

Abstract
Objective
To assess outcomes of prenatal diagnosis of septal leaflet abnormality in fetuses referred for ventriculomegaly (VM).
Methods
In a prospective IRB-approved study with written informed consent, between 7/1/2003 to 5/15/2009, 425 pregnant women with 433 fetuses referred for VM were imaged with US and MRI. Four to six radiologists independently reviewed sonographic and MR images and recorded lateral ventricular diameters at the atrium and frontal horns, ventricular configuration, and the presence of VM and other CNS abnormalities. Final US, MR, and overall prenatal diagnoses were decided by consensus. Fetuses coded for septal leaflet abnormality were identified, and birth outcome, autopsy, postnatal imaging, and postnatal follow-up were obtained. Log-transformed ANCOVA, controlling for GA, was used to compare ventricular dimensions between fetuses with septal leaflet abnormalities and fetuses with isolated VM. Inter-rater agreement of septal leaflet abnormality was assessed with kappa statistics.
Results
23 fetuses had septal leaflet abnormalities and 229 had isolated VM. Atrial and frontal horn diameters, adjusted for GA, were 77% and 98% larger, respectively, in fetuses with septal leaflet abnormalities than in fetuses with isolated VM (p<0.0001). Pre-conference consensus among US readers was moderate (kappa = 0.54) and among MR readers was good (kappa = 0.69). Additional MR CNS findings were seen in 12/23 (52%) fetuses. Eleven pregnancies with septal leaflet abnormalities underwent termination and 12 progressed to live birth with 3 neonatal demise. Neurodevelopmental follow-up was abnormal in all surviving children.
Conclusion
Even when septal leaflet abnormality is an isolated finding, developmental delay can occur postnatally.
Keywords: Agenesis of the septum pellucidum, Septal leaflet abnormality, Septo-optic dysplasia, Fetal MRI, Obstetrical imaging, Prenatal diagnosis
Introduction
The septal leaflets are an important marker of normal fetal brain development. Documentation of the cavum of the septum pellucidum is one of the required views in the AIUM/ACR guidelines for antepartum sonography.[1] Agenesis or dygenesis of the septum pellucidum is relatively rare, estimated to occur in 2–3 per 100,000 births.[2, 3] Often, agenesis of the septum pellucidum is associated with other central nervous system (CNS) anomalies, most commonly anomalies of prosencephalic development such as holoprosencephaly, agenesis of the corpus callosum and septo-optic dysplasia.[4–6] Incomplete visualization or absence of the septal leaflets on obstetrical sonography can be due to a primary or secondary process. Primary septal leaflet agenesis is commonly thought to represent part of a continuum of prosencephalic malformation, whereas a secondary process can be the result of hydrocephalus leading to enlargement of the lateral ventricles and destruction of the septal leaflets.[4, 7–9] When septal leaflet abnormality is associated with other CNS anomalies, postnatal prognosis is largely dependent on the severity of the associated malformation.[8, 10] At times, however, the septal leaflets can be absent without other visualized abnormalities. When septal leaflet agenesis is an isolated CNS finding, outcomes range from asymptomatic to severe pituitary and/or psychiatric abnormalities.[5, 11] The purpose of our study was to assess the postnatal outcomes of septal leaflet abnormalities in a population of fetuses referred for ventriculomegaly (VM).
Materials and Methods
This was a prospective IRB-approved HIPAA-compliant study with written informed consent from pregnant women carrying fetuses referred for VM. Between 7/1/2003 to 5/15/09, 425 women, carrying 433 fetuses, were enrolled in our study. This population included six women carrying twins in which each twin had VM, and two women who were enrolled twice during consecutive pregnancies. Prenatal sonography was performed with a variety of machines during the 6 years of enrollment, including ATL 5000 and IU22 (Philips Healthcare, Andover, MA), Voluson 730 (GE, Waukesha WI) and Siemens Sequoia512 (Siemens Acuson Co., Mountain View, CA) with 2.5–12.0 -MHz transabdominal and transvaginal transducers by one of four radiologists with 12–22 years of experience in ultrasound. When the fetus was in the cephalic position, a transvaginal scan was obtained to better assess intracranial anatomy. Sonographic data included sonographic gestational age, lateral ventricular diameter measured on a transverse image at the atrium, and frontal horn diameter measured on an angled axial or coronal image. The diagnosis of VM (lateral ventricle measurement at atrium > 10 mm) and other CNS anomalies was recorded. The configuration of the lateral ventricles was categorized as normal, angular, fused frontal horns, disproportion of the frontal/occipital horns, parallel configuration of the frontal horns, colpocephaly, too disorganized to characterize, or globally dilated. One or two ultrasound readers, not involved in the initial scanning of the fetus, independently reviewed the images, measured the diameter of the ventricles at the atrium and frontal horns, categorized ventricular appearance, and recorded diagnoses. When more than one descriptor of ventricular appearance was used by reviewers, the most common descriptor was used for data analysis. The median of the larger ventricular measurements obtained by the ultrasound readers was used for data analysis. Final prenatal sonographic diagnosis was decided by consensus. This consensus process for the first 199 fetuses was previously reported.[12]
MRI examinations were performed on a 1.5-T superconducting system (Signa; GE Medical Systems, Milwaukee, WI) using an 8 or 16 element phased-array surface coil with a radiologist monitoring the study as it was being performed. A three-plane scout view was obtained. Single-shot fast spin-echo imaging was performed in the fetal sagittal, coronal, and axial planes. Sequences varied depending on patient and fetal anatomy. A typical sequence used the following parameters: TR/TE, single-shot/60; field of view, from 30 × 30 to 34 × 34 cm; matrix, 256 × 256 or 512 × 512; slice thickness, 3–5 mm; and sequence acquisition time of 29–45 seconds. Each completed sequence was used as the scout for the subsequent sequence. T1-weighted sequences were also obtained. MR examinations were interpreted by an obstetric radiologist (usually the same radiologist who performed the sonogram) and up to 3 pediatric neuroradiologists, all of whom measured the ventricles, categorized the ventricular appearance, and coded diagnoses. Final prenatal MR diagnosis was made by consensus, and has been previously described for the first 199 fetuses.[12]
When disagreements occurred, a final prenatal diagnosis was decided by combined consensus reads of the US and MR studies.
Fetuses coded by at least one reader as having septal leaflet abnormality or alternate similar diagnoses, including septo-optic dysplasia, agenesis of the septum pellucidum or defect in the septum pellucidum, either on prenatal US or MRI, were identified. Fetuses with final prenatal consensus diagnosis of holoprosencephaly and complete agenesis of the corpus callosum were excluded, as abnormal septal leaflets are expected with these diagnoses. Birth outcome, gender, and karyotype were collected on the remaining fetuses. Autopsy follow-up was obtained for terminations and neonatal demises. For live deliveries, postnatal imaging was reviewed. These were interpreted in a similar fashion to prenatal images, and a final US, MR, and postnatal consensus diagnosis was obtained. This interpretation was performed blinded to the prenatal diagnosis as part of the larger study on VM. Postnatal consensus diagnosis was compared to the imaging diagnoses in the written report for the clinical examinations. In addition, information was obtained on the gestational age at delivery, postnatal neurodevelopmental, endocrine and ophthalmologic examinations. Neurodevelopmental follow-up for the first 314 fetuses[13] has been previously reported.
Statistical analysis
Gestational age at the time of prenatal imaging, determined by menstrual dates and by ultrasound biometry, was compared between the fetuses with septal leaflet abnormalities and those with isolated VM using the Wilcoxon two-sample test. Ventricular dimensions at the level of the atrium and the frontal horns were compared similarly. To control the comparison for gestational age, the diameters were log-transformed and compared using ANCOVA adjusted for gestational age. Besides reducing skew in the distribution of diameters, log transformation allowed the results to be expressed as percentage difference. Inter-rater agreement of septal abnormality diagnosis was assessed with kappa statistics with 0–.20 indicating poor agreement, .21–.40 fair agreement, .41–.60 moderate agreement, .61–.80 good agreement, and .81–1.0 excellent agreement.[14, 15] Descriptive statistics for measured variables are provided as mean ± standard deviation (SD). Percentages were compared by the chi-squared test. SAS software (Cary, NC) was used for all computations.
Results
For the entire study cohort of 433 fetuses, gestational age at the time of imaging ranged from 12.1 to 41.0 weeks (25.9 ± 5.8) as determined by dates, and from 15.7 to 39.4 weeks (26.2 ± 6.0) as determined by sonography.
Thirty-one fetuses were coded by at least one reviewer as having septal leaflet abnormality. Four fetuses were excluded due to prenatal consensus diagnosis of either complete agenesis of the corpus callosum (N=2) or holoprosencephaly (N=2). Four fetuses were excluded due to a final prenatal consensus diagnosis that did not include abnormalities of the septal leaflets. These were three fetuses with final diagnosis of isolated VM, and one fetus with hypoplasia of the corpus callosum, Dandy Walker malformation, and a kinked brainstem. After these exclusions, the final sample for analysis included 23 fetuses with prenatal diagnosis of septal leaflet dysplasia.
At the time of imaging, fetuses with a consensus fetal diagnosis of septal leaflet abnormality ranged in age from 20 to 38 weeks (26.2 ± 6.1) by sonography, and from 19 to 35 weeks (25.7 ± 5.7) by menstrual dates. These ages were not significantly different from those of the 229 fetuses with a final prenatal diagnosis of isolated VM, where gestational age ranged from 16 to 38 weeks (26.3 ± 5.7) by sonography and 12 to 41 weeks (25.8 ± 5.4) by dates; p>0.95 by each method.
Ventricular diameter in the group with septal leaflet abnormality was 10–66 mm (23.1 ± 14.1) at the atrium, and 3–32 mm (10.7 ± 7.7) at the frontal horns. Diameters were significantly greater in fetuses with septal leaflet abnormalities than in those with isolated VM, where atrial diameter ranged from 9–22 mm (11.5 ± 1.8) and frontal horn diameter from 2–15 mm (4.9 ± 2.2); p <0.0001 for each measure. Adjusted for gestational age, atrial and frontal horn measurements were 77% and 98% larger, respectively, in the cohort with septal leaflet abnormality than in the cohort with isolated VM.
Eighteen fetuses had septal leaflet abnormality coded on US by at least one reader pre-consensus conference. In the US consensus conference, septal leaflet abnormality was attributed to 15 of those fetuses and to one additional fetus that had not been so coded by any reader pre-consensus conference. Twenty-seven fetuses were coded for septal leaflet abnormality on MR by at least one reader pre-conference, including all 23 in the analysis sample and four among those exclusions listed above. All 23 in the analysis sample were coded with septal leaflet abnormality in the MR consensus conference. Thus seven fetuses received a final prenatal diagnosis of septal leaflet abnormality on MR, but not on US. Pre-conference agreement of all readers in coding septal leaflet abnormality was uncommon, occurring for US in only 6 of the 16 fetuses receiving final US consensus diagnosis of septal leaflet abnormality (38%), for MR in 8 of the 23 with final consensus diagnosis of septal leaflet abnormality (35%), and for both US and MR prior to conference in only 4 of the 23 (17%).
Inter-rater agreement statistics for the diagnosis of septal leaflet abnormality showed moderate pre-conference consensus among US readers (kappa = 0.54) and good pre-conference consensus on MR (kappa = 0.69).
Additional CNS findings, observed by ultrasound and/or MR, associated with the diagnosis of septal leaflet abnormality and the various groupings of prenatal consensus diagnosis are given in Table 1. Five fetuses had no findings other than VM and septal leaflet leaflet abnormality (figure 1). The other 18 fetuses had additional findings, including malformation of the corpus callosum (N= 7), porencephaly (N= 6), cerebellar hypoplasia (N=4), spinal neural tube defects with Chiari malformation (N=3), small brainstem (N=2), encephalocele (N=2), schizencephaly (N=2, figure 2), and one diagnosis each of Dandy Walker malformation (figure 3), migrational abnormalities, arachnoid cyst, tethered cord, nonspecific ventral induction abnormality, microphthalmia (figure 4), and choroid plexus hemorrhage. Pertaining to these diagnoses, there were 12 fetuses with 15 additional abnormalities coded on MR but not on US (Table 1).
Table 1.
Details of subjects with prenatal diagnosis of septal leaflet dysplasia.
| GA | Other CNS Findings (US) | Additional CNS findings (MR) | Outcome | Gender, Karyotype or genetic abnormality | Ventricular Configuration | Autopsy or postnatal imaging | Ventricular-Peritoneal Shunt | Postnatal findings | Last f/u |
|---|---|---|---|---|---|---|---|---|---|
| Pre-conference consensus | |||||||||
| 29 | Live Delivery | M | Globally dilated | Imaging: ASP, thin CC, unilateral optic nerve hypoplasia, aqueductal stenosis, subdural hematoma | Yes | Mild hypotonia, early motor and speech delay, all resolved by age 3 per parental report. Esotropia and 6th nerve palsy | 2y formal evaluation, 3 y parental report | ||
| 31 | Migrational abnormality | Schizencephaly | Live Delivery | F | Normal | Imaging: ASP, schizencephaly, heterotopias, polymicrogyria, arachnoid cyst, dysgenesis CC | No | R hemiplegia, speech delay. Inattentiveness to R visual field | 4.5 y |
| US and MR post-conference consensus | |||||||||
| 19 | CC hypoplasia | Porencephaly | Live Delivery | F, 15q11.2 Deletion | Globally dilated | Imaging: ASP, dygenesis CC, severe VM, porencephaly, parenchymal atrophy, tonsillar herniation | Yes | Hyponatremia, seizures, hypotonia, diffuse weakness. Visual field defect, bilateral choroidal colobomas, cortical visual impairment and hyperopia | 11 m |
| 20 | Termination | F | Globally dilated | Pathology: Macerated fetus | NA | NA | NA | ||
| 20 | Cerebellar hypoplasia | Encephalocoele, porencephaly | Termination | F | Globally dilated | Pathology: Encephalocele | NA | NA | NA |
| 20 | Cerebellar hypoplasia | Porencephaly | Termination | F | Globally dilated | Not performed | NA | NA | NA |
| 20 | Porencephaly | Termination | M | Globally dilated | Pathology: Normal gross examination, parents declined internal autopsy | NA | NA | NA | |
| 21 | Myelomening ocoele, Chiari 2malformation | Termination | F | Angular appearance | Not performed | NA | NA | NA | |
| 21 | Termination | M | Globally dilated | Not performed | NA | NA | NA | ||
| 23 | Arachnoid cyst | Termination | M | Normal | Pathology: Septo-optic dysplasia, postaxial polydactyly, low ears, retrognathia, optic nerve hypoplasia, cataracts | NA | NA | NA | |
| 26 | CC hypoplasia | Live Delivery | M, Xq11 Duplication | Globally dilated | Imaging: ASP, dysplasia CC, midline fusion abnormalities, cerebellar lipoma, subdural hematoma | No | Low oral motor tone and mild expressive language delay, esotropia treated surgically | 2.5 y | |
| 29 | Cerebellar hypoplasia, tethered cord | Hypoplasia CC, small brainstem | Neonatal demise, day 4 | F | Globally dilated | Pathology: ASP, thin CC, VM, microcephaly, diffuse gray- white junction abnormality, small ocular openings, AV canal, coarctation of aorta, duodenal atresia, small mouth, low set ears, radial agenesis, bilobed spleen, hypoplastic kidneys, unicornuate uterus | NA | Comfort care only | NA |
| 31 | CC hypoplasia, encephalocoel e | Secondarily acquired injury | Neonatal demise, day 7 | M, Myotonic dystrophy type 1 mutation | Globally dilated | Imaging: ASP, Severe VM, dysplasia CC, parenchymal atrophy, midline shift | No | Cataracts with no red reflex | NA |
| 33 | Live Delivery | M | Fused frontal horns | Imaging: ASP, polymicrogryia | No | Hypotonia, psychomotor developmental delays, esotropia | 1 y | ||
| 34 | DW malformation, CC hypoplasia | Small brainstem | Live Delivery | M, Factor VII deficiency | Globally dilated | Imaging: ASP, dygenesis CC, severe VM, DW malformation, thin brain parenchyma, choroid plexus hemorrhage, abnormal brainstem | Yes | Intraventricular shunt complicated by infection, hemorrhage, seizures, downward gaze preference | 2 y |
| 35 | CC hypoplasia | Live Delivery | M | Fused frontal horns | Imaging: ASP, absent body of CC, midline fusion anomaly in frontal lobes in holoprosencephal y spectrum, heterotopias | No | Increased tone distally, muscle weakness, delays in motor skills | 3 m | |
| MR post-conference consensus only (not on US consensus) with US/MR consensus of diagnosis | |||||||||
| 20 | Choroid plexus hemorrhage | Cerebellar hypoplasia, porencephaly | Termination | F | Normal | Not performed | NA | NA | NA |
| 21 | Myelomening ocoele, Chiari malformation | Termination | Unknown | Angular appearance | Not performed | NA | NA | NA | |
| 21 | Microopthalmia | Termination | M | Globally dilated | Not performed | NA | NA | NA | |
| 22 | Schizencephaly | Termination | M | Normal | Not performed | NA | NA | NA | |
| 27 | Live Delivery | M | Globally dilated | Imaging: ASP, severe VM, aqueduct stenosis, focal porencephaly, small areas of hemorrhage, cyst | Yes | Poor head control, seizures, proximal muscle weakness | 4 m | ||
| 31 | CC hypoplasia | Ventral induction abnormality | Live Delivery | M | Globally dilated | Imaging: ASP, severe VM, partial ACC, parenchymal atrophy, schizencephaly, subdural effusion | Yes | Decreased hearing, seizures, hypotonia, VSD, micrognathia, ankyloglossia. Afferent pupillary defect, large alternating exotropia, pigment abnormality in both maculae | 1m |
| 34 | Myelomening-ocoele, Chiari malformation | Neonatal demise, day 7 | F | Globally dilated | Imaging: Severe VM, midline shift, CC dysgenesis, Chiari 2 malformation, myelomeningocele | Yes | Floppy tone, no reflexes, fixed pupils | NA | |
GA = gestational age at prenatal US/MRI, F= female, M=male; ASP = absent septum pellucidum; CC= corpus callosum; SP = septum pellucidum; VSD = ventricular septal defect; y=years; m=month
Fig 1.

Fetus at 29 weeks (A–C) and after birth (D,E) with ventriculomegaly, absence of septal leaflets, and postnatal additional diagnosis of dysmorphic corpus callosum. (A) Coronal US shows enlarged frontal horns with absence of the septal leaflets. Coronal (B) and axial (C) T2 weighted MR images show enlarged lateral and third ventricles and absent septal leaflets. Coronal (D) and sagittal (E) head US show the enlarged frontal horns, absence of the septum pellucidum and thin, dysmorphic corpus callosum.
Fig. 2.

Fetus at 31 weeks gestational age with absent septal leaflets and schizencephaly. (A) Coronal T2-weighted image shows absent septal leaflets and open-lipped schizencephaly. (B) Sagittal T2-weighted MRI image shows normal appearance to midline structures, including corpus callosum.
Fig. 3.

Fetus at 34 weeks gestational age (A–D) and postnatal (E–F) with Dandy Walker malformation, hydrocephalus, and absent septal leaflets. (A) Coronal US shows enlarged ventricles, fused frontal horns and absent septal leaflets. Inferiorly the frontal horns appear pointed. (B) Sagittal US shows the enlarged posterior fossa with severe vermian hypoplasia and elevated tentorium consistent with Dandy Walker malformation. Head size was enlarged (biparietal diameter greater than expected for 40 weeks gestational age). Only the anterior portion of the corpus callosum was visualized. Coronal (C) and sagittal (D) T2-weighted MR images show similar findings. Note the mass effect on the brainstem, which appears slightly thin. (E) Coronal postnatal head US shows absent septal leaflets, severely enlarged ventricles, and the anterior portion of the corpus callosum. (F) T1-weighted sagittal view shows similar findings to those seen prenatally, as well as a small brainstem.
Fig. 4.
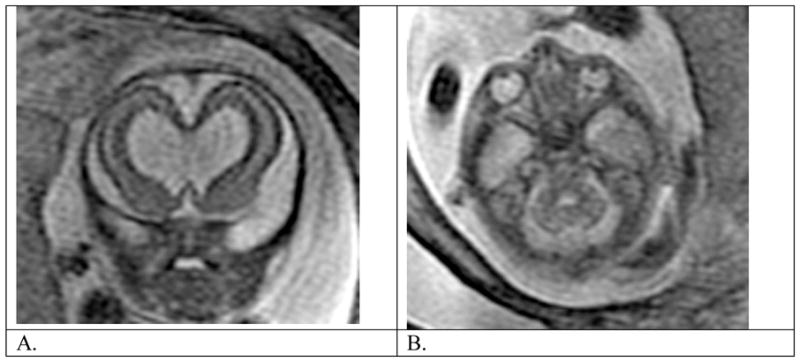
Fetus at 21 weeks gestational age with absent septal leaflets and unilateral microphthalmia. (A) T2 weighted coronal view shows dilated frontal horns with absent septal leaflets. (B) Axial view through orbits shows a smaller globe on the left than on the right.
The description of ventricular configuration on both MR and US varied greatly between fetuses coded for septal leaflet abnormality and those coded for isolated VM (Table 2). Ventricular configuration was characterized as normal in appearance in 200 of the 229 fetuses with isolated VM (87%) and 4 of the 23 fetuses with septal leaflet abnormalities (17% p<0.0001). The majority of fetuses with septal leaflet abnormalities had ventricular configurations described as globally dilated (15/23, 65%), as compared to those with isolated VM (28/229, 12%, p<0.0001). Ten out of the 23 patients with septal leaflet abnormalities were characterized as having fused frontal horns by at least one US or MR reader; but out of these 10 cases, only 2 had fused frontal horns used as the predominant description of ventricular configuration. No fetus with isolated VM had fused frontal horn coded.
Table 2.
Ventricular configuration: majority coded appearance
| Septal Abnormality | Isolated VM | |
|---|---|---|
| Normal configuration | 4 (17.4%) | 200 (87.0%) |
| Angular appearance* | 2 (8.7%) | 0 |
| Disproportion of the frontal/occipital horns | 0 | 1 (0.3%) |
| Parallel configuration of the frontal horns | 0 | 0 |
| Colpocephaly | 0 | 0 |
| Fused frontal horns ** | 2 (8.7%) | 0 |
| Too disorganized to characterize | 0 | 0 |
| Globally dilated | 15 (65.2%) | 28 (12.2%) |
In each case with angular appearance, fetus also had an open NTD
Fused frontal horns coded by any reader in 10 cases with septal leaflet abnormality and 0 cases with isolated VM
Of the 23 fetuses with septal leaflet abnormalities, 11 (47.8%) underwent termination of pregnancy and 12 continued to live birth. Of these 23 fetuses, 13 were male, 9 were female, and one had unclear gender (fetus with cloacal malformation, termination of pregnancy, with autopsy not mentioning gender). The 11 terminations were in the group of 12 fetuses examined prior to 23 weeks gestational age.
Of the 11 terminations, autopsy was performed in 4 cases. Septo-optic dysplasia was diagnosed in one case with findings of optic nerve hypoplasia and retinal pigment epithelial rests, cataracts, post-axial polydactyly, low set ears and retrognathia. A second case had an encephalocele without any comment regarding the septal leaflets. Another fetus was macerated, precluding analysis. A final fetus had a normal surface inspection, but the parents declined an internal autopsy. In another terminated fetus, prenatal MRI showed microphthalmia, but no autopsy was performed.
Three of the 12 live births resulted in neonatal demise at 3–7 days of life, two of whom had postnatal imaging prior to demise, and one of whom had an autopsy. Of the three infants who died, one was diagnosed with myotonic dystrophy. Postnatal MRI findings in this infant revealed severe VM, agenesis of the septum pellucidum, agenesis of the corpus callosum, midline shift and thin parenchyma. Prior to demise, ophthalmic exam revealed bilateral cataracts with absent red reflexes. Another infant had severe intrauterine growth restriction, with autopsy showing absent septum pellucidum, small ocular openings, thin corpus callosum, microcephaly, migrational abnormality, and multiple non-CNS abnormalities. A third neonatal demise occurred in the setting of cerebral herniation due to severe ventriculomegaly. Postnatal US in this infant revealed severe VM, dysgenesis of the corpus callosum, Chiari malformation and midline shift, but did not confirm absence of the septum pellucidum.
Clinical and imaging follow-up were available in the remaining 9 survivors. Follow-up interval ranged from birth to 4.5 years of age. All 9 had imaging confirmation of the diagnosis of septal abnormality (two of these were only described as severe VM in the clinical imaging report, but septal abnormality was coded in the retrospective image review).
Ten of eleven neonates had additional findings on postnatal imaging not seen prenatally, including dysgenesis of the corpus callosum (N=5), cortical heterotopias and other migrational abnormalities (N=3), intracranial hemorrhage (N=3), midline parenchymal fusion abnormalities (N=2), and one finding each of arachnoid cyst, cerebellar lipoma, porencephaly, hemorrhage, microcephaly, parenchymal atrophy, schizencephaly, and optic nerve hypoplasia. Two of the survivors had an abnormal karyotype, one with Xq11 duplication, and one with 15q11.2 deletion. One neonate had factor VII deficiency. None of the surviving children have documented endocrine abnormalities, although only one received a full endocrinological work-up. Cinical ophthalmologic fundoscopic exam was obtained in 5 of the 12 liveborn infants, none of whom showed evidence of optic nerve hypoplasia, but one of whom had an MR showing unilateral optic nerve hypoplasia. In the 9 liveborn infants who survived beyond the neonatal period, 6 had abnormal findings on ophthalmological exam, including esotropia (N=3, one with 6th nerve palsy, one requiring surgery); one with downward gaze preference; one with bilateral choroidal colobomas, cortical visual impairment and hyperopia; and one with afferent pupillary defect, exotropia, and pigment abnormality in the maculae. Six out of the nine surviving children required ventriculotomy procedures postnatally.
Neurodevelopmental follow-up was abnormal on at least one examination in each of the 9 surviving children (Table 1). In one child, however, mild hypotonia and speech delay resolved by 2 years of age.
Discussion
The leaflets of the pellucidum are thin, membranous partitions between the lateral ventricles, bounded dorsally by the body of the corpus callosum, rostrally by the subcallosal gyrus and genu of the corpus callosum, rostroventrally by the nucleus accumbens and subcallosal gyrus, and caudoventrally by the anterior commissure, preoptic area and anterior hypothalamus.[7] Traditionally thought to be composed of two parts, the septum pellucidum is composed in one part of glial cells and fiber bundles, and in another part, known as the septum verum, of multiple fiber systems acting as a central relay station between the diencephalon and limbic system.[2, 4] Two theories exist as to the embryologic formation of the septum pellucidum. In one theory, the leaflets of the septum form through cavitation of the medial inferior commissural plate during the formation of the corpus callosum[16], and in another theory, the septum is formed through the stretching of the commissural plate during the dorsocaudal growth of the corpus callosum.[2] Septal formation is thought to occur during the 7th and 8th weeks of gestation and coincides with the development of the optic nerves, hypophysis and germinal matrix[7, 17], thus offering insight into how dysgenesis of the septum pellucidum is commonly associated with malformations of these structures. In fetal life, the septum pellucidum typically shows a midline cerebrospinal fluid filled cleft known as the cavum of the septum pellucidum, which extends posteriorly as the cavum vergae. This cavum is best visualized by axial and coronal views of the fetal head between 18 to 37 weeks gestational age.[18] Near term, the cavum vergae involutes and the leaves of the septum pellucidum typically “fuse,” creating a single septum.
Agenesis or dysgenesis of the septum pellucidum is a rare congenital malformation characterized by complete or partial absence of the septal leaflets. Septal leaflet abnormality is rarely seen in isolation, and its presence serves as a valuable marker of other congenital neurologic malformations such as holoprosencephaly, schizencephaly, agenesis of the corpus callosum, Chiari II malformation, or other obstructive or destructive processes such as hydrocephalus and porencephaly.[4, 11] Although several cases of isolated septal agenesis have been described in the literature, most reported patients with such findings have been neuropsychiatrically abnormal, suggesting that even if associated gross malformations are not seen, cytoarchitectural disturbances of cortical layers and the limbic system may be present.[11]
Septo-optic dysplasia, also known as de Morsier’s syndrome, is a rare disorder, with an incidence of approximately 1 in 10,000 births, and is in the differential diagnosis of absence of the septal leaflets.[3, 19] Septo-optic dysplasia is a heterogeneous syndrome characterized by agenesis or dysgenesis of septum pellucidum, optic nerve or optic chiasm hypoplasia, and hypothalamic-pituitary abnormalities.[19, 20] It is sometimes associated with midline brain abnormalities and facial manifestation such as bilateral cleft lip and palate.[21, 22] In our series of patients, the final diagnosis of septo-optic dysplasia was made in only one fetus where autopsy revealed optic nerve hypoplasia and retinal pigment epithelial rests in the optic nerve.
Patients with septo-optic dysplasia can display a variety of visual disturbances, ranging from normal vision to nystagmus to complete visual loss.[23] Often, the diagnosis of optic nerve hypoplasia is not easily made through MR and is recognized in only 50% of pediatric patients who are imaged.[24] In the second half of pregnancy, structures such as the optic nerves and the optic chiasm can be studied using MR.[25, 26] However, MR imaging of the fetal optic chiasm is difficult, secondary to the small size of the nerve, slice thickness, gap, and angle of the chiasm with respect to the plane of the section. In addition, optic nerve hypoplasia can be difficult to visualize secondary to chemical shift artifact generated by the interface of orbital fat and the optic nerve. In some patients, optic nerve hypoplasia may not become evident until after birth.[20] Thus, MR imaging of optic nerve hypoplasia is not sensitive to mild hypoplasia and clinical postnatal ophthalmologic exam remains the reference standard for detection. [7] One of the surviving fetuses in our study had optic nerve hypoplasia on postnatal MR, and six of 9 surviving children had ocular abnormalities (albeit some as mild as esotropia) documented after birth.
Hypothalamic-pituitary abnormalities associated with septo-optic dysplasia have been reported in 30–100% of patients.[7, 26, 27] These abnormalities may be detected in utero by low maternal estriol levels which are due to fetal ACTH deficiency[26], but such abnormalities usually manifest at a mean of 4–5 years of age in the form of growth hormone deficiency leading to short stature.[19] Other less common clinical abnormalities include antidiuretic hormone deficiency leading to diabetes insipidus, thyroid hormone abnormalities, or gonadotropin deficiencies.[19, 21] Endocrinopathies may also evolve, with progressive loss of function over time.[19] Only one patient in our series received a full endocrine evaluation at birth for suspected septo-optic dysplasia, and no abnormalities were detected. In general, follow-up in our series was much shorter than the timeframe needed to diagnose endocrine abnormalities associated with septo-optic dysplasia, and thus our conclusions regarding the incidence of this condition in our population are limited.
Morphologically, absence of the septum pellucidum leads to communication between the lateral ventricles, giving the fetal frontal horns a “fused” or square appearance, particularly when other CNS abnormalities such as schizencephaly are not present. The frontal horns were described as having a fused appearance by at least one reviewer in 10 (43%) fetuses with the diagnosis of septal dysplasia and in none of the fetuses with isolated VM. Thus, the appearance of fused frontal horns serves as an important marker in utero, suggesting the diagnosis of septal dysplasia.
Septal dysplasia is often categorized based on whether its presence is thought to be a primary or a secondary process. Primary septal agenesis is often associated with other CNS malformations, of which septo-optic dysplasia is one example. Other associated anomalies can include holoprosencephaly, agenesis of the corpus callosum, Chiari II malformations and encephaloceles. Septal aplasia is well-known to be associated with schizencephaly. Barkovich et. al. proposed that the syndrome of septo-optic dysplasia may actually be composed of two distinct morphologic syndromes, one associated with schizencephaly, and one associated with diffuse white-matter hypoplasia representing a form of mild lobar holoprosencephaly.[7] One subject in our series had schizencephaly, heterotopias and polymicrogyria in addition to septal agenesis, and thus fits this description of primary agenesis.
Abnormalities leading to non-visualization due to secondary destruction of the septum pellucidum were also common in our study. Some of the associated abnormalities in our series included aqueductal stenosis, porencephaly and Chiari 2 malformation. These are well-known conditions causing mechanical necrotic disruption of the septum pellucidum and hence non-visualization of the septal leaflets and thinning of the corpus callosum.[4] The finding of six of nine surviving children needing ventriculostomy procedures attests to ventricular obstruction being an associated factor in our series. The septum pellucidum may not be seen in utero when it is “fenestrated” in association with VM.[8] This was the likely etiology of non-visualization of the septum pellucidum in many of our patients, as shown by the fact that even in this population of fetuses with VM, the ventricular size in fetuses with non-visualization of the septum pellucidum was significantly larger in the group with septal leaflet abnormalities than those without this diagnosis.
Few reports exist pertaining to the prenatal diagnosis and outcome of septal leaflet dysplasia or septo-optic dysplasia,[8, 9, 22, 26] and to the best of our knowledge, our study is the only prospective study. Most of the prior studies are case reports or small case series. The largest series is from a retrospective review by Malinger et al[9] who (once excluding those with a prenatal diagnosis of holoprosencphephy) described 12 fetuses with septal leaflet dysplasia. Of these, 10 resulted in terminations, and 2 continued on to live birth. One prenatally diagnosed with septal leaflet dysplasia and sulcation abnormality was developmentally normal at 6 months, based on clinical follow-up. The other prenatally diagnosed with hydrocephalus and disruption of the septum pellucidum received a VP shunt after birth and was clinically normal at 1 year. Our study has a larger population, systematically employed fetal MR, and has more extensive postnatal follow-up.
In terms of inter-observer variability in our study, abnormality of the septum pellucidum was prenatally diagnosed by at least one reader in 16/23 cases on US and in all 23/23 cases on MR. In 12/23 (52%) fetuses, additional findings were seen on MR that were not seen on US. Thus, as with many CNS abnormalities, it is important to recognize the incremental value of fetal MR when the diagnosis of defective or absent septal leaflets is entertained.
Our study shows that in the population when VM was felt to be the reason the septal leaflets were not seen, outcome still demonstrated developmental delays. Although our sample size is small, this information may be important for parental counseling. The prognosis for prenatally detected isolated septal dysgenesis is not well established. Additional CNS anomalies were present prenatally in 18 of 23 fetuses in our study. This level of association with other CNS anomalies is in accordance with previous literature.[9] In the five fetuses where septal abnormality was the only finding other than VM, three resulted in live deliveries, each with developmental delays. In two of these three infants, however, additional neuropathology was found on postnatal imaging, one showing a thin corpus callosum and aqueductal stenosis, and another showing porencephaly and hemorrhage. It might be that these are progressive changes that occurred later in utero and/or postnally, rather than missed anomalies on fetal imaging. In the one infant who, on postnatal imaging, also showed isolated septal deficiency, postnatal follow-up showed hypotonia and developmental delay. This agrees with earlier reports that isolated septal agenesis is associated with developmental abnormalities.[5, 11]
Our study had limitations. Our sample was biased by lack of follow-up, particularly in the cases where pregnancy was terminated and there was no autopsy. Twelve of the subjects in our study were imaged at less than 24 weeks gestational age, and 11 of these had termination of the pregnancy. Eleven subjects with the diagnosis made after 24 weeks survived to delivery, but three had neonatal demise. Our postnatal follow-up was variable in terms of age at postnatal imaging and neurodevelopmental testing. In each case, however, neurodevelopomental follow-up was abnormal, although in one child the developmental delays resolved by age 2.
In conclusion, agenesis or partial absence of the septum pellucidum is an important marker of fetal brain malformation. Fetal MRI plays an important complementary role in the diagnosis of septal leaflet abnormalities and associated CNS abnormalities. Even when septal leaflet abnormality is an isolated finding on prenatal imaging, developmental delay is common postnatally.
Acknowledgments
This study was funded by NIH NIBIB 01998. Medical student research support was from the Clinical Research Fellowship Program at Harvard Medical School offered by the Doris Duke Charitable Foundation in conjunction with the Harvard PASTEUR Program.
References
- 1.American College of Radiology. ACR practice guidelines and technical standards. Philadelphia, PA: American College of Radiology; 2007. ACR practice guideline for the performance of antepartum obstetrical ultrasound; pp. 1025–1033. [Google Scholar]
- 2.Bruyn G. Agenesis septi pellucidi, cavum septi pellucidi, cavum vergae, and cavum veli interpositi. In: Vinken P, Bruyn G, editors. Handbook of Clinical Neurology Congenital malformations of the brain and skull, part I. North Holland, Amsterdam: Elsevier; 1977. pp. 299–336. [Google Scholar]
- 3.Patel L, McNally RJ, Harrison E, Lloyd IC, Clayton PE. Geographical distribution of optic nerve hypoplasia and septo-optic dysplasia in Northwest England. J Pediatr. 2006;148:85–88. doi: 10.1016/j.jpeds.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 4.Barkovich AJ, Norman D. Absence of the septum pellucidum: a useful sign in the diagnosis of congenital brain malformations. AJR Am J Roentgenol. 1989;152:353–360. doi: 10.2214/ajr.152.2.353. [DOI] [PubMed] [Google Scholar]
- 5.Belhocine O, Andre C, Kalifa G, Adamsbaum C. Does asymptomatic septal agenesis exist? A review of 34 cases. Pediatr Radiol. 2005;35:410–418. doi: 10.1007/s00247-004-1378-2. [DOI] [PubMed] [Google Scholar]
- 6.Kuhn MJ, Swenson LC, Youssef HT. Absence of the septum pellucidum and related disorders. Comput Med Imaging Graph. 1993;17:137–147. doi: 10.1016/0895-6111(93)90056-s. [DOI] [PubMed] [Google Scholar]
- 7.Barkovich AJ, Fram EK, Norman D. Septo-optic dysplasia: MR imaging. Radiology. 1989;171:189–192. doi: 10.1148/radiology.171.1.2928524. [DOI] [PubMed] [Google Scholar]
- 8.Birnholz JC. Septum pellucidum fenestration: visualization by ultrasound. Radiology. 1983;149:122. doi: 10.1148/radiology.149.1.6611915. [DOI] [PubMed] [Google Scholar]
- 9.Malinger G, Lev D, Kidron D, Heredia F, Hershkovitz R, Lerman-Sagie T. Differential diagnosis in fetuses with absent septum pellucidum. Ultrasound Obstet Gynecol. 2005;25:42–49. doi: 10.1002/uog.1787. [DOI] [PubMed] [Google Scholar]
- 10.Williams J, Brodsky MC, Griebel M, Glasier CM, Caldwell D, Thomas P. Septo-optic dysplasia: the clinical insignificance of an absent septum pellucidum. Dev Med Child Neurol. 1993;35:490–501. doi: 10.1111/j.1469-8749.1993.tb11679.x. [DOI] [PubMed] [Google Scholar]
- 11.Supprian T, Sian J, Heils A, Hofmann E, Warmuth-Metz M, Solymosi L. Isolated absence of the septum pellucidum. Neuroradiology. 1999;41:563–566. doi: 10.1007/s002340050805. [DOI] [PubMed] [Google Scholar]
- 12.Levine D, Feldman HA, Tannus JF, et al. Frequency and cause of disagreements in diagnoses for fetuses referred for ventriculomegaly. Radiology. 2008;247:516–527. doi: 10.1148/radiol.2472071067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beeghly M, Ware J, Soul J, et al. Neurodevelopmental outcomes of fetuses referred for ventriculomegaly. Ultrasound Obstet Gynecol. doi: 10.1002/uog.7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kraemer H, Periyakoil V, Noda A. Kappa coefficients medical research. Statistical Medicine. 2002;21:2109–2129. doi: 10.1002/sim.1180. [DOI] [PubMed] [Google Scholar]
- 15.Szklo M, Nieto F. Epidemiology: Beyond the Basics. Gaithersburg, MD: Aspen Publishers, Inc; 1999. pp. 375–388. [Google Scholar]
- 16.Yakovlev PI. Pathoarchitectonic studies of cerebral malformations. III. Arrhinencephalies (holotelencephalies) J Neuropathol Exp Neurol. 1959;18:22–55. doi: 10.1097/00005072-195901000-00003. [DOI] [PubMed] [Google Scholar]
- 17.Barkovich AJ, Norman D. MR imaging of schizencephaly. AJR Am J Roentgenol. 1988;150:1391–1396. doi: 10.2214/ajr.150.6.1391. [DOI] [PubMed] [Google Scholar]
- 18.Falco P, Gabrielli S, Visentin A, Perolo A, Pilu G, Bovicelli L. Transabdominal sonography of the cavum septum pellucidum in normal fetuses in the second and third trimesters of pregnancy. Ultrasound Obstet Gynecol. 2000;16:549–553. doi: 10.1046/j.1469-0705.2000.00244.x. [DOI] [PubMed] [Google Scholar]
- 19.Kelberman D, Dattani MT. Septo-optic dysplasia - novel insights into the aetiology. Horm Res. 2008;69:257–265. doi: 10.1159/000114856. [DOI] [PubMed] [Google Scholar]
- 20.Volpe P, Campobasso G, De Robertis V, Rembouskos G. Disorders of prosencephalic development. Prenat Diagn. 2009;29:340–354. doi: 10.1002/pd.2208. [DOI] [PubMed] [Google Scholar]
- 21.Fitz CR. Holoprosencephaly and septo-optic dysplasia. Neuroimaging Clin N Am. 1994;4:263–281. [PubMed] [Google Scholar]
- 22.Pilu G, Sandri F, Cerisoli M, Alvisi C, Salvioli GP, Bovicelli L. Sonographic findings in septo-optic dysplasia in the fetus and newborn infant. Am J Perinatol. 1990;7:337–339. doi: 10.1055/s-2007-999517. [DOI] [PubMed] [Google Scholar]
- 23.Skarf B, Hoyt CS. Optic nerve hypoplasia in children. Association with anomalies of the endocrine and CNS. Arch Ophthalmol. 1984;102:62–67. doi: 10.1001/archopht.1984.01040030046032. [DOI] [PubMed] [Google Scholar]
- 24.Barkovich AJ. Congenital malformations of the brain and skull. In: Barkovich AJ, editor. Pediatric Neuroimaging. Philadephia, PA: Lippincott Williams & Wilkins; 2000. pp. 251–382. [Google Scholar]
- 25.Pilu G, Tani G, Carletti A, Malaigia S, Ghi T, Rizzo N. Difficult early sonographic diagnosis of absence of the fetal septum pellucidum. Ultrasound Obstet Gynecol. 2005;25:70–72. doi: 10.1002/uog.1786. [DOI] [PubMed] [Google Scholar]
- 26.Lepinard C, Coutant R, Boussion F, et al. Prenatal diagnosis of absence of the septum pellucidum associated with septo-optic dysplasia. Ultrasound Obstet Gynecol. 2005;25:73–75. doi: 10.1002/uog.1807. [DOI] [PubMed] [Google Scholar]
- 27.Hellstrom A, Aronsson M, Axelson C, et al. Children with septo-optic dysplasia -how to improve and sharpen the diagnosis. Horm Res. 2000;53 (Suppl 1):19–25. doi: 10.1159/000053200. [DOI] [PubMed] [Google Scholar]