

Abstract
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disease characterized by a reciprocal translocation between long arms of chromosomes 9 and 22 t(9;22) that generates the BCR-ABL fusion gene. If left untreated, newly diagnosed chronic phase CML patients finally progress to accelerated and blastic phase. After the introduction of tyrosine kinase inhibitors (TKIs), treatment strategies of CML changed dramatically. However, the development of resistance to TKIs started to create problems over time. In this review, the current information about CML biology before and after imatinib mesylate treatment is summarized.
Keywords: Chronic myeloid leukemia, moleculer biology, imatinib mesylate
Introduction
Chronic myeloid leukemia (CML) is a malignant myeloproliferative clonal disorder of haematopoietic stem cells resulting from a translocation between chromosomes 9 and 22 t(9;22)(q34;q11) or its variants t(V;9;22) [1]. This reciprocal translocation generates the shortened 22q known as the Philadelphia (Ph) chromosome and the new fusion oncogene is called as BCR-ABL (Breakpoint Cluster Region-Abelson Leukaemia). This oncogene encodes a chimeric 210 kD Bcr-Abl protein that incorporates an activated Abl tyrosine kinase domain, which is a well documented underlying reason for the malignant transformation in CML [1-3]. CML accounts for 15-20% of the newly diagnosed cases of adult leukemias. Most of the CML patients have been diagnosed in chronic phase (CP), but as a result of genomic instability, it progresses to ill-defined unstable accelerated phase (AP) and then to the terminal blastic crisis phase (BP) over time, becoming increasingly resistant to therapy [4].
Implementation of tyrosine kinase inhibitor therapy
Before 2000s, CML has been treated with hydroxyurea and interferon therapy that provide temporary disease control but do not alter progression to advanced disease with a median survival ranging 45-55 months from diagnosis [4]. Therefore, the most effective treatment strategy was allogeneic stem cell transplantation (ASCT). The recognition of the BCR-ABL oncogene and the corresponding protein led to the synthesis of small-molecule drugs, designed to interfere with BCR-ABL tyrosine kinase activation [5]. After the introduction of targeted treatment with tyrosine kinase inhibitors (TKIs), treatment strategies and outcomes were changed dramatically. Imatinib is the first used, generally well tolerated TKI that targets the tyrosine kinase activity of Bcr-Abl in CP-CM [6]. Imatinib was introduced into clinical practice in 1998. Imatinib is an ABL tyrosine kinase inhibitor of the 2‑phenylamino pyrimidine class blocking the inactive conformation of BCR-ABL protein. This prevents the transfer of phosphate group from adenosine triphosphate (ATP) to substrates, and blocks the downstream signal transduction pathways. Thus, imatinib voids the inhibition of proliferation and induction of apoptosis [7]. Imatinib became the first choice for the treatment of CP-CML because of its high efficacy, low toxicity and ability to maintain durable hematological and cytogenetic responses. In the International Randomized Interferon versus STI571 (IRIS) trial, the 8-year follow-up data revealed an estimated overall survival of 85% for imatinib suggesting a high and persistent efficacy of this TKI in CML-CP [8]. Despite high rates of hematologic and cytogenetic responses, primary refractory disease and drug resistance have been observed in 25% of patients with imatinib monotherapy, which is often caused by the domain mutations of BCR-ABL kinase, that prevent imatinib binding [4].
Current definitions of biology of chronic myeloid leukemia after imatinib
Imatinib has been shown to induce a complete haematologic response in CP-CML patients [9]. However, imatinib has been unable to completely eliminate BCR-ABL-expressing leukemic cells [10,11]. It has been shown that imatinib prolongs survival of mice with BCR-ABL-induced CML, but does not cure the disease [12]. Shortly after the introduction of imatinib, the impressive success of the drug as afront-line therapy in CML has been tempered by problems such as disease persistence or relapse arising from different mechanisms, including duplications, mutations in the kinase domain of the BCR-ABL protein and mechanisms independent from BCR-ABL activity. Growing evidence has also suggested a pivotal role of persistent leukemic cancer stem cells, characterized by high self-renewal and pluripotency, in CML maintenance and/or relapse [13]. Stem cells may escape imatinib-mediated apoptosis due to the inability of imatinib to attack quiescent stem cells [10]. Although the heterogeneous development of imatinib resistance is challenging, the fact that BCR-ABL is active in many resistant patients suggests that the chimeric oncoprotein remains as a good therapeutic target. However, patients with clonal evolution are more likely to have BCR-ABL-independent mechanisms of resistance [14]. Imatinib-resistance mechanisms are shown in Table 1.
Table 1.
Imatinib-resistance mechanisms
| Kinase domain mutations | Mutation-independent | Duplication | Other targets |
|---|---|---|---|
| ● T315I | ● PI3K/Akt | ● Amplification in Abl sequence | ● p53 |
| ● P-Loop | ● MDR-1, Pgp | ● bcl-2 | |
| - M244V | ● HIF-1α | ● JAK-2/STAT-5 | |
| - G250E | ● Aberrant ceramide metabolism | ● OCT-1 | |
| - Y253F/H | |||
| - E255K/V | |||
| ● M351T | |||
| ● F359V | |||
| ● SH2, SH3 | |||
| ● Cap |
Duplications
The development of imatinib-resistance was firstly described in 2000 through BCR/ABL oncogene amplification [15]. Mahon, Weisberg and le Coutre demonstrated an amplification in the Abl sequence in vitro by generating imatinib-resistant cell lines using BCR-ABL-transformed murine hematopoietic cells and BCR-ABL-positive human cell lines [16-18]. The same groups identified elevated Abl kinase activity due toa several-fold increase in the amount of BCR-ABL protein, but the value of these data was limited since these results were obtained in vitro.
Mutations
Mutations in the tyrosine kinase inhibitor binding site of BCR/ABL is another important mechanism of drug resistance. The kinase domain mutation frequency is 23% in naive CML patients [19]. 40-60% of CML patients under prolonged imatinib exposure that have clinical resistance, harbour BCR-ABL kinase domain mutations [20]. Point mutations were observed in approximately 35-70% of patients displaying imatinib resistance, either spontaneously or through the evolutionary pressure of imatinib [21]. If mutations are detected prior toor early after administration of imatinib treatment, they generally predict theclinical imatinib-resistance and progression. Soverini et al reported the frequency of mutations according to disease phase at the time of diagnosis and they found that 52% of patients with AP, 75% of BP and 27% of CP CML patients had mutations. Thus, mutational frequencies appear to increase in imatinib resistance and progress from CP to BP [15]. Mutations were observed in the critical contact points of imatinib to BCR-ABL or the most relevant biological consequence is theincapabilitiy of imatinib to inhibit the kinase activity of BCR-ABL due to prevention of BCR-ABL from adopting the inactive conformation. These mutations may also lead to disturbed function of BCR-ABL that would lead to death of the cell and would not be detectable, resulting in restoration of the BCR-ABL function and clonal selection of mutated cells resulting in reduced kinase activity. This is sufficient to allow cellular survival with imatinib-resistance and mutations of the activation loop which may result in an activated conformation that is insensitive to inhibition by imatinib [14,22,23].
Hochaus and Shindler et al firstly demonstrated four regions that clustered to acquired mutations which lead to substitutions of amino acids that are important for specific binding of imatinib. These mutations were P-loop, a highly conserved region responsible for phosphate binding; T315, a non-conserved residue that is in part responsible for the selective inhibition of ABL by imatinib; and M351 and E355, mutations of the activation loop, resulting in an activated conformation of ABL insensitive to imatinib [7,14]. Soverini et al demonstrated that 85% of all imatinib-resistant mutations are associated with amino acid substitutions at seven residues (P-loop: M244V, G250E, Y253F/H and E255K/V; contact site: T315I; and catalytic domain: M351T and F359V) [22]. In recent papers, >90 different amino acid substutions were identified in imatinib-resistant patients as shown in Figure 1 [24].
Figure 1.
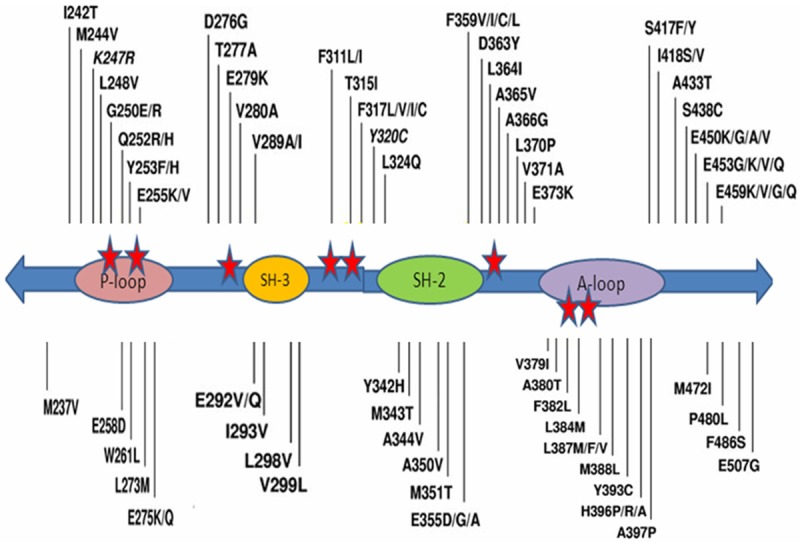
Map of all the amino acid substitutions in the Bcr-Abl kinase domain identified in clinical samples from patients reported to be resistant to imatinib in published papers [24].
The most frequently observed mutation identified in imatinib-resistant CML patients is, T315I mutation [25]. T315I mutation is a single C→T nucleotide substitution at position 944 of the Abl gene, resulting in a threonine to isoleucine substitution at amino acid 315 (Th315→Ile315; T315I) in the Bcr/Abl protein [26]. It was shown that the T315I mutation is associated with poor prognosis by increasing oncogenicity and promoting progression, or related to the pleiotropic resistance to TKI, in several studies [27-29].
In the GIMEMA study, P-loop mutations were found in 43% of patients. The frequency of P-loop mutations clearly increases in AP and BC as well as with disease duration. Therefore, patients with CML in these phases tend to develop imatinib-resistance mutations. P-loop mutations in Y253F and E255K exhibited an increased transformation potency that correlates with intrinsic BCR-ABL kinase activity [30]. P-loop mutations were reported as detectable 2,8 months before the development of resistance in patients having imatinib treatment as compared to 6,3 months for T315I mutations and 10,8 months for M351T mutations [31]. P-Loop mutations have also been suggested to cause worse outcome in terms of time to progression and inferior overall survival [20,22]. Thus, earlier detection of the P-loop mutations may provide clinical benefit for patients by earlier reconsideration of the therapeutic interventions [32]. Mutational frequencies appear to increase the progress from CP to BP in several studies [15,22]. Hochhaus et al did not find any difference in the setting of hematologic resistance between patients with and without a mutation regarding the time to progression [14].
Capdeville et al reported that 40% of late CP CML patients fail to reach a major cytogenetic response (MCgR) on imatinib therapy, and these patients experienced the disease progression to BC more quickly than those who obtain cytogenetic remission [33]. This poor prognosis was also confirmed in terms of survival [22].
In vitro studies suggested that different mutations confer different degrees of resistance to imatinib [34]. Although some mutations like T315I confer a true resistant phenotype, thereby suggesting withdrawal of imatinib in favor of alternative treatment options, others (ie, M351T) might be overcome by dose escalation.
Mutation-independent resistance to imatinib
Imatinib-resistance arises very rarely in patients that are treated with imatinib in early CP-CML. This implies that BCR-ABL independent factors such as the cellular context of BCR-ABL expression and stage of the disease decisively control the evolution of imatinib-resistance [20].
The Bcr-abl protein is composed of many domains. The Abl sequences encode a tyrosine kinase domain as well as Src-homology (SH3 and SH2) domains, a DNA binding domain, nuclear localisation signals and a nuclear export signal. The BCR/ABL fusion protein acts as an oncoprotein by activating several signaling pathways that lead to transformation. Myc, Ras, c-Raf, MAP/ERK, SAPK/JNK, STAT, nucleer factor kappa-B (NF-κB), phosphoinositole 3-kinase (PI-3K) and c-Jun are included as signal cascade molecules regulated by the Bcr/Abl activity [35]. It has also been shown in vitro that mutations outside the kinase domain in the neighbouring linker, SH2, SH3, and Cap domains can confer imatinib-resistance. In the context of ABL, these domains have an autoinhibitory effect on the the kinase activity and mutations in this region can activate the enzyme. BCR-ABL independent survival in the presence of IM reflects the inherent/innate or acquired features of BCR-ABL+ leukemic populations. Whereas acquired, the BCR-ABL independent resistance refers to signalling pathways that are activated in response to imatinib exposure [22]. Some additional mutations in substrate binding site, C-helix, A-loop, and C-terminal lobe have been characterized throughout the Abl sequence [22,30,36,37].
Burchet et al also demonstrated a heterogeneous Akt-signaling-cascade activation during the manifest of imatinib-resistance independently from BCR/ABL-kinase mutations [38]. Activation of the PI-3K/Akt pathway is crucial for survival and proliferation of leukemic cells in CML [39]. Imatinib treatment activated the PI-3K/Akt/mammalian target of rapamycin (mTOR) pathway in BCR/ABL positive LAMA cells in a CP CML patient in vivo. In fact, the PI-3K/Akt activation critically mediated survival during the early phase of imatinib-resistance development before the manifestation of BCR/ABL dependent strong imatinib resistance such as through a kinase mutation [38]. Activation of the PI-3K/mTOR signaling increased the formation of reactive oxygen species and thereby contributing to BCR/ABL transformation. Thus, selective mTOR pathway activation may translate into a differential effectiveness of mTOR inhibitors in overcoming the imatinib-resistance [38].
Expression of the MDR-1 gene that encodes themultidrug resistance protein, P-glycoprotein (Pgp), was implicated in the mechanism of drug resistance in most chemotherapies. Mahon et al demonstrated that reduced imatinib intakeis also mediated by theoverexpression of the Pgp efflux pump in cells derived from a BP-CML patients [16].
Zhao et al demonstrated that when BCR-ABL transformed cells were selected for imatinib-resistance in vitro, the cells that grew out displayed higher BCR-ABL expression comparable to an increase observed in accelerated forms of the disease. BCR-ABL transformation is associated with cell-autonomous proliferation. This enhanced expression of BCR-ABL was associated with an increased rate of glycolysis but a decreased rate of proliferation. The higher levels of BCR/ABL expression in the selected cells correlated with a non-hypoxic induction of HIF-1α that was required for cells to tolerate enhanced BCR/ABL signaling. HIF-1α induction resulted in an enhanced rate of glycolysis but reduced glucose flux through both the TCA cycle and the oxidative arm of the pentose phosphate pathway (PPP). The reduction in oxidative PPP mediated ribose synthesis was compensated by the HIF-1α-dependent activation of the non-oxidative PPP enzyme, transketolase, in imatinib-resistant CML cells. The increased glycolysis and decreased cell proliferation in resistant cells were both found to depend on the non-hypoxic activation of HIF-1α [40].
Other target pathways
Kinase domain mutations constitute 30-50% of thereason for imatinib-resistance. There are different molecular determinants that contribute to the sensitivity and resistance of tumour cells to imatinib-induced apoptosis. It has recently been suggested that p53 may be an important mediator of the imatinib induced apoptotic response, and, a deficiency in p53-signaling pathway antagonizes this response and mediates imatinib-resistance [41]. Increased expression of bcl-2 due to loss of ICSBP also mediates imatinib-resistance [20]. Sun et al showed that BCR/ABL independent activation of the JAK-2/STAT-5 signal transduction cascade causes imatinib-resistance due to antiapopitotic effect of this cascade [42]. JAK-2 also plays a role in cytokine induced BCR/ABL-independent imatinib-resistance [43]. The organic cation transporter-1 (OCT-1) is the major active influx pump responsible for the transport of imatinib into target BCR/ABL positive cells. Imatinib is a substrate for OCT-1, and thus alterations in the expression and function of OCT-1 have a role in imatinib-resistance in progenitors [44,45].
Bioactive sphingolipids are important second messengers regulating important celullar functions such as growth, proliferation, cell cycle, apoptosis, drug resistance, senescence and quinescence [46]. It was shown by our group that there were significant increases in expression levels of glucosyle ceramide synthase (GCS) and sphingosine kinase-1 (SK-1), converting the apoptotic ceramide to antiapoptotic glucosyle ceramide and sphingosine-1-phosphate (S1P), respectively, in imatinib resistant cells as compared to sensitive CML cells [47]. Interestingly, we have also shown that sphingosine kinase-1 regulates the expression levels of BCR/ABL and its protein stability through S1P receptor-2 (S1PR-2) [48]. More importantly, we have shown that inhibition of GCS and SK-1 by molecular or biochemical approaches restored imatinib sensitivity [47-49]. Interestingly, downregulation of SK-1 and S1PR2 by siRNAs inhibited the expression levels of BCR/ABL and induced apoptosis both in wild type and mutant form of BCR/ABL positive CML cells [48]. On the other hand, imatinib by itself increased the intracellullar concentrations of apoptotic ceramide through theinduction of the ceramide synthase genes [47].
CML stem cell: is it real?
CML was the first cancer shown to be initiated at the hematopoietic stem cell (HSCs) level by BCR/ABL, and thus it is a hallmark for understanding the molecular evolution of cancer. Stem cells have some functional properties such as the capacity to become dormant, acquire multi-lineage differentiation potential, survival and self-renewal [50]. Granulocyte-macrophage progenitors have been identified as potential leukaemic stem cells for human CML myeloid blastic crisis [51,52]. The function of the BCR/ABL expressing HSCs as the stem cells in mice was tested by Hu and coworkers. When C57BL/6 (B6) bone marrow cells transduced with BCR/ABL retrovirus were sorted into two separate populations (Sca-1- or Sca-1+), only the BCR/ABLtransduced Sca-1+ cells transferred lethal CML to secondary B6 recipient mice, suggesting that early bone marrow progenitors contain CML stem cells [53]. Discontinuation of imatinib even in the rare patients with complete molecular response causes recurrence of thedisease [54]. This suggests the existence of stem cells in CML. Malignant bone marrow progenitors play an integral role in disease progression and TKI resistance in CML by reprogramming of theprogenitors [55]. Abnormal activation of thesignal transduction pathways and epigenetic events that regulate survival, differentiation and self-renewal can occur with this reprograming (Figure 2).
Figure 2.
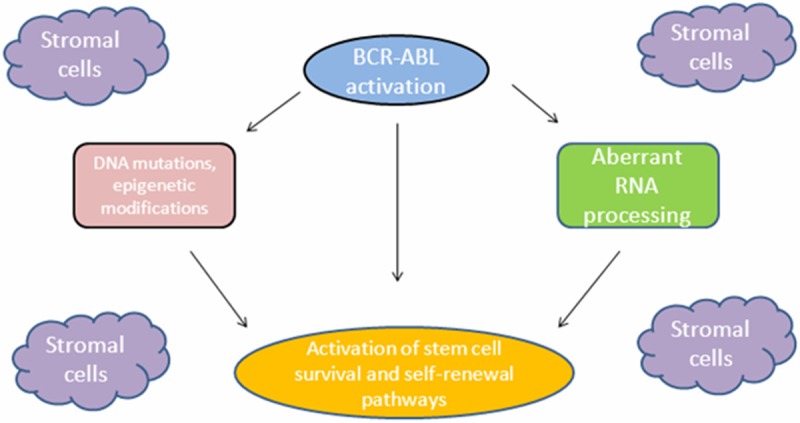
Diagram of malignant reprogramming and CML stem cell generation [51].
Strategies for overcoming CML stem cell: where are we now?
Discovery of the TKIs targeting the BCR/ABL1 kinase, revolutionized CML therapy. However, TKIs are still unable to eradicate the disease due to the presence of a drug-insensitive stem cell population that sustains continued growth of the malignant cells [56]. Although the kinase domain mutations are infrequently detected in newly diagnosed CP CML patients, these mutations were found in a substantial number of patients when the CD34+ stem cells were analyzed [57]. Corbin et al reported that human CML stem cells do not depend on BCR/ABL activity for survival and are thus less responsive to imatinib therapy and act as a reservoir for the emergence of imatinib-resistant subclones. Imatinib inhibited the BCR/ABL activity to the same degree in all stem (CD34+CD38-CD133+) and progenitor (CD34+CD38+) cells, and in quiescent and cycling progenitors from newly diagnosed CML patients [58]. This is an important step towards understanding how to approach the persistent disease with the ultimate goal of leukemic stem cell eradication as a means to achieve a cure. When imatinib therapy is discontinued in patients with undetectable BCR/ABL levels by RT-PCR, they usually experience recurrence of active leukemia. Leukemic cells persist in most patients even when the disease burden is reduced below the detectable limits. Therefore, the current recommendation is lifelong continuance of the therapy [53].
The detection of pre-existing mutations in primitive stem/progenitor (CD34+) cells may have therapeutic and prognostic implications and is likely to be helpful in optimizing the management of CML patients.
Conclusion and future perspectives
TKIs implentation is a breakthrough in the treatment of CML but the resistance is still a significant clinical problem. TKI resistance mechanisms are therefore are well investigated. Resistance resulting from CML stem cell is becoming more important over time. Therefore, the determination of the molecular mechanisms of TKI resistance can provide an important avenue for the more effective treatment of CML patients through biochemical or molecular targeting of these mechanisms that provide survival advantage for the leukemic cells.
Disclosure of conflict of interest
None of the authors have any interests which might influence the compilation of the current literature in this subject. We apologize to the authors whose valuable studies were not included here due to space limitations and the concentrated scope of the review.
References
- 1.Rowley J. A new consistent chromosomal abnormality in chronic myelogenous leukemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;43:290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 2.Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–183. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- 3.Jørgensen HG, Holyoake TL. Characterization of cancer stem cells in chronic myeloid leukaemia. Biochem Soc Trans. 2007;35:1347–1351. doi: 10.1042/BST0351347. [DOI] [PubMed] [Google Scholar]
- 4.Fausel C. Targeted chronic myeloid leukemia therapy: Seeking a cure. Am J Health Syst Pharm. 2007;64:s9–15. doi: 10.2146/ajhp070482. [DOI] [PubMed] [Google Scholar]
- 5.Gora-Tybor J, Robak T. Targeted drugs in chronic myeloid leukemia. Curr Med Chem. 2008;15:3036–3051. doi: 10.2174/092986708786848578. [DOI] [PubMed] [Google Scholar]
- 6.Manley PW, Cowan-Jacob SW, Mestan J. Advances in the structural biology, design and clinical development of Bcr-Abl kinase inhibitors for the treatment of chronic myeloid leukaemia. Biochimica et Biophysica Acta. 2005;1754:3–13. doi: 10.1016/j.bbapap.2005.07.040. [DOI] [PubMed] [Google Scholar]
- 7.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 8.Deininger M, O’Brien SG, Guilhot F, Goldman JM, Hochhaus A, Hughes TP, Radich JP, Hatfield AK, Mone M, Filian J, Reynolds J, Gathmann I, Larson RA, Druker BJ. International Randomized Study of Interferon Vs STI571 (IRIS) 8-year follow up: sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. Blood. 2009;114:462. [abstract 1126] [Google Scholar]
- 9.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 10.Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, Holyoake TL. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99:319–25. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- 11.Marley SB, Deininger MW, Davidson RJ, Goldman JM, Gordon MY. The tyrosine kinase inhibitor STI571, like interferon-alpha, preferentially reduces the capacity for amplification of granulocyte-macrophage progenitors from patients with chronic myeloid leukemia. Exp Hematol. 2000;28:551–557. doi: 10.1016/s0301-472x(00)00142-9. [DOI] [PubMed] [Google Scholar]
- 12.Li S, Li D. Stem cell and kinase activity-independent pathway in resistance of leukaemia to BCR-ABL kinase inhibitors. J Cell Mol Med. 2007;11:1251–1262. doi: 10.1111/j.1582-4934.2007.00108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stefanachi A, Leonetti F, Nicolotti O, Catto M, Pisani L, Cellamare S, Altomare C, Carotti A. New strategies in the chemotherapy of leukemia: eradicating cancer stem cells in chronic myeloid leukemia. Curr Cancer Drug Targets. 2012;12:571–596. doi: 10.2174/156800912800673239. [DOI] [PubMed] [Google Scholar]
- 14.Hochhaus A, Kreil S, Corbin AS, Rose’e PL, Müller MC, Lahaye T, Hanfstein B, Schoch C, Cross NC, Berger U, Gschaidmeier H, Druker BJ, Hehlmann R. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 2002;16:2190–2196. doi: 10.1038/sj.leu.2402741. [DOI] [PubMed] [Google Scholar]
- 15.Bixby D, Talpaz M. Mechanisms of resistance to tyrosine kinase inhibitors in chronic myeloid leukemia and recent therapeutic strategies to overcome resistance. Hematology Am Soc Hematol Educ Program. 2009:461–476. doi: 10.1182/asheducation-2009.1.461. [DOI] [PubMed] [Google Scholar]
- 16.Mahon FX, Deininger MW, Schultheis B, Chabrol J, Reiffers J, Goldman JM, Melo JV. Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: diverse mechanisms of resistance. Blood. 2000;96:1070–1079. [PubMed] [Google Scholar]
- 17.Weisberg E, Griffin JD. Mechanism of resistance to the ABL tyrosine kinase inhibitor STI571 in BCR/ABLtransformed hematopoietic cell lines. Blood. 2000;95:3498–3505. [PubMed] [Google Scholar]
- 18.le Coutre P, Tassi E, Varella-Garcia M, Barni R, Mologni L, Cabrita G, Marchesi E, Supino R, Gambacorti-Passerini C. Induction of resistance to the Abelson inhibitor STI571 in human leukemic cells through gene amplification. Blood. 2000;95:1758–1766. [PubMed] [Google Scholar]
- 19.Willis SG, Lange T, Demehri S, Otto S, Crossman L, Niederwieser D, Stoffregen EP, McWeeney S, Kovacs I, Park B, Druker BJ, Deininger MW. High-sensitivity detection of BCR-ABL kinase domain mutations in imatinib-naive patients: correlation with clonal cytogenetic evolution but not response to therapy. Blood. 2005;106:2128–2137. doi: 10.1182/blood-2005-03-1036. [DOI] [PubMed] [Google Scholar]
- 20.Burchert A. Roots of imatinib resistance: a question of self-renewal? Drug Resist Updat. 2007;10:152–161. doi: 10.1016/j.drup.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 21.Jabbour E, Kantarjian H, Jones D, Talpaz M, Bekele N, O’Brien S, Zhou X, Luthra R, Garcia-Manero G, Giles F, Rios MB, Verstovsek S, Cortes J. Frequency and clinical significance of BCR-ABL mutations in patients with chronic myeloid leukemia treated with imatinib mesylate. Leukemia. 2006;20:1767–1773. doi: 10.1038/sj.leu.2404318. [DOI] [PubMed] [Google Scholar]
- 22.Soverini S, Colarossi S, Gnani A, Rosti G, Castagnetti F, Poerio A, Iacobucci I, Amabile M, Abruzzese E, Orlandi E, Radaelli F, Ciccone F, Tiribelli M, di Lorenzo R, Caracciolo C, Izzo B, Pane F, Saglio G, Baccarani M, Martinelli G GIMEMA Working Party on Chronic Myeloid Leukemia. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin Cancer Res. 2006;12:7374–7379. doi: 10.1158/1078-0432.CCR-06-1516. [DOI] [PubMed] [Google Scholar]
- 23.Von Bubnoff N, Schneller F, Peschel C, Duyster J. BCR-ABL gene mutations in relation to clinical resistance of Philadelphiachromosome-positive leukaemia to STI571: a prospective study. Lancet. 2002;359:487–491. doi: 10.1016/S0140-6736(02)07679-1. [DOI] [PubMed] [Google Scholar]
- 24.Soverini S, Hochhaus A, Nicolini FE, Gruber F, Lange T, Saglio G, Pane F, Müller MC, Ernst T, Rosti G, Porkka K, Baccarani M, Cross NC, Martinelli G. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: an expert panel on behalf of recommendations from European LeukemiaNet. Blood. 2011;118:1208–1215. doi: 10.1182/blood-2010-12-326405. [DOI] [PubMed] [Google Scholar]
- 25.Apperley JF. Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007;8:1018–1029. doi: 10.1016/S1470-2045(07)70342-X. [DOI] [PubMed] [Google Scholar]
- 26.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCRABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 27.Nicolini FE, Ibrahim AR, Soverini S, Martinelli G, Müller MC, Hochhaus A, Dufva IH, Kim DW, Cortes J, Mauro MJ, Chuah C, Labussière H, Morisset S, Roche-Lestienne C, Lippert E, Hayette S, Peter S, Zhou W, Maguer-Satta V, Michallet M, Goldman J, Apperley JF, Mahon FO, Marin D, Etienne G. The BCR-ABLT315I mutation compromises survival in chronic phase chronic myelogenous leukemia patients resistant to tyrosine kinase inhibitors, in a matched pair analysis. Haematologica. 2013 doi: 10.3324/haematol.2012.080234. doi:10.3324/haematol.2012.080234. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skaggs BJ, Gorre ME, Ryvkin A, Burgess MR, Xie Y, Han Y, Komisopoulou E, Brown LM, Loo JA, Landaw EM, Sawyers CL, Graeber TG. Phosphorylation of the ATP-binding loop directs oncogenicity of drug-resistant BCR-ABL mutants. Proc Natl Acad Sci U S A. 2006;103:19466–19471. doi: 10.1073/pnas.0609239103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol. 2008;15:1109–1118. doi: 10.1038/nsmb.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Hare T, Eide CA, Deininger MW. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood. 2007;110:2242–2249. doi: 10.1182/blood-2007-03-066936. [DOI] [PubMed] [Google Scholar]
- 31.Ernst T, Erben P, Müller MC, Paschka P, Schenk T, Hoffmann J. Dynamics of BCR-ABL mutated clones prior to hematologic or cytogenetic resistance to imatinib. Haematologica. 2008;93:186–192. doi: 10.3324/haematol.11993. [DOI] [PubMed] [Google Scholar]
- 32.Cang S, Liu D. P-loop mutations and novel therapeutic approaches for imatinib failures in chronic myeloid leukemia. J Hematol Oncol. 2008;1:15. doi: 10.1186/1756-8722-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Capdeville R, Silberman S. Imatinib: A targeted clinical drug development. Semin Hematol. 2003;40:15–20. doi: 10.1053/shem.2003.50037. [DOI] [PubMed] [Google Scholar]
- 34.Corbin AS, Rosee PL, Stoffregen EP, Druker BJ, Deininger MW. Several Bcr-Abl kinase domain mutants associated with imatinib mesylate resistance remain sensitive to imatinib. Blood. 2003;101:4611–4614. doi: 10.1182/blood-2002-12-3659. [DOI] [PubMed] [Google Scholar]
- 35.Inokuchi K. Chronic Myelogenous Leukemia: From Moleculer Biology to Clinical Aspects and Novel Targeted Therapies. J Nippon Med Sch. 2006;73:178–192. doi: 10.1272/jnms.73.178. [DOI] [PubMed] [Google Scholar]
- 36.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL. Multiple BCRABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 37.Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, Laï JL, Philippe N, Facon T, Fenaux P, Preudhomme C. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood. 2002;100:1014–1018. doi: 10.1182/blood.v100.3.1014. [DOI] [PubMed] [Google Scholar]
- 38.Burchert A, Wang Y, Cai D, von Bubnoff N, Paschka P, Müller-Brüsselbach S, Ottmann OG, Duyster J, Hochhaus A, Neubauer A. Compensatory PI3-kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia. 2005;19:1774–1782. doi: 10.1038/sj.leu.2403898. [DOI] [PubMed] [Google Scholar]
- 39.Markova B, Albers C, Breitenbuecher F, Melo JV, Brümmendorf TH, Heidel F, Lipka D, Duyster J, Huber C, Fischer T. Novel pathway in Bcr-Abl signal transduction involves Akt-independent, PLC-gamma1-driven activation of mTOR/p70S6-kinase pathway. Oncogene. 2010 Feb 4;29:739–51. doi: 10.1038/onc.2009.374. [DOI] [PubMed] [Google Scholar]
- 40.Zhao F, Mancuso A, Bui TV, Tong X, Gruber JJ, Swider CR, Sanchez PV, Lum JJ, Sayed N, Melo JV, Perl AE, Carroll M, Tuttle SW, Thompson CB. Imatinib-resistance associated with BCR-ABL upregulation is dependent on HIF-1α-induced metabolic reprogramming. Oncogene. 2010;29:2962–2972. doi: 10.1038/onc.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wendel HG, de Stanchina E, Cepero E, Ray S, Emig M, Fridman JS, Veach DR, Bornmann WG, Clarkson B, McCombie WR, Kogan SC, Hochhaus A, Lowe SW. Loss of p53 impedes the antileukemic response to BCR-ABL inhibition. Proc Natl Acad Sci U S A. 2006;103:7444–7449. doi: 10.1073/pnas.0602402103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun X, Layton JE, Elefanty A, Lieschke GJ. Comparison of effects of the tyrosine kinase inhibitors AG957, AG490, and STI571 on BCR-ABL-expressing cells, demonstrating synergy between AG490 and STI571. Blood. 2001;97:2008–2015. doi: 10.1182/blood.v97.7.2008. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Cai D, Brendel C, Barett C, Erben P, Manley PW, Hochhaus A, Neubauer A, Burchert A. Adaptive secretion of granulocyte-macrophage colony-stimulating factor (GM-CSF) mediates imatinib and nilotinib resistance in BCR/ABL+ progenitors via JAK-2/STAT-5 pathway activation. Blood. 2007;109:2147–2155. doi: 10.1182/blood-2006-08-040022. [DOI] [PubMed] [Google Scholar]
- 44.White DL, Radich J, Soverini S, Saunders VA, Frede AK, Dang P, Cilloni D, Lin P, Mongay L, Woodman R, Manley P, Slader C, Kim DW, Pane F, Martinelli G, Saglio G, Hughes TP. Chronic phase chronic myeloid leukemia patients with low OCT-1 activity randomized to high-dose imatinib achieve better responses and have lower failure rates than those randomized to standard-dose imatinib. Haematologica. 2012;97:907–914. doi: 10.3324/haematol.2011.056457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Engler JR, Frede A, Saunders VA, Zannettino AC, Hughes TP, White DL. Chronic Myeloid Leukemia CD34+ cells have reduced uptake of imatinib due to low OCT-1 Activity. Leukemia. 2010;24:765–770. doi: 10.1038/leu.2010.16. [DOI] [PubMed] [Google Scholar]
- 46.Ekiz HA, Baran Y. Therapeutic Applications of Bioactive Sphingolipids in Hematological Malignancies. Int J Cancer. 2010;127:1497–506. doi: 10.1002/ijc.25478. [DOI] [PubMed] [Google Scholar]
- 47.Baran Y, Salas A, Senkal CE, Bielawski J, Gunduz U, Obeid LM, Ogretmen B. Alterations of human longevity assurance gene 1 (LASS1)/Sphingosine Kinase-1-dependent ceramide generation and metabolism involve in the regulation of imatinib-induced apoptosis and resistance in K562 human chronic myeloid leukemia (CML) cells. J Biol Chem. 2007;282:10922–10934. doi: 10.1074/jbc.M610157200. [DOI] [PubMed] [Google Scholar]
- 48.Salas A, Ponnusamy S, Senkal CE, Meyers MA, Selvam SP, Saddoughi SA, Apohan E, Sentelle RD, Smith C, Gault CR, Obeid LM, El-Shewy HM, Oaks J, Ramasamy S, Marcucci G, Baran Y, Mahajan S, Fernandes D, Stuart RK, Perrotti D, Ogretmen B. Sphingosine kinase-1 and sphingosine 1-phosphate receptor 2 mediate Bcr-Abl1 stability and drug resistance by modulation of protein phosphatase 2A. Blood. 2011;117:5941–5952. doi: 10.1182/blood-2010-08-300772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baran Y, Bielawski J, Ogretmen B, Gunduz U. Inhibition of Glucosylceramide Synthase by PDMP Resensitizes Multidrug-Resistant Human Chronic Myeloid Leukemia Cells to Imatinib. J Cancer Res Clin Oncol. 2011;137:1535–1544. doi: 10.1007/s00432-011-1016-y. [DOI] [PubMed] [Google Scholar]
- 50.Jamieson C. The MLLgnant consequences of reverting to an embryonic transcriptional program. Cell Stem Cell. 2009;4:97–98. doi: 10.1016/j.stem.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 51.Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–414. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- 52.Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR 3rd, Nusse R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- 53.Hu Y, Swerdlow S, Duffy TM, Weinmann R, Lee FY, Li S. Targeting multiple kinase pathways in leukemic prognitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc Natl Acad Sci U S A. 2006;103:16870–16875. doi: 10.1073/pnas.0606509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crews LA, Jamieson CHM. Chronic Myeloid Leukemia Stem Cell Biology. Curr Hematol Malig Rep. 2012;7:125–132. doi: 10.1007/s11899-012-0121-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cortes J, O’Brien S, Kantarjian H. Discontinuation of imatinib therapy after achieving a molecular response. Blood. 2004;104:2204–2205. doi: 10.1182/blood-2004-04-1335. [DOI] [PubMed] [Google Scholar]
- 56.Ito T. Stem cell maintenance and disease progression in chronic myeloid leukemia. Int J Hematol. 2013 doi: 10.1007/s12185-013-1318-8. DOI.10.1007\s12185-o13-1318-8. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 57.Jiang X, Forrest D, Nicolini F, Turhan A, Guilhot J, Yip C, Holyoake T, Jorgensen H, Lambie K, Saw KM, Pang E, Vukovic R, Lehn P, Ringrose A, Yu M, Brinkman RR, Smith C, Eaves A, Eaves C. Properties of CD34+ CML stem/progenitor cells that correlate with different clinical responses to imatinib mesylate. Blood. 2010;116:2112–2121. doi: 10.1182/blood-2009-05-222471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121:396–409. doi: 10.1172/JCI35721. [DOI] [PMC free article] [PubMed] [Google Scholar]