

Abstract
Inherited mutations in the folliculin (FLCN) gene cause the Birt–Hogg–Dubé syndrome of familial hair follicle tumours (fibrofolliculomas), lung cysts and kidney tumours. Though folliculin has features of a tumour suppressor, the precise function of the FLCN gene product is not well characterized. We identified plakophilin-4 (p0071) as a potential novel folliculin interacting protein by yeast two-hybrid analysis. We confirmed the interaction of folliculin with p0071 by co-immunoprecipitation studies and, in view of previous studies linking p0071 to the regulation of rho-signalling, cytokinesis and intercellular junction formation, we investigated the effect of cell folliculin status on p0071-related functions. Folliculin and p0071 partially co-localized at cell junctions and in mitotic cells, at the midbody during cytokinesis. Previously, p0071 has been reported to regulate RhoA signalling during cytokinesis and we found that folliculin deficiency was associated with increased expression and activity of RhoA and evidence of disordered cytokinesis. Treatment of folliculin-deficient cells with a downstream inhibitor of RhoA signalling (the ROCK inhibitor Y-27632) reversed the increased cell migration phenotype observed in folliculin-deficient cells. Deficiency of folliculin and of p0071 resulted in tight junction defects and mislocalization of E-cadherin in mouse inner medullary collecting duct-3 renal tubular cells. These findings suggest that aspects of folliculin tumour suppressor function are linked to interaction with p0071 and the regulation of RhoA signalling.
INTRODUCTION
Germline mutations in the folliculin (FLCN) gene cause Birt Hogg Dubé (BHD) syndrome, an inherited cancer predisposition disorder, characterized by benign tumours of the hair follicles (fibrofolliculomas) on the face and upper torso, renal cell carcinoma (RCC) and lung cysts and pneumothorax (1,2). In addition, an increased risk of colonic polyps and cancers has been described in a subset of BHD syndrome families (3–5). The FLCN gene, mapped to chromosome 17p11.2, is a tumour suppressor gene and biallelic FLCN inactivation has been described in RCC from patients with BHD syndrome (6,7). Germline FLCN mutations have also been described in patients with inherited RCC and familial spontaneous pneumothorax (8,9). FLCN encodes a 64 kDa protein, folliculin (FLCN), which has no significant homology to other known proteins and is highly conserved throughout evolution.
Elucidation of the molecular functions of folliculin is critical for understanding the role of FLCN inactivation in neoplasia and for developing novel therapeutic strategies for BHD syndrome. Currently, however, the functions of the FLCN gene product are poorly understood. Folliculin, through its C terminus, has been reported to bind two relatively uncharacterized proteins, FNIP1 and FNIP2, and to indirectly interact with AMPK (AMP-activated protein kinase), thus linking folliculin to mammalian target of rapamycin (mTOR) signalling (10,11). However, the precise role of folliculin in the regulation of mTOR signalling is still under debate as although a folliculin null cell line derived from a BHD patient and FLCN knockout mice demonstrated up-regulation of mTOR signalling (10,12,13), other reports have suggested a more complex, context-dependent effect on mTOR activity (14,15). Folliculin has also been reported to play a role in regulating transforming growth factor beta signalling (16,17) and increase hypoxia inducible factor transcriptional activity (18), although the molecular mechanisms for these effects have not been elucidated.
We investigated whether the identification of novel folliculin-interacting proteins might provide insights into the mechanisms of folliculin tumour suppressor activity. We performed a yeast two-hybrid screen with a full-length folliculin as bait and identified plakophilin-4 (PKP4, p0071) as a candidate novel interactor. Previously, p0071 has been linked to the regulation of Rho-signalling, cytokinesis and intercellular junction formation, and we proceeded to investigate whether folliculin could also regulate these functions.
RESULTS
P0071 and FLCN interact in vitro
p0071 (PKP4) was detected as a potential interacting partner of folliculin in a yeast two-hybrid screen using full-length folliculin as a bait against a human foetal brain cDNA library. Four independent clones mapping to exons 7–10 of p0071 were identified and p0071 was one of the seven potential interactors detected more than once. To confirm the yeast two-hybrid results which suggested a direct interaction between p0071 and folliculin, co-immunoprecipitation studies were undertaken to confirm the interaction in vitro. p0071 co-immunoprecipitated with folliculin in Hek293 cells transfected with Flag-FLCN (Fig. 1A) and also with endogenous folliculin in untransfected cells (Fig. 1C). Similarly, when p0071 was pulled down, folliculin was also detected by immunoblotting (Fig. 1B and C). Immunoprecipitation with control antibody produced no visible FLCN or p0071 signal. Co-immunoprecipitation was also confirmed in the kidney cancer cell line (ACHN) (data not shown).
Figure 1.

FLCN and p0071 interact in vitro: flag-tagged FLCN was stably transfected into Hek293 cells. Cell lysates were immunoprecipitated (IP) with Flag (A) or p0071 (B) and blotted with Flag and p0071. (C) Endogenous interaction was confirmed in Hek293 parental cells. Lysates were IP with FLCN, p0071 or control IgG and blotted for p0071 and FLCN. In both IP's p0071 and FLCN together, there was no pull down with control IgG. Arrows indicated the size of bands (kDa).
p0071 and FLCN co-localize and interact at cell junctions during interphase
To identify the cellular context of the FLCN and p0071 interaction, we investigated the intracellular localization of both proteins using immunofluorescence microscopic analysis. Interphase breast cancer cell line-7 (MCF-7), co-transfected with Flag-tagged FLCN and dsRed-tagged p0071 showed a co-localization of folliculin and p0071 at cell contacts and also a more disperse co-localization pattern throughout the cytoplasm (Fig. 2A). The co-localization at cell junctions was also observed at endogenous levels in MCF-7 cells, although the pattern of folliculin signal is more consistent with a transient role for folliculin at cell junctions rather than as a major structural component (Fig. 2B). Bimolecular fluorescence complementation (BiFC) analysis confirmed the association between folliculin and p0071 in living cells (Hela), with evidence of a strong interaction at cell junctions and also at regions within the cytoplasm (Fig. 2C).
Figure 2.

p0071 and folliculin are co-localized and interact at cell–cell junctions. (A) MCF-7 cells were co-transfected with flag-tagged FLCN and dsRed-tagged p0071, fixed and stained for Flag and 4′,6-diamidino-2-phenylindole (DAPI). Folliculin and p0071 co-localize at cell junctions and in a disperse pattern at certain points within the cytoplasm. Manders overlap co-efficient = 0.968. (B) Confluent MCF-7 cells were fixed and stained for endogenous folliculin and p0071. Both p0071 and FLCN localized to cell junctions displaying a co-localized pattern. FLCN and p0071 co-localized pixels were identified using ImageJ software and are displayed in white on the zoomed images. Manders overlap co-efficient = 0.599. (C) BiFC analysis was used to confirm/identify the location of the interaction between FLCN and p0071 in living cells. Hela cells co-expressed with pVen-1-Flag-p0071 and pVen2-HA-FLCN display a clear BiFC signal at cell junctions and throughout the cytoplasm.
p0071 and FLCN co-localize and interact at the midbody during cytokinesis
p0071 localization is reported to differ depending on the cell cycle stage, with a cell junctional localization in the interphase cell, differing in mitotic cells when p0071 becomes localized in the midzone region in anaphase and telophase, then tightly localized at the midbody during cytokinesis (19). We hypothesized that folliculin may also interact with p0071 at these regions in mitotic cells. FTC-133 cells stably transfected with folliculin were fixed and stained for folliculin, p0071 and α-tubulin (to identify cells in the late stages of mitosis). A co-localization pattern of folliculin and p0071 was observed at the midbody during cytokinesis (Fig. 3A). A concentrated folliculin and p0071 staining at the midbody was also observed at endogenous levels in HaCat cells (Fig. 3B). Using BiFC analysis, we observed a direct interaction of folliculin and p0071 by BiFC at the midzone during late telophase/cytokinesis in Hela cells (Fig. 3C).
Figure 3.

FLCN and p0071 co-localize and interact at the midbody during telophase/cytokinesis. (A) FTC-133 cells transfected with FLCN were stained for FLCN, endogenous p0071 and α-tubulin. Enlargements of midbodies are displayed below the original images with only FLCN and p0071 channels displayed. P0071 and FLCN are concentrated at the midbody (stained with α-tubulin) at cytokinesis. FLCN and p0071 co-localized pixels are displayed in white on the zoomed images. Manders overlap co-efficient = 0.625. (B) HaCat cells were fixed and stained for FLCN, p0071 and α-tubulin. Both proteins show concentrated localization at the midbody at endogenous levels. FLCN and p0071 co-localized pixels are displayed in white on the zoomed images. Manders overlap co-efficient = 0.687. (C) Hela cells were co-expressed with pVen-1-Flag-p0071 and pVen2-HA-FLCN and fixed after 24 h. Cells were stained for HA (to identify transfected cells) and α-tubulin (to identify cells in the late stages of cytokinesis). Association was detected by BiFC, displaying a focused signal at the midzone of a telophase cell.
Loss of FLCN deregulates RhoA signalling/activity
Previously, p0071 has been reported to regulate RhoA signalling during cytokinesis by binding to RhoA and also Ect2, a Rho guanine exchange factor (GEF) essential for cytokinesis. We postulated that as folliculin interacts with p0071 at cell junctions and during cytokinesis, it might also impinge upon RhoA signalling. Comparison of paired isogenic cell lines demonstrated that folliculin deficiency was associated with increased expression of RhoA (Fig. 4A). In the ACHN cell line, levels of folliculin knockdown by stable shRNA were inversely correlated with RhoA levels in a dose-dependent manner.
Figure 4.

Loss of FLCN induces the deregulation of RhoA signalling. (A) Three BHD cell models were tested for levels of RhoA. RhoA levels significantly increased in FTC133 cells (FLCN-negative) compared with FLCN re-expressing clone. In both renal cell models Hek293 and kidney cancer cell line (ACHN), transient and stable FLCN knockdown, respectively, caused an increase in RhoA levels. (B) GTP bound (active) RhoA is increased in FLCN null cells (FTC-133) compared with FLCN re-expressing cells (∼2-fold). Results normalized to FLCN expressing FTC-133 cells (n = 3). (C) Active RhoA is increased in ACHN cells with stable FLCN knockdown (∼30% increase). Repeats normalized to the activation of ACHN scrambled control (n = 3).
RhoA is a small GTPase which is inactive when bound to guanosine diphosphate (GDP) and is converted to its active guanosine triphosphate (GTP) bound from when catalysed by RhoGTPase-specific guanine nucleotide exchange factors (Rho GEF's). Conversion back to the GDP bound, inactive form is stimulated by Rho-specific GTPase activating proteins. To test if folliculin status influenced cellular RhoA activity, we used the G-LISA RhoA activation assay to determine the levels of GTP-bound RhoA in folliculin expressing and folliculin-deficient cells. In the FTC-133 cell line, the levels of active RhoA were significantly higher (>2-fold increase) in folliculin-deficient cells than in those in which folliculin had been re-expressed (Fig. 4B). Stable knockdown of FLCN in ACHN cells also caused a significant increase in RhoA activation (∼30%; Fig. 4C).
Loss of FLCN causes cytokinesis defects resulting in multinucleation
Previously, depletion of p0071 had been reported to causes severe cytokinesis defects and to induce a multinucleation phenotype (19). These effects were correlated with deregulation of RhoA signalling during cytokinesis. As we had found that loss of folliculin increased flux through RhoA signalling, we investigated if folliculin loss influenced levels of multinucleation in our cellular models. In the folliculin-deficient FTC-133 cell line, >16% of cells were found to be multinucleated but the reintroduction of folliculin in the FTC-133 cell line significantly reduced this number to ∼2% multinucleated cells (P < 0.005), indicating that folliculin is required for cells to complete cytokinesis correctly (Fig. 5).
Figure 5.
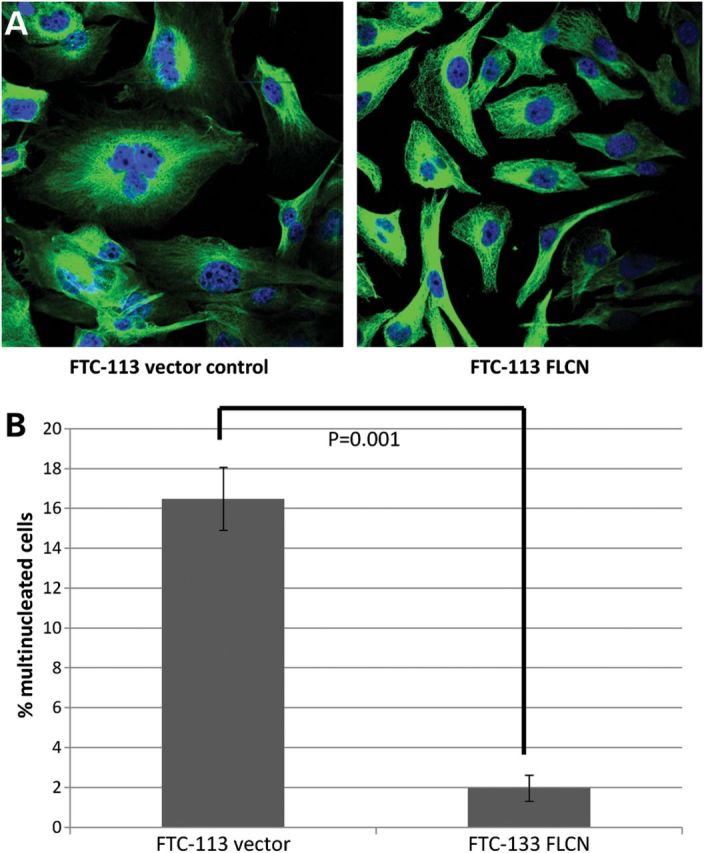
Reintroduction of FLCN rescues the multinucleation phenotype in FTC-133 cells. (A) Representative images of FLCN+/− FTC-133 cells stained with tubulin and DAPI. (B) Graph of percentage of multinucleated cells in FTC-133 vector expressing and FLCN expressing cells (P < 0.01; at least 400 cells counted from at least three independent experiments). Error bars represent the SD.
Reintroduction of FLCN into null metastatic cells ameliorates the migratory phenotype
Altered expression and activity of RhoA has been previously shown to correlate with a number of metastatic diseases (20,21). In FTC-133 cell lines (which are derived from a metastatic thyroid carcinoma), folliculin inactivation was associated with increased RhoA expression and activity (Fig. 4). We hypothesized that this increased RhoA activity might be associated with a more migratory phenotype. This was investigated by a wound healing assay, in which folliculin expressing FTC-133 cells migrated significantly more slowly than folliculin-deficient (empty vector expressing) cells (P < 0.05; Fig. 6A and C). The Rho-associated kinases (ROCK 1 and ROCK 2) function downstream of RhoA and can be specifically targeted by the compound Y-27632, a well-characterized ROCK inhibitor (22). We postulated that inhibiting signalling downstream of RhoA in folliculin-deficient FTC-133 cells might ameliorate the migratory phenotype and phenocopy the re-expression of folliculin. Addition of 10 µm Y-27632 every 12 h significantly reduced the migratory ability of the cells (P < 0.005; Fig. 6B and C).
Figure 6.

FTC-133 cells lacking FLCN are more migratory due to increased RhoA signalling. A serum-starved monolayer of confluent FTC-133 cells was scratched with a sterile pipette and left to migrate for 96 h. (A) Graph displaying % of scratch healed every 24 h for FLCN re-expressing FTC-133 cells (red line) and FTC-133 empty vector expressing cells (blue line). (B) Graph displaying % of scratch healed for FTC-133 empty vector expressing cells treated with a 12 h, 10 μm dose of Y27632 (Rock inhibitor; green line) compared with the same cells treated with water control (blue line). Error bars represent the standard deviation calculated from three-independent experiments. (C) Representative pictures of migrations at 0, 48 and 96 h after scratching. (D) Folliculin expressing cells migrate significantly more slowly through a porous membrane towards a chemotactic stimulus than those expressing empty vector (P = 0.02). Treatment of FTC-133 cells (no FLCN expressed) with Y27632, significantly reduces migratory ability (P = 0.007). In neither assay was there a significant effect of Y27632 treatment on cells expressing folliculin (data not shown). Significance tested by independent t-tests from three independent experiments and error bars represent the SD.
Similarly, when tested in a Boyden chamber, folliculin expressing FTC-133 cells migrated towards a chemotactic stimulus [fetal bovine serum (FBS)] significantly more slowly than those null for folliculin (P < 0.02) and treatment of folliculin null FTC-133 cells with Y-27632 significantly inhibited cell migration (P < 0.01; Fig. 6D).
Both knockdown of folliculin and p0071 disrupt cell junctional formation
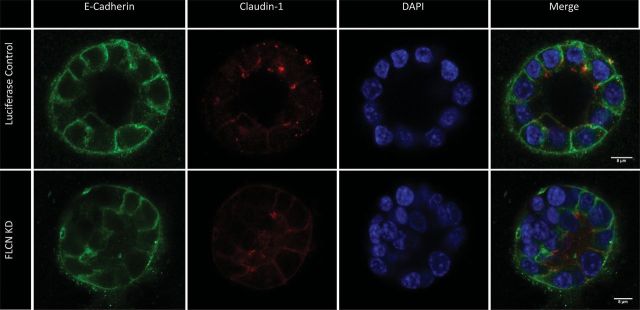
To investigate the functional impact of the interaction between FLCN and p0071 at cell junctions, we used the polarized, renal epithelial cell line mouse inner medullary collecting duct-3 (IMCD-3), cultured on Transwell permeable supports. siRNA oligonucleotides designed for p0071 and FLCN, produced 55 and 46% transient knockdown, respectively, at day 4, tested by quantitative real-time polymerase chain reaction (PCR) (Fig. 7A). Trans-epithelial electrical resistance (TEER) experiments were carried out at day 8 to test whether the knockdown of FLCN or p0071 affected the integrity of the cell monolayer. Cells treated with either FLCN or p0071 siRNA showed significantly reduced TEER at day 8 (FLCN knockdown, 52.5% residual TEER P = 0.004; p0071 knockdown, 72.1% residual TEER, P = 0.005), suggesting a delay in tight junction formation (Fig. 7B). To investigate the cause of the reduction in TEER, the cells were stained for a number of cell junctional proteins. Claudin-1 (tight-junction component) staining was reduced and disordered in both FLCN and p0071 knockdown cells, as was E-cadherin (adherens junction component; Fig. 7C). However, no abnormality was detected for either knockdown in the staining pattern of ZO-1 (Supplementary Material, Fig. S1), which is located on the cytoplasmic surface of intercellular tight junctions. In addition, IMCD3 cells treated with either FLCN or luciferase (control) siRNA were grown for 4 days in 2% Geltrex (Invitrogen, UK). Cells treated with FLCN siRNA displayed a significantly larger number of incorrectly formed colonies (the absence of a lumen or multilayered structures, P < 0.0001; Fig. 8).
Figure 7.

Cell junctional defects in IMCD-3 cells with reduced FLCN or p0071. Cells were seeded onto transwell supports to induce cell polarization. (A) Knockdown of FLCN and p0071 was confirmed by quantitative real-time polymerase chain reaction (PCR). (B) There is a significantly reduced TEER in cells depleted of FLCN or p0071 at day 8 (P < 0.005). (C) There is reduced expression of both E-cadherin and claudin-1 at cell junctions in cells treated with FLCN or p0071 siRNA.
Figure 8.
FLCN knockdown disrupts cell polarization in a three-dimensional culture. IMCD3 cells treated with either FLCN or control siRNA were grown in the Geltrex matrix. The cells with FLCN knockdown did not form a normal lumen and formed multilayered structures in 49 of 100 structures (compared with 9 of 100 in the control knockdown cells; Fisher's exact test, P < 0.0001).
DISCUSSION
To further elucidate the mechanisms of folliculin tumour suppression, we undertook a yeast two-hybrid screen and identified PKP4 (p0071) as a potential folliculin interacting protein. p0071 is a member of the armadillo superfamily and is part of a subtype of armadillo proteins of which p120ctn is the prototype. p120ctn proteins include p0071, δ-catenin, ARCF (armadillo repeat protein deleted in velo-cardio-facial syndrome) and the more distantly related PKP1–3. The characteristic armadillo-repeat domain shared by p120 family proteins is crucial in the interaction of p120 proteins with classical type I and II cadherins, resulting in the stabilization of cadherins at membrane adherens junctions (23). p0071 has also been shown to bind desmosomal proteins (desmocollin 3a, plakoglobin and desmoplakin) (24,25). In addition, this family of proteins are multifunctional, also displaying cytoplasmic and nuclear localization and a signalling function as well as their well-characterized role in cell–cell contacts. Recent studies have provided insights into the range of p0071 functions and demonstrated a role for p0071 in regulating Rho signalling during cytokinesis (19). p0071 localizes at the centrosomes and cell junctions during interphase, however during mitosis, the protein localizes at the spindle poles until anaphase becoming focused at the midbody during cytokinesis (the last step of mitosis) (26). Both loss and overexpression of p0071 result in defects in cytokinesis that correlate with deregulation of RhoA signalling (through an interaction between both proteins and the Rho GEF Ect2) (19). To determine whether there was an association between FLCN and PKP4 transcript expression in renal tumours from patients with BHD syndrome, we interrogated gene expression in six BHD tumours reported previously by Klomp et al. (27). This demonstrated that PKP4 expression was significantly down-regulated in BHD renal tumours (−0.536-fold change, P = 0.007).
The interaction between folliculin and p0071 was confirmed in vitro at both exogenous and endogenous levels by co-immunoprecipitation. The smallest region covered by all clones of p0071, which were identified in the yeast two-hybrid screen is from amino acids 374 to 493, representing a section of the head domain which likely binds folliculin (early indications by deletion mapping suggest this finding to be accurate). We proceeded to define the cellular context of the folliculin/p0071 interaction by performing comprehensive immunofluorescence analysis using a number of different cell lines. When both proteins were overexpressed, co-localization was clearly observed at cell–cell contacts and at regions throughout the cytoplasm. By re-expressing FLCN in FTC-133 cells (no endogenous expression of FLCN), we observed tight co-localization of Flag-tagged FLCN and endogenous p0071 at the midbody in dividing cells. Similarly, co-localization was clearly visible at cell contacts and at the midbody in the mitotic cell, by endogenous staining of p0071 and FLCN. BiFC analysis for which folliculin and p0071 was fused to separate halves of yellow fluorescent protein (YFP) and co-expressed in Hela cells, demonstrated a folliculin/p0071 interaction at both cell junctions and also a concentrated BiFC signal between dividing cells during late telophase/cytokinesis.
Disordered cytokinesis is a frequent feature of human cancers and RhoA (and its regulators) have been demonstrated to be critical for normal cytokinesis (28). RhoA is a small GTPase which functions as a molecular switch in diverse cellular processes, including cell adhesion, migration and cell cycle progression (29,30). Previously, p0071 was shown to regulate Rho activity through a direct association with RhoA and with Ect2 (a Rho guanine-nucleotide exchange factor essential for cytokinesis) (19). We found that, in addition to folliculin co-localizing with p0071 at the midbody, folliculin deficiency was associated with the deregulation of Rho signalling and cytokinesis defects with significantly increased numbers of multinucleated cells.
Though folliculin function has not previously been linked to the cell junctions, we found that folliculin co-localized with p0071 at cell junctions. Loss of cell polarity and cell–cell adhesion is a frequent feature of epithelial cancers and, while linked to tissue invasion and metastasis, can also occur early in tumourigenesis (31). As cancer cell lines may harbour multiple genetic and epigenetic events that might disrupt epithelial polarity, we investigated the effects of folliculin inactivation on epithelial cell polarization in IMCD-3 cells which are widely used to investigate the physiology of epithelial polarization. We found that the knockdown of FLCN expression in IMCD-3 cells disrupted the ability of cells to form tight junctions and was associated with a significant reduction in trans-epithelial resistance at day 8. This observation, phenocopied the effects of knocking down p0071, consistent with a functional role for the folliculin/p0071 complex in the formation of intercellular cell junctions. Consistent with the TEER results, both FLCN and p0071 knocked down cells displayed disordered patterns of the Claudin-1 tight junction protein by immunostaining. p0071 binds classical cadherins through its armadillo repeat region and is believed to regulated cadherin stability (23). We postulated that as the adhesive activity of E-cadherin is required for the assembly of tight junctions (32,33), the disruption of Claudin-1 staining might result from abnormal E-cadherin localization. Both FLCN and p0071 knocked down cells showed an abnormal pattern of E-cadherin staining, thus suggesting that both FLCN and p0071 are required for the formation and maintenance of adherens junctions. This is interesting as loss of E-cadherin is a frequent finding in epithelial cancers and loss of the major clear cell, RCC tumour suppressor gene, Von Hippel-Lindau, is associated with the down-regulation of E-cadherin expression (34).
In the interphase cell, Rho induces actin cytoskeletal stress fibres and facilitates cell migration at both the leading edge and rear of cells (35,36). RhoA is often overexpressed in clinical cancers, and given its role in cell adhesion and migration, it has been implicated in invasion and metastasis (21). Though fibrofolliculomas are benign tumours, BHD-associated renal cancers may metastasise (2,37). We investigated the potential impact of disordered RhoA signalling associated with folliculin inactivation on the migratory ability of the metastasis-derived cancer cell line FTC-133. We found that cells not expressing folliculin migrated significantly more quickly than folliculin expressing cells in both cell scratch and Boyden chamber assays. Furthermore, the treatment of FTC-133 folliculin null cells with Y-27632 (an inhibitor of ROCK—a downstream effector of Rho) significantly reduced the migratory phenotype. Previously, the inhibition of ROCK was shown to reduce cancer cell motility in vivo (38) and our findings suggest a novel potential therapeutic approach to the treatment of metastatic BHD tumours with deregulated Rho signalling.
The investigation of rare familial forms of kidney cancer can (as exemplified by Von Hippel–Lindau disease) provide important insights into the pathogenesis of more common forms of kidney cancer (39). To date, the functions of the BHD syndrome gene product folliculin have been relatively poorly characterized. Through identifying a novel folliculin-binding partner, we have linked folliculin inactivation to loss of epithelial cell polarity and normal intercellular junction formation, disordered cytokinesis and dysregulated RhoA signalling and so provided novel insights into the mechanisms of folliculin tumour suppressor activity. Furthermore, the cellular consequences of the folliculin/p0071 complex inactivation are germane to the pathogenesis of many human cancers.
MATERIALS AND METHODS
DNA constructs, antibodies and RNA interference
Human FLCN cloned into the pFLAG-CMV vector (Sigma-Aldrich, St Louis, MO, USA) has been described previously (40). p0071 cloned into pDSRed-N1 has been described previously (19).
For BiFC analysis, the p0071 vectors [p.Ven1-Flag-p0071 wild-type (WT), p.Ven1-Flag-p0071-Rep] have been described previously (19). FLCN was cloned into the corresponding Venus 2 plasmid to form a fusion of FLCN tagged with HA to the N-terminus of Venus 2 (p.Ven-HA-FLCN).
Monoclonal antibodies used for immunoprecipitation and western blotting include FLCN (Rabbit mAb Cell Signalling), p0071 (Mouse mAb, Progen Biotechnik, Heidelberg, Germany), RhoA (Mouse mAb, Abcam, Cambridge, UK), Flag (monoclonal ANTI-FLAG M2, Sigma-Aldrich) and β-actin (mouse monoclonal, Sigma-Aldrich).
Antibodies used for immunofluorescence microscopy analysis include FLCN (Rabbit mAb Cell Signalling), p0071 (Guinea Pig antibody), Flag (M2, Sigma-Aldrich), HA (rabbit polyclonal, Rockland) and α-tubulin (mouse monoclonal, Sigma-Aldrich)]. Rabbit polyclonal antibodies against ZO-1 and claudin-1 and a mouse monoclonal antibody against E-cadherin were from Invitrogen.
siRNA sequences were purchased from Applied Biosystems. The human FLCN siRNA sequence was GGAAGGUGGCAUUCAGAUG. The mouse FLCN siRNA sequence was CCUCAGCAAGUAUGAG, and mouse p0071 sequence was CAGACAGCAUUGUAUCGC.
Yeast two-hybrid screen
The yeast two-hybrid assay was conducted at the DFKZ Cancer Research Centre, using a human foetal brain library as a source of prey.
Cell lines and culture conditions
The thyroid carcinoma cell line FTC-133 harbours an FLCN inactivating mutation and does not express WT FLCN and so was used to determine the effects of folliculin in an isogenic background. FTC-133 cells stably transfected with FLCN or empty vector have been described previously (41). FTC-133 cells were maintained in 50% Dulbecco's modified Eagle's medium and 50% Ham's F12, supplemented with 0.5 µg/ml G418 solution (PAA, Pasching, Austria). To confirm the effects of folliculin deficiency in another cell line, we used ACHN renal carcinoma cells with stable knockdown of FLCN (by shRNA) and a stable, scrambled control that were kindly donated by Professor Arnim Pause (McGill University, Montreal, Canada) and have been described in Hudon et al. (15). These cells were grown in 100% Dulbecco's modified Eagle medium (DMEM) supplemented with 1 µg/ml puromycin. Three cell lines (MCF-7, HaCat and Hela cells) that were most appropriate for high-resolution imaging studies were investigated. IMCD-3 cells were used to investigate the effects of folliculin on cell junction formation as the other cell lines are derived from cancers and may harbour multiple mutations in cell adhesion protein genes. MCF-7, HaCat and Hela cells were grown in 100% DMEM (PAA) and Hek293 and IMCD-3 in 100% DMEM, all without selective antibiotic. IMCD-3 cells were plated and grown on 0.4 µm-pore Transwell permeable supports (Corning), at 1 × 105 cells per well to allow polarization of cells.
All cell culture reagents were purchased from Sigma-Aldrich unless otherwise stated.
Protein extraction
Cells were grown in either 100 mm dishes or 6-well plates, washed with phosphate buffered saline and scraped into cell lysis buffer [Tris, pH 7.4, NaCl 150 mm, ethylenediaminetetraacetic acid (EDTA) 0.5 mm, 1% Triton] containing protease inhibitors (Roche Applied Sciences). Lysates were centrifuged (at 13 300 × g, 4°C for 20 min) and levels of total cellular protein determined using the DC protein assay kit according to the manufacturer's instructions (Bio-Rad Laboratories, Ltd).
Co-immunoprecipitation
For co-immunoprecipitation, 4 µg of antibody was covalently conjugated to 20 µl of Dynabeads Protein G (Invitrogen) using triethanolamine and dimethyl pilimidate (except in the case of anti-FLCN in which case 0.5 µg was used). For semi-exogenous, co-immunoprecipitation 200 µg and endogenous co-immunoprecipitation 250 µg of the total cellular protein was incubated with 20 µl of antibody-bound Dynabeads at 4°C for 3 h. The complexes were washed three times with lysis buffer [Tris, pH 7.4, NaCl 300 mm, EDTA 0.5mm, 1% Triton X-100, protease inhibitor cocktail (Roche)]. Pulled down proteins were eluted by boiling in 2× sample buffer, Laemmi (Sigma-Aldrich) and loaded onto 8% SDS–PAGE gels.
Protein blotting
For immunoblotting, eluted protein (co-immunoprecipitate) or 15–25 µg of the extracted total cellular protein were separated using 8% SDS–PAGE gels (for p0071 and FLCN) or 12.5% gels for all other proteins. For primary antibody details, see above. Secondary antibodies were purchased from Dako, and signal detected using the enhanced chemiluminescence (ECL, Amersham) western blot analysis system.
Immunofluorescence microscopy analysis
Primary antibodies used for immunofluorescence are given above. Cells were cultured on poly-l-lysine-coated glass coverslips, fixed with 3.7% formaldehyde at 4°C and permeablized with 0.5% Triton X-100. Secondary antibodies were Cy-3-coupled anti-rabbit, anti-mouse and anti-guinea pig (The Jackson Laboratory), Dylight 649 anti-mouse and anti-rabbit, Dylight 488 anti-mouse and anti-guinea Pig (Dianova). DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen-Molecular probes). Images were taken with either a Nikon Eclipse 600 or a Zeiss, Axio Observer .Z1 microscope with ApoTome.
BiFC analysis
Hela cells were co-transfected with pVen1-Flag-p0071 and p.Ven2-HA-FLCN using a calcium precipitation transfection procedure. Twenty-four hours after transfection, yellow fluorescence of the cells (indicative of interaction between the proteins and reconstitution of YFP) was detected and imaged using a Nikon Eclipse 600 microscope. Cells were also stained for HA (using anti HA-rabbit) and Flag (anti-Flag mouse) to identify co-transfected cells.
G-LISA RhoA activation assay
To measure the levels of GTP-bound (active) RhoA, the G-LISA activation assay was used (Cytoskeleton, Inc.). The assay uses a 96-well plate, with a specific Rho-GTP-binding protein linked to each well. Active RhoA in cell lysates bind to the well of the G-LISA plate, while the inactive RhoA (GDP bound) is removed by washing. The bound active RhoA is then detected with a RhoA-specific antibody and a horseradish peroxidase conjugated secondary antibody. After the addition of a detection reagent, the levels of RhoA activity were measured using a Victor X3 Multilabel plate reader (PerkinElmer) detecting absorbance at 490 nm.
Cell migration assays
Cell migration was assayed using in vitro cell-scratch migration assays and transwell migration assays. Briefly, folliculin expressing FTC-133 cells and FTC-133 cells expressing an empty vector were grown to full confluency in 6-well plates. Cells were starved of serum overnight and a scratch introduced using a p200 pipette tip (and also a perpendicular scratch for reference). Cells were treated with either 10 µm Y-27632 (Rock inhibitor) or distilled water control (media changed every 12 h). Images of the scratch were captured at 0 h and every 24 h until the scratch was healed. The percentage of scratch healed was determined from the images using ImageJ software (http://rsb.info.nih.gov/ij/). Experiments were carried out in triplicate.
Effects of folliculin expression and Y-27632 treatment on chemotaxis were tested using the CytoSelect™ 24-Well Cell Migration Assay kit from Cell Biolabs according to the manufacturer's protocol. FTC-133 cells expressing folliculin or an empty vector (6 × 104 per assay), and treated with either 10 µm Y-27632 or ddH2O, were serum starved for 2 h and then seeded, in serum-free media, into an upper chamber, and were left for 18 h to migrate towards serum-positive media (10% FBS) in the lower chamber. Migratory abilities were quantified with a Perkin Elmer VICTOR™ X3 fluorescence plate reader at 480/520 nm.
Real-time quantitative PCR analysis
Quantitative TaqMan PCR was used to confirm FLCN and p0071 knockdown in IMCD3 cells. mRNA was extracted from cell pellets using the TRIzol Plus RNA Purification Kit (Ambion), and cDNA synthesized. TaqMan gene expression assays for murine p0071 and FLCN were purchased from Applied Biosystems.
Trans-epithelial resistance assay and investigation of cell polarization defects
IMCD3 cells were treated with either p0071, FLCN or Luciferase siRNA and seeded onto Transwell supports. TEER was measured after 8 days using an EVOM AC square wave current resistance meter (World Precision Instruments).
At day 8, filters were mounted between coverslips and fixed with 4% formaldehyde. Cells were stained for E-cadherin, claudin-1 and ZO-1 to assess junctional formation and were visualized using a Leica SP5 Laser Scanning Confocal Microscope.
For three-dimensional culture of cells, IMCD3 cells were treated with siRNA to either FLCN or luciferase (control) and were grown in 2% Geltrex (Invitrogen) for 4 days at 37°C. Cells were then fixed and stained for E-cadherin and claudin-1 and were visualized by Confocal microscope. One hundred cell colonies were counted for both control and FLCN knockdown with the absence of a lumen and multilayered structures noted, and significance tested by Fisher's exact test.
Statistical methods
Error bars displayed represent the standard deviation. Statistical significance was determined by comparing data from at least three independent experiments, using the Student's t-test unless otherwise stated. Probability values are located in corresponding figure legends.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by the Myrovlytis Trust.
REFERENCES
- 1.Menko F.H., van Steensel M.A., Giraud S., Friis-Hansen L., Richard S., Ungari S., Nordenskjold M., Hansen T.V., Solly J., Maher E.R. Birt-Hogg-Dube syndrome: diagnosis and management. Lancet Oncol. 2009;10:1199–1206. doi: 10.1016/S1470-2045(09)70188-3. [DOI] [PubMed] [Google Scholar]
- 2.Toro J.R., Wei M.H., Glenn G.M., Weinreich M., Toure O., Vocke C., Turner M., Choyke P., Merino M.J., Pinto P.A., et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dube syndrome: a new series of 50 families and a review of published reports. J. Med. Genet. 2008;45:321–331. doi: 10.1136/jmg.2007.054304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nahorski M.S., Lim D.H., Martin L., Gille J.J., McKay K., Rehal P.K., Ploeger H.M., van Steensel M., Tomlinson I.P., Latif F., et al. Investigation of the Birt-Hogg-Dube tumour suppressor gene (FLCN) in familial and sporadic colorectal cancer. J. Med. Genet. 2010;47:385–390. doi: 10.1136/jmg.2009.073304. [DOI] [PubMed] [Google Scholar]
- 4.Rongioletti F., Hazini R., Gianotti G., Rebora A. Fibrofolliculomas, tricodiscomas and acrochordons (Birt-Hogg-Dube) associated with intestinal polyposis. Clin. Exp. Dermatol. 1989;14:72–74. doi: 10.1111/j.1365-2230.1989.tb00890.x. [DOI] [PubMed] [Google Scholar]
- 5.Khoo S.K., Giraud S., Kahnoski K., Chen J., Motorna O., Nickolov R., Binet O., Lambert D., Friedel J., Levy R., et al. Clinical and genetic studies of Birt-Hogg-Dube syndrome. J. Med. Genet. 2002;39:906–912. doi: 10.1136/jmg.39.12.906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nickerson M.L., Warren M.B., Toro J.R., Matrosova V., Glenn G., Turner M.L., Duray P., Merino M., Choyke P., Pavlovich C.P., et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. 2002;2:157–164. doi: 10.1016/s1535-6108(02)00104-6. [DOI] [PubMed] [Google Scholar]
- 7.Vocke C.D., Yang Y., Pavlovich C.P., Schmidt L.S., Nickerson M.L., Torres-Cabala C.A., Merino M.J., Walther M.M., Zbar B., Linehan W.M. High frequency of somatic frameshift BHD gene mutations in Birt-Hogg-Dube-associated renal tumors. J. Natl Cancer Inst. 2005;97:931–935. doi: 10.1093/jnci/dji154. [DOI] [PubMed] [Google Scholar]
- 8.Woodward E.R., Ricketts C., Killick P., Gad S., Morris M.R., Kavalier F., Hodgson S.V., Giraud S., Bressac-de Paillerets B., Chapman C., et al. Familial non-VHL clear cell (conventional) renal cell carcinoma: clinical features, segregation analysis, and mutation analysis of FLCN. Clin. Cancer Res. 2008;14:5925–5930. doi: 10.1158/1078-0432.CCR-08-0608. [DOI] [PubMed] [Google Scholar]
- 9.Painter J.N., Tapanainen H., Somer M., Tukiainen P., Aittomaki K. A 4-bp deletion in the Birt-Hogg-Dube gene (FLCN) causes dominantly inherited spontaneous pneumothorax. Am. J. Hum. Genet. 2005;76:522–527. doi: 10.1086/428455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baba M., Hong S.B., Sharma N., Warren M.B., Nickerson M.L., Iwamatsu A., Esposito D., Gillette W.K., Hopkins R.F., 3rd, Hartley J.L., et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc. Natl Acad. Sci. USA. 2006;103:15552–15557. doi: 10.1073/pnas.0603781103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hasumi H., Baba M., Hong S.B., Hasumi Y., Huang Y., Yao M., Valera V.A., Linehan W.M., Schmidt L.S. Identification and characterization of a novel folliculin-interacting protein FNIP2. Gene. 2008;415:60–67. doi: 10.1016/j.gene.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J., Futami K., Petillo D., Peng J., Wang P., Knol J., Li Y., Khoo S.K., Huang D., Qian C.N., et al. Deficiency of FLCN in mouse kidney led to development of polycystic kidneys and renal neoplasia. PLoS one. 2008;3:e3581. doi: 10.1371/journal.pone.0003581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baba M., Furihata M., Hong S.B., Tessarollo L., Haines D.C., Southon E., Patel V., Igarashi P., Alvord W.G., Leighty R., et al. Kidney-targeted Birt-Hogg-Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J. Natl Cancer Inst. 2008;100:140–154. doi: 10.1093/jnci/djm288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Slegtenhorst M., Khabibullin D., Hartman T.R., Nicolas E., Kruger W.D., Henske E.P. The Birt-Hogg-Dube and tuberous sclerosis complex homologs have opposing roles in amino acid homeostasis in Schizosaccharomyces pombe. J. Biol. Chem. 2007;282:24583–24590. doi: 10.1074/jbc.M700857200. [DOI] [PubMed] [Google Scholar]
- 15.Hudon V., Sabourin S., Dydensborg A.B., Kottis V., Ghazi A., Paquet M., Crosby K., Pomerleau V., Uetani N., Pause A. Renal tumour suppressor function of the Birt-Hogg-Dube syndrome gene product folliculin. J. Med. Genet. 2010;47:182–189. doi: 10.1136/jmg.2009.072009. [DOI] [PubMed] [Google Scholar]
- 16.Hong S.B., Oh H., Valera V.A., Stull J., Ngo D.T., Baba M., Merino M.J., Linehan W.M., Schmidt L.S. Tumor suppressor FLCN inhibits tumorigenesis of a FLCN-null renal cancer cell line and regulates expression of key molecules in TGF-beta signaling. Mol. Cancer. 2010;9:160. doi: 10.1186/1476-4598-9-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cash T.P., Gruber J.J., Hartman T.R., Henske E.P., Simon M.C. Loss of the Birt-Hogg-Dube tumor suppressor results in apoptotic resistance due to aberrant TGFbeta-mediated transcription. Oncogene. 2011;30:2534–2546. doi: 10.1038/onc.2010.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Preston R.S., Philp A., Claessens T., Gijezen L., Dydensborg A.B., Dunlop E.A., Harper K.T., Brinkhuizen T., Menko F.H., Davies D.M., et al. Absence of the Birt-Hogg-Dube gene product is associated with increased hypoxia-inducible factor transcriptional activity and a loss of metabolic flexibility. Oncogene. 2011;30:1159–1173. doi: 10.1038/onc.2010.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolf A., Keil R., Gotzl O., Mun A., Schwarze K., Lederer M., Huttelmaier S., Hatzfeld M. The armadillo protein p0071 regulates Rho signalling during cytokinesis. Nat. Cell Biol. 2006;8:1432–1440. doi: 10.1038/ncb1504. [DOI] [PubMed] [Google Scholar]
- 20.Chan C.H., Lee S.W., Li C.F., Wang J., Yang W.L., Wu C.Y., Wu J., Nakayama K.I., Kang H.Y., Huang H.Y., et al. Deciphering the transcriptional complex critical for RhoA gene expression and cancer metastasis. Nat. Cell Biol. 2010;12:457–467. doi: 10.1038/ncb2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Narumiya S., Tanji M., Ishizaki T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 2009;28:65–76. doi: 10.1007/s10555-008-9170-7. [DOI] [PubMed] [Google Scholar]
- 22.Liu S., Goldstein R.H., Scepansky E.M., Rosenblatt M. Inhibition of rho-associated kinase signaling prevents breast cancer metastasis to human bone. Cancer Res. 2009;69:8742–8751. doi: 10.1158/0008-5472.CAN-09-1541. [DOI] [PubMed] [Google Scholar]
- 23.Hatzfeld M. The p120 family of cell adhesion molecules. Eur. J. Cell Biol. 2005;84:205–214. doi: 10.1016/j.ejcb.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 24.Hatzfeld M., Green K.J., Sauter H. Targeting of p0071 to desmosomes and adherens junctions is mediated by different protein domains. J. Cell Sci. 2003;116:1219–1233. doi: 10.1242/jcs.00275. [DOI] [PubMed] [Google Scholar]
- 25.Hatzfeld M., Nachtsheim C. Cloning and characterization of a new armadillo family member, p0071, associated with the junctional plaque: evidence for a subfamily of closely related proteins. J. Cell Sci., 1996;109(Pt 11):2767–2778. doi: 10.1242/jcs.109.11.2767. [DOI] [PubMed] [Google Scholar]
- 26.Keil R., Wolf A., Huttelmaier S., Hatzfeld M. Beyond regulation of cell adhesion: local control of RhoA at the cleavage furrow by the p0071 catenin. Cell Cycle. 2007;6:122–127. doi: 10.4161/cc.6.2.3741. [DOI] [PubMed] [Google Scholar]
- 27.Klomp J.A., Petillo D., Niemi N.M., Dykema K.J., Chen J., Yang X.J., Saaf A., Zickert P., Aly M., Bergerheim U., et al. Birt-Hogg-Dube renal tumors are genetically distinct from other renal neoplasias and are associated with up-regulation of mitochondrial gene expression. BMC Med. Genomics. 2010;3:59. doi: 10.1186/1755-8794-3-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piekny A., Werner M., Glotzer M. Cytokinesis: welcome to the Rho zone. Trends Cell Biol. 2005;15:651–658. doi: 10.1016/j.tcb.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 29.Jaffe A.B., Hall A. Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 30.Burridge K., Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 31.Tervonen T.A., Partanen J.I., Saarikoski S.T., Myllynen M., Marques E., Paasonen K., Moilanen A., Wohlfahrt G., Kovanen P.E., Klefstrom J. Faulty epithelial polarity genes and cancer. Adv. Cancer Res. 2011;111:97–161. doi: 10.1016/B978-0-12-385524-4.00003-9. [DOI] [PubMed] [Google Scholar]
- 32.Watabe M., Nagafuchi A., Tsukita S., Takeichi M. Induction of polarized cell-cell association and retardation of growth by activation of the E-cadherin-catenin adhesion system in a dispersed carcinoma line. J. Cell Biol. 1994;127:247–256. doi: 10.1083/jcb.127.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tunggal J.A., Helfrich I., Schmitz A., Schwarz H., Gunzel D., Fromm M., Kemler R., Krieg T., Niessen C.M. E-cadherin is essential for in vivo epidermal barrier function by regulating tight junctions. EMBO J. 2005;24:1146–1156. doi: 10.1038/sj.emboj.7600605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Evans A.J., Russell R.C., Roche O., Burry T.N., Fish J.E., Chow V.W., Kim W.Y., Saravanan A., Maynard M.A., Gervais M.L., et al. VHL promotes E2 box-dependent E-cadherin transcription by HIF-mediated regulation of SIP1 and snail. Mol. Cell Biol. 2007;27:157–169. doi: 10.1128/MCB.00892-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Timpson P., McGhee E.J., Morton J.P., von Kriegsheim A., Schwarz J.P., Karim S.A., Doyle B., Quinn J.A., Carragher N.O., Edward M., et al. Spatial regulation of RhoA activity during pancreatic cancer cell invasion driven by mutant p53. Cancer Res. 2011;71:747–757. doi: 10.1158/0008-5472.CAN-10-2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Machacek M., Hodgson L., Welch C., Elliott H., Pertz O., Nalbant P., Abell A., Johnson G.L., Hahn K.M., Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461:99–103. doi: 10.1038/nature08242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pavlovich C.P., Grubb R.L., Hurley K., Glenn G.M., Toro J., Schmidt L.S., Torres-Cabala C., Merino M.J., Zbar B., Choyke P., et al. Evaluation and management of renal tumors in the Birt-Hogg-Dube syndrome. J. Urol. 2005;173:1482–1486. doi: 10.1097/01.ju.0000154629.45832.30. [DOI] [PubMed] [Google Scholar]
- 38.Wyckoff J.B., Pinner S.E., Gschmeissner S., Condeelis J.S., Sahai E. ROCK- and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr. Biol. 2006;16:1515–1523. doi: 10.1016/j.cub.2006.05.065. [DOI] [PubMed] [Google Scholar]
- 39.Maxwell P.H., Wiesener M.S., Chang G.W., Clifford S.C., Vaux E.C., Cockman M.E., Wykoff C.C., Pugh C.W., Maher E.R., Ratcliffe P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 40.Nahorski M.S., Reiman A., Lim D.H., Nookala R.K., Seabra L., Lu X., Fenton J., Boora U., Nordenskjold M., Latif F., et al. Birt Hogg-Dube syndrome associated FLCN mutations disrupt protein stability. Hum. Mutat. 2011 doi: 10.1002/humu.21519. [DOI] [PubMed] [Google Scholar]
- 41.Lu X., Wei W., Fenton J., Nahorski M.S., Rabai E., Reiman A., Seabra L., Nagy Z., Latif F., Maher E.R. Therapeutic targeting the loss of the birt-hogg-dube suppressor gene. Mol. Cancer Ther. 2011;10:80–89. doi: 10.1158/1535-7163.MCT-10-0628. [DOI] [PubMed] [Google Scholar]