 receptors58, while suppression of prostaglandin synthesis by non-steroidal anti-inflammatory drugs (NSAIDs) is associated with GI damage49. In our present study, the capacity of Ly6Chi monocytes to control neutrophil function, and more particularly their production of TNF-α and ROS, sheds new light on the potential impact of prostaglandins in multiple gut pathologies. Intriguingly, treatment of IL-10−/− animals with NSAIDs rapidly leads to the development of severe colitis59. This PGE2/IL-10 interaction may occur at the level of the inflammatory monocyte as PGE2 has been demonstrated to favor IL-10 production by macrophages in vitro60. However, despite the extensive use in the clinic of NSAIDs, such as aspirin, the mode of action and cellular targets of these inhibitors remains unclear. Notably, our present data suggest that in these settings increased mucosal inflammation may be directly associated with uncontrolled responses to the microbiota and neutrophil activation. Moreover these results support the idea that modulating PGE2 metabolism and/or monocyte recruitment in inflamed tissues need to be considered in a highly contextual manner.
Overall, our findings identify PGE2 as a critical mediator of commensal-induced regulation during inflammation, adding an additional layer to our understanding of the complex actions of lipid mediators in modulating the immune response. Further our data suggest that modulating PGE2 metabolism may represent an important future therapeutic approach in the treatment of mucosal inflammatory diseases.
Methods
Mice
Female C57BL/6 (WT) mice were purchased from Taconic Farms or Jackson Laboratories. B6.129S4-Ccr2tm1Ifc/J (Ccr2−/−) mice, and B6.129P(Cg)-Ptprca Cx3cr1tm1Litt/LittJ (CX3CR1-GFP) mice were purchased from Jackson Laboratories. Lysozyme MeGFP transgenic mice (C57BL/6NTac-Lysozyme, Tac Line 342), and CD11cYFP (C57BL/6-[Tg]CD11c:EYFP, Tac Line 307) transgenic mice, were obtained through the Taconic NIAID exchange. Animals were bred in house to generate the LysMeGFPxCD11cYFPstrain. Germ-free C57BL/6 mice were bred at Taconic Farms and maintained in the NIAID gnotobiotic animal facility. All mice were bred and maintained under pathogen-free conditions at an American Association for the Accreditation of Laboratory Animal Care-accredited animal facility at the NIAID and housed in accordance with the procedures outlined in the Guide for the Care and Use of Laboratory Animals. All experiments were performed at the NIAID under an animal study proposal approved by the NIAID Animal Care and Use Committee. Gender- and age-matched mice between 8-16 weeks of age were used.
Human Subjects
Whole blood was obtained from healthy donors under Institutional Review Board (IRB)-approved protocols from the Department of Transfusion Medicine (Clinical Center, National Institutes of Health (NIH), Bethesda, MD). All subjects signed written informed consent.
Parasite and Infection Protocol
ME-49 clone C1 of T. gondii (kindly provided by Dr M.E. Grigg, NIAID/NIH) was obtained by electroporation of the parental ME-49 type II strain (ATCC 50840) with red fluorescent protein (RFP) and was used for production of tissue cysts in C57BL/6 mice. Tissue cysts used in experiments were obtained from female mice that were perorally inoculated with 10 cysts 2-3 months earlier. Animals were killed and their brains removed and homogenized in 1 ml of PBS pH 7.2. Cysts were counted under an inverted fluorescent microscope on the basis of two aliquots of 20 μl. For experiments, female mice were either infected by intragastric gavage with 10 cysts of ME-49 C1, or intraperitoneally injected with 20 cysts of the same parasite. Soluble Toxoplasma Antigen (STAg) was prepared as previously described61.
Antibody Treatment
For depletion of neutrophils, and inhibition of TNF-α, Ccr2−/− mice were injected intraperitoneally (i.p.) with anti-Gr1 antibody (clone RB6-8C562, 500μg), anti-TNF-α antibody (clone MP6-XT2263, 1 mg) or rat IgG isotype control on day 5 and day 8 p.i.
Antibiotic Treatment
Female 6-8 week old Ccr2−/− mice were provided ampicillin (1 g/l), vancomycin (500 mg/l), neomycin trisulfate (1 g/l), and metronidazole (1 g/l) in drinking water as previously described64. All antibiotics were purchased from Sigma-Aldrich.
Quantitation of Parasite Tissue Loads by Plaque Assay
Cultures of human fibroblasts (Hs27; ATCC no. CRL-1634) were used for parasite burden quantification as previously described65,66. Single-cell tissue suspensions (106 cells or 100 μl of sample) were added to the fibroblast monolayer cultures and titrated for plaque formation. The results of these titrations are reported in Plaque Forming Units (PFUs).
Phenotypic Analysis
Cells from lamina propria (Lp), or spleen, were prepared as previously described14. Single cell suspensions were incubated with an anti-FcγIII/II (93) receptor, purified rat immunoglobulin (Jackson ImmunoResearch), and mouse serum (Jackson ImmunoResearch). Cells were stained with fluorochrome-conjugated antibodies against surface markers CD11c (N418), CD11b (M1/70), MHCII (I-A/I-E) (M5/114), Ly6C (HK1.4), Ly6G (1A8), CD103 (2E7), and F4/80 (BM8) in PBS containing 1% FBS (FACS Buffer) for 15 min on ice and then washed. DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) (Sigma) was used to exclude dead cells. All antibodies were purchased from BD Biosciences or eBioscience. Cell acquisition was performed on an LSRII machine using FACSDiVa software (BD Biosciences). For each sample, at least 300,000 events were collected. Data were analyzed with FlowJo software (TreeStar).
In Vitro Restimulation of Innate Cells and Intracellular Cytokine Detection
RPMI 1640 supplemented with 10% FBS, penicillin, streptomycin, HEPES, glutamine, nonessential amino acids, and 50 mM of β-mercaptoethanol (Complete Media) was used for in vitro restimulation. For ex vivo cytokine detection lamina propria (Lp), or spleen, single cell suspensions were cultured at 5×105-1×106 cells/well in a 96-well round-bottom plates in the presence of brefeldin A alone (GolgiPlug, BD Biosciences). In some cases, single cell suspensions were stimulated with commensal or parasite-derived ligands in combination with brefeldin A. After 3 hrs, cells were surface stained for Ly6C (HK1.4), Ly6G (1A8), MHCII (I-A/I-E) (M5/114), and CD11b (M1/70), then washed twice with FACS buffer, and fixed in a solution of 2% paraformaldehyde (Electron Microscopy Sciences). Prior to fixation Live/Dead Fixable Blue Cell Stain Kit (Invitrogen) was used to exclude dead cells. Cells were then stained with fluorochrome-conjugated antibodies against inducible nitric oxide synthase (iNOS) (Polyclonal Rabbit), TNF-α (MP6-XT22), IL-1α (ALF-161), IL-1β (NJTEN3), IL-6 (MP5-20F3), and IL-10 (JES5-16E3), or their appropriate isotype controls Rat IgG1, Rat IgG2b, or Armenian Hamster IgG, in the presence of anti-FcεIII/II receptor for 60 min in FACS buffer containing 0.5% saponin. All antibodies were purchased from BD Biosciences or eBioscience.
Ly6Chi Inflammatory Monocyte, Macrophage, and Neutrophil Purification
Monocytes, macrophages, and neutrophils were purified from pooled single cell suspensions of naïve or T. gondii infected blood, spleen, or Lp. Blood was lysed and washed. Spleen or Lp digests were passed through 70 μM and 40 μM cell strainers, centrifugated and cells were resuspended in 1.077 g/cm3 iso-osmotic NycoPrep medium (Accurate Chemical & Scientific Corp.) and overlayed with RPMI 1640. The low-density fraction was collected after centrifugation at 1650 g for 15 min. Cells were incubated with mixture of monoclonal antibodies (mAb) containing anti-FcγIII/II and 7-AAD viability staining solution.
Stimulation of FACS purified populations
Purified Ly6Chi inflammatory monocytes, and neutrophils, were cultured in complete media at 2×104-2×105 cells per well in a 96-well round-bottom plate. In some cases, cells were stimulated with E. coli lysate (5 μg/ml), Soluble Toxoplasma Antigen (STAg) (5 μg/ml), or commercially available TLR ligands Pam2CSK4, LPS, Flagellin, CpG (all from Invivogen) (1 μg/ml), or Profilin (Alexis Biochemicals) (1 μg/ml) for 18 hrs. Supernatants were assayed for IL-10, or TNF-α using the DuoSET ELISA system (R&D Systems), or PGE2 using an enzyme-immunoassay (EIA) (Cayman Chemicals), as per manufacturers’ instructions.
Human Monocyte and Neutrophil Isolation and Culture
Peripheral venous blood was obtained from healthy adult subjects. Monocytes were isolated from the peripheral blood mononuclear cell (PBMC) layer. An ammonium chloride based lysis was performed to remove erythrocytes. PBMC were then stained in FACS buffer containing mouse serum with monoclonal antibodies to CD14 (M5E2) and HLA-DR (G46-6) for sorting. For neutrophil isolation, after isolation on a Ficoll gradient, erythrocytes were removed by two rounds of hypotonic lysis with ice-cold ddH2O to keep the granulocytes within physiological condition. Neutrophils were stained with CD16 (3GB) for FACS sorting. Purified monocytes and neutrophils were stimulated at 1×106 cells/ml overnight with or without E. coli lysate (5 μg/ml). PGE2 production was assessed in supernatants by EIA.
Human Neutrophil Suppression by PGE2
Human neutrophil production of ROS was assessed by flow-cytometry after stimulation with N-Formyl-Met-Leu-Phe (fMLP) (10μM) (Sigma) in the presence of increasing concentrations of PGE2. Alternatively neutrophils were cultured overnight at 1×106 cells/ml and stimulated with E. coli lysate (5 μg/ml) in the presence or absence of PGE2. After overnight culture, TNF-α was measured in supernatants using a human TNF-α DuoSet ELISA system (R&D Systems).
Detection of Reactive Oxygen Species (ROS)
Single cell suspensions from small intestine lamina propria (SILp), or human blood, FACS purified neutrophils were plated at 5×105 or 3-5×104 cells, respectively, in 96 well round-bottom plates, and resuspended in complete media containing 2 ng/ml of dihydrorhodamine 123 (Invitrogen). Cells were incubated for 30 mins at 37°C, in the presence or absence of fMLP (10 μM), and then washed with FACS buffer prior to being stained with the surface markers Ly6G (1A8), Ly6C (HK1.4) and CD11b (M1/70) to identify neutrophils. DAPI was used to exclude dead cells. Cell acquisition was performed on an LSRII machine using FACSDiVa software (BD Biosciences). For each sample, at least 300,000 events were collected. Data were analyzed with FlowJo software (TreeStar).
Ly6Chi Inflammatory Monocyte Suppression Assay
Ly6Chi inflammatory monocytes were FACS purified from the SILp of day 8 T. gondii infected mice, and then stimulated overnight with E. coli lysate (5 μg/ml) in complete media. The COX-1/-2 inhibitor indomethacin (10 μM) (Sigma) was added to some cultures. Overnight supernatants were removed and used to treat FACS purified bone-marrow neutrophils that were concurrently activated with E. coli lysate (2 μg/ml) in the presence of brefeldin A. In some cases, antibodies against cytokines (α-TGF-β (1D11) (10 μg/ml)) and the cytokine receptors α-IL-10R (1B1.3a) (10 μg/ml), α-IL-27R (10 μg/ml)), inhibitors of adenosine receptor signaling xanthine amine congener (XAc) (1μM), or inhibitors of PGE2-receptors EP2 (AH-6809) (10 μM) and EP4 (AH-23848) (10 μM) (Cayman Chemicals), were included at the start of the assay. After 3 hrs stimulation, neutrophils were fixed and stained for intracellular cytokines.
In Vivo Neutrophil Suppression Assay
Mice were infected intraperitoneally (i.p.) with 20 cysts C1-RFP T. gondii. At day 4 p.i. 1-2×106 FACS sorted bone marrow Ly6Chi monocytes, isolated from day 6 orally infected animals, were transferred i.p. Prior to transfer monocytes were stimulated for 30 mins with E. coli lysate in the presence of indomethicin (10 μM), celecoxib (25 μM), or DMSO vehicle control. Post monocyte transfer peritoneal cells were activated by i.p. E. coli lysate injection (2 μg/mouse). Mice were lavaged 30 mins later with 6 ml of ice cold PBS and neutrophil production of TNF-α assessed by ICS after 3 hrs culture with brefeldin A.
16,16-dimethyl Prostaglandin E2 (diMePGE2), Indomethacin, and Celecoxib Treatment
Animals that were orally infected with 10 cysts T. gondii were treated once daily starting day 6 p.i. with an intraperitoneal injection of diMePGE2 (12 μg/kg) (Cayman Chemicals), indomethacin (3 mg/kg), or celecoxib (15 mg/kg) (Sigma). Untreated animals received a DMSO vehicle control.
Pathology Assessment
Mice were euthanized at day 8–10 post-infection. Small intestines were removed and sections of the jejunum and ileum immediately fixed in a 10% formalin solution. Paraffin-embedded sections were cut longditudinally at 0.5 μm and stained with hematoxylin and eosin. The sections were examined histologically and a semi-quantitative 15-point assessment combining scores for inflammatory infiltrate, crypt architecture, and mucosal surface involvement was made. Scoring was evaluated on the following modified scale9,15. Inflammatory infiltrate score: 0 = occasional or no infiltrate, 1 = minimal inflammatory leukocyte or granulocyte infiltration locally or multifocally, 2 = mild inflammatory leukocytic or granulocytic infiltrates within the lamina propria, 3 = mild to moderate inflammatory infiltrates in the lamina propria and submucosa, 4 = moderate to marked inflammatory infiltrates admixed with cellular debris within the lamina propria and submucosa, 5 = marked to severe inflammation diffusely with abundant inflammatory infiltrates extending transmurally. Crypt architecture score: 0 = occasional or no crypt involvement, 1 = one-third of lower crypt involved in inflammation, 2 = two-third of crypt involved, 3 = entire length of crypt involved, 4 = entire length of crypt involved with loss of crypt separation 5 = complete loss of normal histologic crypt structure with deep mucosal ulceration present. Mucosal surface involvement score: 0 = no involvement, 1 = 1–5% of area scored, 2 = 5–10% of area scored, 3 = 10–20% of area scored, 4 = 20–50% of area scored, 5 = >50% of area scored. Grading was performed in a blinded fashion by Dr. I. Fuss. For each animal at least two sections, obtained at 100 μm distance were evaluated.
Fluoresecence In Situ Hybridization (FISH)
For Fluorescence In Situ Hybridization (FISH), the small intestines were prepared by fixation in 60% Methanol, 30% Chloroform, 10% Acetic acid, washed in 70% ethanol and embedded in paraffin. 5 μm longitudinal sections were hybridized to a bacterial 16S rRNA gene probe: [AminoC6+Alexa488]-GCTGCCTCCCGTAGGAGT-[AmC7_Q+Alexa488] as previously published67 and counterstained with DAPI. Sections were then visualized on a Leica TCS SP8 microscope.
Confocal Analysis
Jejunal and ileal sections, from T. gondii infected LysMeGFP×CD11cYFP mice, were whole mounted for imaging. Briefly, the small intestines were removed, cut open longitudinally, and washed in PBS. Intestinal tissue was fixed overnight in 4% paraformaldehyde solution (Electron Microscopy Sciences), and then stained with Hoechst 33342 (Invitrogen) followed by mounting with Prolong Gold (Invitrogen). Images were collected on a Leica TCS SP5 microscope, and processing performed using Imaris software (Bitplane).
Microarray Gene-expression Profiling and Data Normalization
Ly6Chi SILp inflammatory monocytes or Ly6Chi blood monocytes were purified by FACS from the SILp and blood of day 8 T. gondii infected mice. Three biological replicates were sorted for each sample, containing 5×104–1×105 cells. Post-sort purity was >95%. Cells were resuspended in TRIzol (Invitrogen) and then mRNA isolated and amplified before being hybridized to an Illumina MouseRef BeadChip. Density plots and principle components analysis were used to check the data for any outlier chips or samples. Global median normalization was used to remove array and sample effects from the expression data. Probes with mean log2 expression less than or equal to 6, or log2 expression standard deviations less than or equal to 0.5 were removed to eliminate any probes that were below detection threshold or potentially uninformative housekeeping genes. When multiple probes represented the same gene symbol, the probes were averaged together to simplify graphics and statistical results. Log2 expression values of Ly6Chi blood monocytes (Mo) were plotted against log2 expressions of Ly6Chi SILp Mo. Differential expression of Ly6Chi blood monocytes / Ly6Chi SILp monocytes was tested with Student’s t-test, an empirical Bayes moderated t-test, and significance analysis of microarrays (SAM). Significant genes were identified by volcano plot and heatmap.
Statistical Analysis
Groups were compared with Prism software (GraphPad) using a two-tailed unpaired Student’s t test. Data are presented as mean ± SEM.
Supplementary Material
Acknowledgments
This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases. We thank, members of the Belkaid Lab for thoughtful discussion and critical reading of the manuscript; Dr. Kevin Holmes, Elena Stregevsky, and the NIAID Sorting Facility; Kim Beacht and Dr. Joanne Konkel for technical assistance; Dr. Charlie Brown for assistance with tissue staining; Dr. Owen Schwartz and the NIAID Biological Imaging Facility for assistance with confocal microscopy; Dr. Tim Meyers and the NIAID Genomic Technologies Section for microarray processing; Jeff Skinner and the NIAID Bioinformatics and Computational Bioscience Branch for assistance with microarray analysis and Dr. Lora Hooper, UT Southwestern, for sharing protocols.
Footnotes
Author Contributions: J.R.G., E.A.W., M.H.A., N.B., F.L. and Y.B. designed and performed experiments; J.R.G., E.A.W., M.H.A., I.D.C.F, I.F. L.K and Y.B. analyzed the data; J.R.G., N.B. and Y.B. wrote the manuscript.
References
- 1.Gill SR, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Littman DR, Pamer EG. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell host & microbe. 2011;10:311–323. doi: 10.1016/j.chom.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall JA, et al. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity. 2008;29:637–649. doi: 10.1016/j.immuni.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benson A, Pifer R, Behrendt CL, Hooper LV, Yarovinsky F. Gut commensal bacteria direct a protective immune response against Toxoplasma gondii. Cell Host Microbe. 2009;6:187–196. doi: 10.1016/j.chom.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slack E, et al. Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science. 2009;325:617–620. doi: 10.1126/science.1172747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cerutti A. The regulation of IgA class switching. Nat Rev Immunol. 2008;8:421–434. doi: 10.1038/nri2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vaishnava S, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hand TW, et al. Acute gastrointestinal infection induces long-lived microbiota-specific T cell responses. Science. 2012;337:1553–1556. doi: 10.1126/science.1220961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heimesaat MM, et al. Gram-negative bacteria aggravate murine small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii. Journal of immunology. 2006;177:8785–8795. doi: 10.4049/jimmunol.177.12.8785. [DOI] [PubMed] [Google Scholar]
- 10.Man SM, Kaakoush NO, Mitchell HM. The role of bacteria and pattern-recognition receptors in Crohn’s disease. Nature reviews Gastroenterology & hepatology. 2011;8:152–168. doi: 10.1038/nrgastro.2011.3. [DOI] [PubMed] [Google Scholar]
- 11.Bogunovic M, et al. Origin of the lamina propria dendritic cell network. Immunity. 2009;31:513–525. doi: 10.1016/j.immuni.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hadis U, et al. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity. 2011;34:237–246. doi: 10.1016/j.immuni.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 13.Coombes JL, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun CM, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oldenhove G, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benson A, et al. Microbial infection-induced expansion of effector T cells overcomes the suppressive effects of regulatory T cells via an IL-2 deprivation mechanism. J Immunol. 188:800–810. doi: 10.4049/jimmunol.1100769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rivollier A, He J, Kole A, Valatas V, Kelsall BL. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med. 2012;209:139–155. doi: 10.1084/jem.20101387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zigmond E, et al. Ly6C(hi) Monocytes in the Inflamed Colon Give Rise to Proinflammatory Effector Cells and Migratory Antigen-Presenting Cells. Immunity. 2012;37:1076–1090. doi: 10.1016/j.immuni.2012.08.026. [DOI] [PubMed] [Google Scholar]
- 20.Kim YG, et al. The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity. 2011;34:769–780. doi: 10.1016/j.immuni.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iijima N, Mattei LM, Iwasaki A. Recruited inflammatory monocytes stimulate antiviral Th1 immunity in infected tissue. Proc Natl Acad Sci U S A. 2011;108:284–289. doi: 10.1073/pnas.1005201108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hohl TM, et al. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe. 2009;6:470–481. doi: 10.1016/j.chom.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dunay IR, et al. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity. 2008;29:306–317. doi: 10.1016/j.immuni.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 25.Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal immunology. 2012;5:354–366. doi: 10.1038/mi.2012.24. [DOI] [PubMed] [Google Scholar]
- 26.Gazzinelli RT, et al. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J Immunol. 1996;157:798–805. [PubMed] [Google Scholar]
- 27.Geissmann F, et al. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scharton-Kersten TM, Yap G, Magram J, Sher A. Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii. J Exp Med. 1997;185:1261–1273. doi: 10.1084/jem.185.7.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jebbari H, Roberts CW, Ferguson DJ, Bluethmann H, Alexander J. A protective role for IL-6 during early infection with Toxoplasma gondii. Parasite Immunol. 1998;20:231–239. doi: 10.1046/j.1365-3024.1998.00152.x. [DOI] [PubMed] [Google Scholar]
- 30.Suzuki Y, et al. IL-10 is required for prevention of necrosis in the small intestine and mortality in both genetically resistant BALB/c and susceptible C57BL/6 mice following peroral infection with Toxoplasma gondii. J Immunol. 2000;164:5375–5382. doi: 10.4049/jimmunol.164.10.5375. [DOI] [PubMed] [Google Scholar]
- 31.El Kasmi KC, et al. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol. 2008;9:1399–1406. doi: 10.1038/ni.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katz JB, Muller AJ, Prendergast GC. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol Rev. 2008;222:206–221. doi: 10.1111/j.1600-065X.2008.00610.x. [DOI] [PubMed] [Google Scholar]
- 33.O’Banion MK, Sadowski HB, Winn V, Young DA. A serum- and glucocorticoid-regulated 4-kilobase mRNA encodes a cyclooxygenase-related protein. J Biol Chem. 1991;266:23261–23267. [PubMed] [Google Scholar]
- 34.Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol. 2012;188:21–28. doi: 10.4049/jimmunol.1101029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Molloy MJ, Bouladoux N, Belkaid Y. Intestinal microbiota: shaping local and systemic immune responses. Semin Immunol. 2012;24:58–66. doi: 10.1016/j.smim.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi C, et al. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity. 2011;34:590–601. doi: 10.1016/j.immuni.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dunay IR, Fuchs A, Sibley LD. Inflammatory monocytes but not neutrophils are necessary to control infection with Toxoplasma gondii in mice. Infect Immun. 2010;78:1564–1570. doi: 10.1128/IAI.00472-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naito Y, Takagi T, Yoshikawa T. Molecular fingerprints of neutrophil-dependent oxidative stress in inflammatory bowel disease. J Gastroenterol. 2007;42:787–798. doi: 10.1007/s00535-007-2096-y. [DOI] [PubMed] [Google Scholar]
- 39.Liesenfeld O, et al. TNF-alpha, nitric oxide and IFN-gamma are all critical for development of necrosis in the small intestine and early mortality in genetically susceptible mice infected perorally with Toxoplasma gondii. Parasite Immunol. 1999;21:365–376. doi: 10.1046/j.1365-3024.1999.00237.x. [DOI] [PubMed] [Google Scholar]
- 40.Cassatella MA. The neutrophil: one of the cellular targets of interleukin-10. Int J Clin Lab Res. 1998;28:148–161. doi: 10.1007/s005990050036. [DOI] [PubMed] [Google Scholar]
- 41.Fridlender ZG, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Troy AE, et al. IL-27 regulates homeostasis of the intestinal CD4+ effector T cell pool and limits intestinal inflammation in a murine model of colitis. J Immunol. 2009;183:2037–2044. doi: 10.4049/jimmunol.0802918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cronstein BN, Daguma L, Nichols D, Hutchison AJ, Williams M. The adenosine/neutrophil paradox resolved: human neutrophils possess both A1 and A2 receptors that promote chemotaxis and inhibit O2 generation, respectively. J Clin Invest. 1990;85:1150–1157. doi: 10.1172/JCI114547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wertheim WA, et al. Regulation of neutrophil-derived IL-8: the role of prostaglandin E2, dexamethasone, and IL-4. Journal of immunology. 1993;151:2166–2175. [PubMed] [Google Scholar]
- 45.Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282:11613–11617. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- 46.Goldszmid RS, et al. NK cell-derived interferon-gamma orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity. 2012;36:1047–1059. doi: 10.1016/j.immuni.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.FitzGerald GA, Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med. 2001;345:433–442. doi: 10.1056/NEJM200108093450607. [DOI] [PubMed] [Google Scholar]
- 48.Ohno H, Morikawa Y, Hirata F. Studies on 15-hydroxyprostaglandin dehydrogenase with various prostaglandin analogues. J Biochem. 1978;84:1485–1494. doi: 10.1093/oxfordjournals.jbchem.a132272. [DOI] [PubMed] [Google Scholar]
- 49.Lanas A. Nonsteroidal antiinflammatory drugs and cyclooxygenase inhibition in the gastrointestinal tract: a trip from peptic ulcer to colon cancer. The American journal of the medical sciences. 2009;338:96–106. doi: 10.1097/MAJ.0b013e3181ad8cd3. [DOI] [PubMed] [Google Scholar]
- 50.Medina-Contreras O, et al. CX3CR1 regulates intestinal macrophage homeostasis, bacterial translocation, and colitogenic Th17 responses in mice. J Clin Invest. 2011;121:4787–4795. doi: 10.1172/JCI59150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuhl AA, et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology. 2007;133:1882–1892. doi: 10.1053/j.gastro.2007.08.073. [DOI] [PubMed] [Google Scholar]
- 52.Anderson CF, Oukka M, Kuchroo VJ, Sacks D. CD4(+)CD25(-)Foxp3(-) Th1 cells are the source of IL-10-mediated immune suppression in chronic cutaneous leishmaniasis. J Exp Med. 2007;204:285–297. doi: 10.1084/jem.20061886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jankovic D, et al. Conventional T-bet(+)Foxp3(-) Th1 cells are the major source of host-protective regulatory IL-10 during intracellular protozoan infection. J Exp Med. 2007;204:273–283. doi: 10.1084/jem.20062175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cuenca AG, et al. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol Med. 2011;17:281–292. doi: 10.2119/molmed.2010.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nature reviews Immunology. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Libioulle C, et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet. 2007;3:e58. doi: 10.1371/journal.pgen.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berg DJ, et al. Rapid development of colitis in NSAID-treated IL-10-deficient mice. Gastroenterology. 2002;123:1527–1542. doi: 10.1053/gast.2002.1231527. [DOI] [PubMed] [Google Scholar]
- 60.Wall EA, et al. Suppression of LPS-induced TNF-alpha production in macrophages by cAMP is mediated by PKA-AKAP95-p105. Sci Signal. 2009;2:ra28. doi: 10.1126/scisignal.2000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grunvald E, et al. Biochemical characterization and protein kinase C dependency of monokine-inducing activities of Toxoplasma gondii. Infect Immun. 1996;64:2010–2018. doi: 10.1128/iai.64.6.2010-2018.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tepper RI, Coffman RL, Leder P. An eosinophil-dependent mechanism for the antitumor effect of interleukin-4. Science. 1992;257:548–551. doi: 10.1126/science.1636093. [DOI] [PubMed] [Google Scholar]
- 63.Mohan VP, et al. Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: possible role for limiting pathology. Infect Immun. 2001;69:1847–1855. doi: 10.1128/IAI.69.3.1847-1855.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 65.Roos DS, Donald RG, Morrissette NS, Moulton AL. Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol. 1994;45:27–63. doi: 10.1016/s0091-679x(08)61845-2. [DOI] [PubMed] [Google Scholar]
- 66.Pfefferkorn ER, Pfefferkorn LC. Toxoplasma gondii: isolation and preliminary characterization of temperature-sensitive mutants. Exp Parasitol. 1976;39:365–376. doi: 10.1016/0014-4894(76)90040-0. [DOI] [PubMed] [Google Scholar]
- 67.Ismail AS, et al. Gammadelta intraepithelial lymphocytes are essential mediators of host-microbial homeostasis at the intestinal mucosal surface. Proc Natl Acad Sci U S A. 2011;108:8743–8748. doi: 10.1073/pnas.1019574108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.