

Abstract
Argininemia is a rare hereditary disease due to a deficiency of hepatic arginase, which is the last enzyme of the urea cycle and hydrolyzes arginine to ornithine and urea. The onset of the disease is usually in childhood, and clinical manifestations include progressive spastic paraparesis and mental retardation. Liver involvement is less frequent and usually not as severe as observed in other UCDs. For this reason, and because usually there is a major neurological disease at diagnosis, patients with argininemia are rarely considered as candidates for OLT despite its capacity to replace the deficient enzyme by an active one. We report on long-term follow-up of two patients with argininemia. Patient 1 was diagnosed by the age of 20 months and despite appropriate conventional treatment progressed to spastic paraparesis with marked limp. OLT was performed at 10 years of age with normalization of plasmatic arginine levels and guanidino compounds. Ten years post-OLT, under free diet, there is no progression of neurological lesions. The second patient (previously reported by our group) was diagnosed at 2 months of age, during a neonatal cholestasis workup study. OLT was performed at the age of 7 years, due to liver cirrhosis with portal hypertension, in the absence of neurological lesions and an almost-normal brain MRI. After OLT, under free diet, there was normalization of plasmatic arginine levels and guanidino compounds. Twelve years post-OLT, she presents a normal neurological examination. We conclude that OLT prevents progressive neurological impairment in argininemia and should be considered when appropriate conventional treatment fails.
Abbreviations
- CSF
Cerebrospinal fluid
- MRI
Magnetic resonance imaging
- OLT
Orthotopic liver transplantation
- UCDs
Urea cycle disorders
Introduction
Argininemia (MIM #207800) is a rare inborn error of urea cycle, due to arginase A1 deficiency. This enzyme is codified by ARG1 gene and is also known as hepatic arginase because it is responsible for 98% of arginase activity in the liver. The clinical symptoms of argininemia become apparent in early childhood, usually between 2 and 4 years of age. They include progressive spastic paraparesis (with marked degree of spasticity of the four extremities) and severe mental deterioration, which are characteristic of this disease (and are not observed in other UCDs) (Brusilow and Horwich 2005; Scaglia and Lee 2006). The neurological symptoms referred to here cannot be explained merely as a consequence of arginine levels and hyperammoniemia (which is observed intermittently), since neurological impairment sometimes progresses despite good compliance with treatment (presenting stable arginine and ammonia plasmatic levels). So it was suspected that other factors, such as the presence of guanidino compounds, might be implicated in the neurological impairment (Mizutani et al. 1987), since the endogenous production of these substances may remain significant even when arginine is normalized (Marescau et al. 1990).
In the past, we demonstrated that OLT can normalize arginine, ammonia, and guanidino compounds levels in patients with argininemia, allowing them to follow a free diet for life (Santos Silva et al. 2001). Nevertheless, it remains to be clarified if in patients with already existing neurological lesions, OTL can prevent its progression, and in the ones who have not yet developed such symptoms, whether OLT will be able to prevent them in the long term.
Here, we report on two patients with argininemia, with more than 10 years follow-up after OLT, which support the evidence that this therapeutic strategy can prevent long-term development and progression of neurological impairment in patients with such an inborn error of metabolism.
Material and Methods
Amino acids profile was determined by liquid ion-exchange chromatography.
Guanidino compounds in plasma and urine were determined by high-performance liquid chromatography.
Case Reports
Case 1 – The patient is the first daughter of a non-consanguineous couple with irrelevant family history. Pregnancy and delivery were unremarkable. Birth weight was 4,100 g and height 51 cm. No neonatal jaundice was reported. She was overweight since the second year of life, and the motor milestones and cognitive development were normal.
She was diagnosed with infectious mononucleosis by 20 months of age, followed by persistent rise of transaminases and asymptomatic coagulation abnormalities. Biochemical data included total bilirubin 12 μmol/L, conjugated bilirubin 2.5 μmol/L, AST 170 UI/L (n<56), ALT 200 UI/L (n<39), GGT 10 UI/L (n<45), total cholesterol 4.5 mmol/L (n:3.4–6.0), thromboplastin time 46.6 s (n<35s), and prothrombin time 31 s (n<12s). Amino acids profile showed an elevated arginine concentration (481 μmol/l, n<140). Blood ammonia and urinary orotic acid were both within the normal range. Argininemia diagnosis was confirmed by the absence of arginase A1 activity in red blood cells and by molecular analysis of ARG1 gene (homozygous for R21X mutation). Other diagnoses have been excluded by appropriate testing, namely, alpha-1-antitrypsin deficiency, hepatitis B and C, autoimmune hepatitis, Wilson’s disease, myopathy, and other aminoacidopathies. After diagnosis, she started immediately on a low-protein and arginine-restricted diet and oral sodium benzoate 300 mg/kg/day.
Outcome was spastic paraparesis with marked limp, despite good treatment compliance and lowering of plasmatic arginine levels (200–300 μmol/L). She benefited from physiotherapy and Vulpius surgery at 7 years of age. Meanwhile brain MRI, performed 1 year later, showed increased T2 signal intensity within the peritrigonal white matter (Fig. 1a) and increased T2 signal intensity within the semioval center, more prominent in the left hemisphere (Fig. 1b).
Fig. 1.
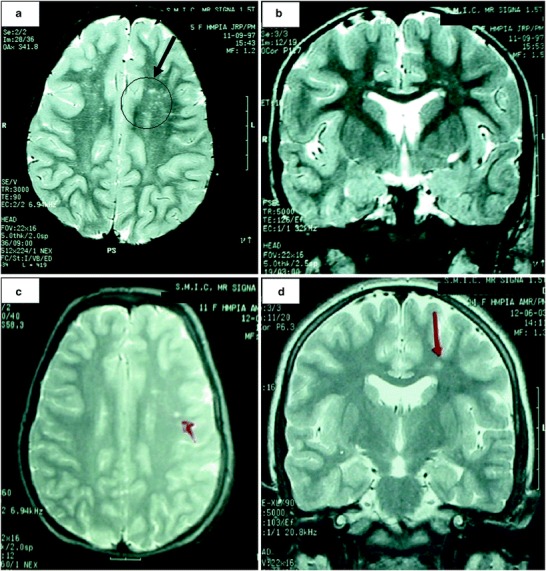
Brain MRI studies in Patient 1. Before OLT we detected (a) an increased T2 signal intensity within the peritrigonal white matter (black circle and arrow) and (b) increased T2 signal intensity within the semioval center, more prominent in the left hemisphere. Later, 15 months post-OLT we found (c) an increased T2 signal intensity within the peritrigonal white matter, and (d) within the semioval center (more prominent in the left hemisphere), but less severe than before OLT (red arrows)
School performance was satisfactory (Wechsler scale GIQ – 97), growth was appropriate (height p50), but overweight was always an unsolved problem. She had raised transaminases, mild coagulation abnormalities, and liver steatosis (ultrasound). By the age of 8 years, she started oral phenylbutyrate 250 mg/kg/day, without much success in decreasing more effectively the levels of blood arginine. By this time, guanidino compounds in plasma and urine were markedly elevated (Fig. 2).
Fig. 2.
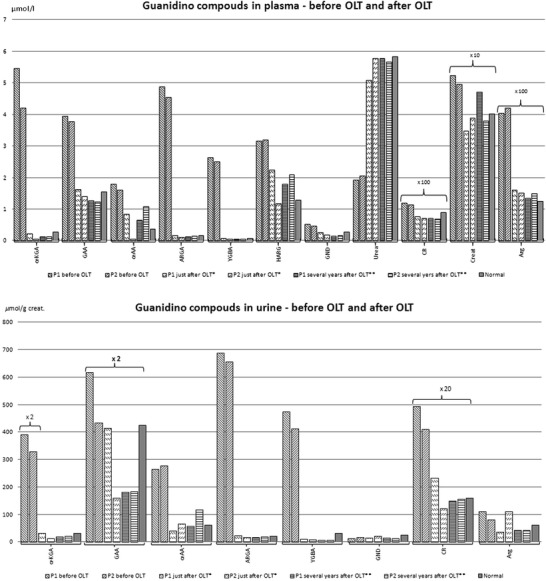
Plasmatic and urinary levels of guanidino compounds in patients with argininemia. Patient 1 first control post-OLT at * 20 months and later ** at 8 years post-OLT. Patient 2 first control post-OLT at * 26 months and later ** at 4.5 years post-OLT. α-KVA α-keto-δ guanidinovaleric acid, CR creatine, GAA guanidinoacetic acid, αAA α-N-acetylarginine, ARGA argininic acid, Creat creatinine, YGBA γ-guanidinobutyric acid, Arg arginine, HARG homoarginine, GND guanidine
As spastic paraparesis worsened, it was proposed to the family to perform a liver transplant and thus cure the hepatic enzymatic deficiency. OLT was performed on the patient when she was 10 years old (at the Liver Transplantation Center of Coimbra – Portugal) with complete normalization of plasmatic arginine levels 24 h after OLT, and since then she has remained on an unrestricted diet. After OLT, plasmatic and urinary guanidine compounds also became normal (Fig. 2) and progression of the neurological disease stopped immediately. Ten years later, there has been no progression of neurological features or new MRI abnormalities (Fig. 1c and d).
Case 2– This patient was previously reported upon (Braga et al. 1997; Santos Silva et al. 2001; Martins et al. 2011). Diagnosis of argininemia was made by 2 months of age during the workup of a neonatal cholestasis (plasma arginine 1,756 μmol/L, n: 22–88), and was confirmed by the absence of arginase A1 activity in blood red cells and by molecular analysis of ARG1 gene (homozygous for R21X mutation) (Cardoso et al. 1999). She was put on a low-protein and arginine-restricted diet, and oral sodium benzoate. The outcome was liver cirrhosis with portal hypertension, marked hypercholesterolemia (18.0 mmol/L, N:3.4–6.0), xanthomatosis, and pruritus. Despite an extensive investigation, no other cause was established for this kind of liver involvement. By the age of 7 years, she had terminal liver cirrhosis, growth development delay (P<3), General Quotient Development = 80 (Griffiths method), and normal neurological features. Despite suitable dietary compliance, arginine plasmatic levels were around 300 μmol/l and guanidino compounds were markedly raised (Fig. 2). Brain MRI studies showed increased T1 signal intensity in the pallidum nucleus and no T2 signal abnormalities in the peritrigonal white matter. At this point, she underwent OLT at the Liver Transplantation Center of Coimbra – Portugal. We observed a slight and transient hyperammoniemia with complete normalization of arginine and ammonia blood levels 24 h after OLT. When the patient started a free diet, blood arginine and ammonia levels remained normal and those of guanidino compounds normalized (Fig. 2). Twelve years after OLT, we note that neurological features, plasmatic arginine levels, and guanidino compounds remain normal. She has a healthy and unaffected sibling, born July 2000, after molecular prenatal diagnosis.
Discussion
In several metabolic diseases, liver transplant, despite correcting the hepatic enzymatic deficiency, fails in stopping long-term consequences of the disease. As no data is available on the long-term surveillance of patients submitted to liver transplant for argininemia, this report is imperative.
Argininemia treatments that successfully lower arginine and guanidino compounds levels should prevent neurological symptoms, but although arginine plasmatic levels may normalize under conventional treatment, the arginine’s catabolites can remain elevated (Marescau et al. 1990), and some of these guanidino compounds are neurotoxic (De Deyn et al. 2009; Hiramatzu, 2003). In both of our patients, arginine concentration decreased significantly under conventional treatment (but never normalized), and guanidine compounds were markedly elevated in plasma and urine. However, after OLT all these parameters normalized completely (Fig.2).
It would have been very interesting to evaluate the guanidino compounds also in CSF, however, that was not possible because Patient 1 never cooperated, and Patient 2 had terminal liver disease, with portal hypertension, thrombocytopenia, and coagulation abnormalities, which made the procedure unwise.
Patient 1 had a relatively early diagnosis (<2 years old) compared with the cohort of Portuguese symptomatic patients (12 patients, age of diagnosis > 3 years in the majority of cases). At diagnosis, she had subclinical liver disease (occasional finding) and was neurologically asymptomatic. Furthermore, she started conventional treatment immediately, with good compliance, and success in lowering the plasmatic levels of arginine. Despite everything, she developed progressive paraparesis and marked limp. Patient 2 had an uncommon form of presentation of argininemia (Braga et al. 1997; Martins et al. 2011) and benefited from an unusual early diagnosis and treatment (by 2 months of age). As in Patient 1, arginine plasmatic levels decreased significantly but never got completely normal. This patient never developed neurological impairment. Both patients had markedly elevated guanidino compounds pre-OLT. Perhaps the explanation for the different neurological outcome in these two patients is that vast brain growth with organizational changes takes place during the first two postnatal years (Knickmeyer et al. 2008). Thus, any toxic factor acting on the brain during this period of time will be more harmful and leave the worst possible sequels. Patient 1 was much more exposed to neurotoxicity in this crucial period for brain development than Patient 2.
When Patient 1 started to show signs of neurological impairment, we already had the experience of following other patients, under conventional treatment, with inexorable progression to spastic tetraparesis and severe mental retardation (unpublished data). But this time, we also had the experience of the first OLT performed in a patient with argininemia (Santos Silva et al. 2001). The encouraging result of OLT in Patient 2 prompted us to transplant Patient 1, in order to stop progression of neurological impairment. This aim was completely achieved. In Patient 1, progression of neurological disease stopped immediately, and there was even some regression in MRI features (Fig.1). This effect has been sustained 10 years after OLT. Patient 2 remains free of neurological disease 12 years post-OLT.
These two clinical observations also suggest that although the highest aggression for the brain may occur during the first 2 years of life, it may continue while no complete normalization of biochemical parameters is achieved, mainly of guanidino compounds levels. They also show that once normalization happens, the progression of the neurological disease stops. Based on such observations, it is recommended that patients with argininemia should be carefully monitored clinically (and biochemically) for early detection of the development of neurological symptoms.
Argininemia is the second most common urea cycle disease in Portugal, after ornithine transcarbamylase deficiency (1/215,000 live newborns) (Martins et al. 2011). Since 2004, it has been included in the Portuguese expanded newborn screening (3–6 day of life, arginine level in dried blood spot cutoff = 37 μM). If arginine is above the cutoff level, a second blood sample is requested. If the second blood analysis shows arginine plasmatic level > 140 μmol/L, the neonate is called for full assessment at a reference center. Based on this approach, we have, until now, diagnosed four cases of argininemia in newborns (1/181,550 live newborns) (Personal communication, E. Martins 2011), allowing the affected patients to start conventional treatment during the first month of life (median age 16 days), and thus protecting their brains during infancy and childhood from exposure to high amounts of neurotoxic compounds. The follow-up of such patients will further help to clarify the long-term outcome of the disease in ideally treated patients. Meanwhile, we know that OLT is able to prevent progressive neurological impairment in patients with argininemia, when appropriate conventional treatment fails, and should be offered to these patients before severe neurological impairment occurs.
Concise Sentence Take-Home Message
OLT prevents progressive neurological impairment in argininemia and should be considered when appropriate conventional treatment fails.
Reference to Electronic Databases
Urea cycle disorders, argininemia, liver transplantation, neurological impairment.
Conflict of Interest
There is no conflict of interest.
Footnotes
Competing interests: None declared
Contributor Information
E. Santos Silva, Email: ermelinda.rss@gmail.com.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Braga AC, Vilarinho L, Ferreira E, Rocha H. Hyperargininemia presenting as persistent neonatal jaundice and hepatic cirrhosis. J Pediatr Gastroenterol Nutr. 1997;24:218–221. doi: 10.1097/00005176-199702000-00018. [DOI] [PubMed] [Google Scholar]
- Brusilow SW, Horwich AL, et al. Urea cycle enzymes. In: Scriver CR, et al., editors. Metabolic and molecular bases of inherited disease. 8. New York: McGraw-Hill; 2005. [Google Scholar]
- Cardoso ML, Martins E, Vasconcelos R, et al. Identification of a novel R21X mutation in the liver-type arginase gene (ARG1) in four Portuguese patients with argininemia. Hum Mutation. 1999;14:355–356. doi: 10.1002/(SICI)1098-1004(199910)14:4<355::AID-HUMU20>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- De Deyn PP, Vanholder R, Eloot S, Glorieux G. Guanidino compounds as uremic (neuro)toxins. Semin Dial. 2009;22(4):340–5. doi: 10.1111/j.1525-139X.2009.00577.x. [DOI] [PubMed] [Google Scholar]
- Hiramatzu M. A role for guanidine compounds in the brain. Mol Cell Biochem. 2003;244:57–62. doi: 10.1023/A:1022491419813. [DOI] [PubMed] [Google Scholar]
- Knickmeyer RC, Gouttard S, Kang C, Evans D, Wilber K, Smith JK, et al. A structural MRI study of human brain development from birth to 2 years. Journal of Neuroscience. 2008;28(47):12176–12182. doi: 10.1523/JNEUROSCI.3479-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marescau B, De Deyn PP, Lowenthal A, Qureshi IA, Antonozzi I, Bachmann C, Cederbaum SD, Cerone R, Chamoles N, Colombo JP, et al. Guanidino compound analysis as a complementary diagnostic parameter for hyperargininemia: follow-up of guanidino compound levels during therapy. Pediatr Res. 1990;27(3):297–303. doi: 10.1203/00006450-199003000-00020. [DOI] [PubMed] [Google Scholar]
- Martins E, Santos Silva E, Vilarinho S, Saudubray JM, Vilarinho L (2011) Neonatal cholestasis: an uncommon presentation of hyperargininemia. J Inherit Metab Dis. 2011; DOI 10.1007/s10545- 010-9263-7. [DOI] [PubMed]
- Mizutani N, Hayakawa C, Ohya Y, Watanabe K, Watanabe Y, Mori A. Guanidino compounds in hyperargininemia. Tohoku J Exp Med. 1987;153:197–205. doi: 10.1620/tjem.153.197. [DOI] [PubMed] [Google Scholar]
- Santos Silva E, Martins E, Cardoso ML, Barbot C, Vilarinho L, Medina M. Liver transplantation in a case of argininemia. J inherit Metab Dis. 2001;24:885–887. doi: 10.1023/A:1013960712516. [DOI] [PubMed] [Google Scholar]
- Scaglia M, Lee B. Clinical, biochemical and molecular spectrum of hyperargininemia due to arginase I deficiency. Am J Med Genet. 2006;142C:113–120. doi: 10.1002/ajmg.c.30091. [DOI] [PMC free article] [PubMed] [Google Scholar]