

Abstract
β-Mannosidosis results from a functional deficiency of the lysosomal enzyme, β-mannosidase. While being a well recognised, naturally occurring disease in both goats and cattle, it is an extremely rare disorder in humans with the first cases only being recorded in 1986. Until now the severity of the human disease has not mirrored that of its bovine or caprine counterparts, whose presentation is typically in the neonatal period with both altered skull morphology and seizures. Human β-mannosidosis has previously appeared to be a more indolent disease, with its only consistent phenotypic feature being developmental delay of varying severity. We report a patient, homozygous for a private mutation, who presented in the immediate perinatal period with seizures and who subsequently developed communicating hydrocephalus at 2 years of age.
These are two new phenotypic features for β-mannosidosis. The first being the neonatal onset of the seizures, for while seizures have been seen in 3 out of the previous 20 documented cases, they have never before manifested prior to 6 months of age. However, as in the previous documented cases, the seizures were difficult to control and were associated with severe developmental delay.
The second unique feature about this case was the development of communicating hydrocephalus. We discuss the possible mechanisms of its development.
In summary, β-mannosidosis must thus now be considered in the differential diagnosis of neonatal onset seizures, and the potential for the development of hydrocephalus should be monitored during subsequent clinical follow-up.
Introduction
β-Mannosidosis in man is a rare panethnic lysosomal storage disorder which results from a deficiency in the function of β-mannosidase, an enzyme that regulates the removal of N-linked mannose moieties during oligosaccharide catabolism. Currently there is a lack of a common clinical phenotype (Bedilu et al. 2002) which, when coupled to the absence of many of the overt hallmarks of classical storage disorders, i.e. organomegaly, dysostosis multiplex, ophthalmological changes or lymphocytic vacuolation (Thomas 2001), makes its clinical recognition difficult. We report an infant who presented in the immediate perinatal period with recurrent seizures who subsequently developed hydrocephalus. This presentation reflects a severity of disease that is more in keeping with the naturally occurring caprine or bovine diseases, than that previously documented in man.
Case Report
The patient, a girl, is the first child of consanguineous South Asian parents (the parents being 1st cousins). Three cousins have developmental delay and epilepsy of unknown aetiology. Antenatally there were no concerns, and she was born in good condition at term by vaginal delivery with forceps. However, at 24 h of age, she was noted to have abnormal clonic movements of her limbs and was poorly responsive. A full septic screen including lumbar puncture was normal, as was a cranial ultrasound and routine biochemistry. She was started on phenobarbitone and discharged home, pending further investigations. Prior to discharge, auditory brainstem response evaluation was performed which indicated she had impaired sensoneural hearing.
At 3 weeks of age, she developed recurrent generalised tonic-clonic seizures. Examination at that time revealed an increased truncal tone, brisk reflexes and a subtle facial dysmorphism with slight hypertelorism and flattening of the nasal bridge. Brain MRI at 4 weeks of age showed prominent ventricles and cerebrospinal fluid volume in the subarachnoid spaces, but no focal lesions were noted and myelination was age appropriate (see Fig. 1). The frequency of the seizures increased, despite the introduction of sodium valproate and a trial of pyridoxine, necessitating her transfer to the regional neurological centre for further assessment. A further panel of infective and metabolic investigations to investigate the cause of this perinatal onset of seizures was performed. This included both viral and bacterial plasma and CSF PCRs, urine organic acids, urine aminoadipic semialdehyde, urinary sulphite, urinary glycosaminoglycans, plasma electrolytes including calcium, paired plasma and CSF amino acids, ammonia, purine analysis, vacuolated lymphocytes, biotindase, very long chain fatty acids, CSF neurotransmitters and tetrahydrofolate and CSF/plasma glucose ratios. All of these investigations were unremarkable. The patient was also noted to have a degree of stridor at this time with laryngomalacia being confirmed on microlaryngobronchoscopy. Seizure control was established by the use of topiramate and phenytoin at 3 months of age.
Fig. 1.
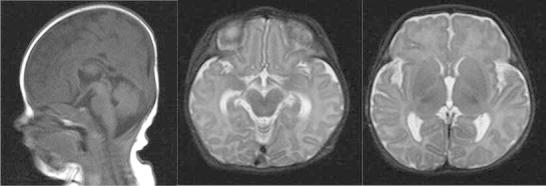
Initial MRI performed at 4 weeks of age
At discharge at 3 months she had mild persistent stridor and pronounced gastroesophageal reflux, and required feeding by nasogastric tube. However, her seizures appeared to be well controlled. She made some progress over the next 2 months with a degree of resolution of her stridor and the development of some head control. However at 5 months of age, following further seizures, the diagnosis of β-mannosidosis was established by the demonstration of a leucocyte β-mannosidase activity of 0.2 nmol/h/mg of protein (normal range 100–187). This enzymic study was undertaken as urinary oligosaccharide analysis as a screening test for the glyocoproteinoses is not routinely available at our centre. Subsequent urine analysis has confirmed the presence of the typical disaccharide. Mutation analysis subsequently revealed homozygosity for c.293T>A, a private mutation in exon 3 of MANBA which encodes for a stop codon.
Her seizures again became more prominent at 8 months of age where despite the use of clobabazm, topiramate and midazolam she continued to have breakthrough seizures. She also developed diarrhoea requiring the use of a hypoallergenic infant formula (Neocate); loperamide was required for symptomatic relief. She remained severely developmentally delayed with signs of visual impairment.
At her current age of 23 months, she is unable to sit, has visual inattention and only babbling vocalisation. Her ears show retracted tympanic membranes suggestive of a mild ear effusion, with tympanometry confirming middle ear dysfunction.
Given her extremely poor clinical state, the slow increase in her head circumference (9th centile at birth, 75th at 9th months, to the 91st centile at 23 months of age) was not initially investigated. However at 22 months a further increase in her seizure activity, together with unequal upper limb tone, raised the possibility of raised intracranial pressure or focal pathology. Brain MRI was thus undertaken and demonstrated a communicating hydrocephalus. However shortly after the MRI, her clinical state stabilised without further intervention.
Now at 25 months of age, her head circumference is static on the 91st centile and her seizures are controlled by topiramate with additional midazolam when required. Although neurological stable, she remains severely neurological affected with generalised increased tone in all 4 limbs requiring baclofen. She also requires exclusively feeding by NG tube, due to the extent of her dysphagia. She continues to have only mild hepatosplenomegaly and minimal facial coarsening but has now also suffered a fracture of her left femur with bone mineral density scanning revealing a generalised decrease in bone mineral density.
Discussion
β-Mannosidosis is an extremely uncommon lysosomal storage disease with an estimated incidence of 0.1 per 100,000 (Poorthuis et al. 1999; Poupetova et al. 2010). It results from a deficiency in the activity of the lysosomal enzyme β-mannosidase which cleaves the unique β(1-4)-linked mannose sugar found in all N-linked oligosaccharides of glycoproteins (Thomas 2001). To the best of our knowledge, there have been 20 cases in 16 families reported in the medical literature, since it was first described in humans in 1986 by 2 independent groups (Cooper et al. 1986; Wenger et al. 1986). While the age of presentation ranges from a few weeks (Dorland et al. 1988) to adulthood (Gort et al. 2006), overall the disease has been thought to generally be less severe than that found naturally occurring in goats or cows (Bedilu et al. 2002), where frank dysmorphism and severe neonatal onset neurological disease is the norm (Jones et al. 1983; Abbitt et al. 1991). This has in part been attributed to the fact that β-mannosidosis deficient ruminants accumulate the trisaccharide Man(β1-4) GlcNAc (β1-4) GlcNAc to a greater extent than the disaccharide Man(β1-4) GlcNAc (Jones et al. 1992). However humans who, like other non-ruminants, have an additional lysosomal enzyme chitobiase (Zhu et al. 2006) accumulate the disaccharide (Cooper et al. 1988). This potential correlation between phenotype and predominating oligosaccharide is strengthened by the fact that the mouse model, where predominately disaccharides accumulate, also exhibits a mild phenotype with no obvious dysostosis (Zhu et al. 2006).
In a review of the literature, Bedilu found that the main phenotypic traits in man were developmental delay and hearing loss (Bedilu et al. 2002), although many of the more recent cases do not have the hearing impairment. Both of these features were present in our patient, with the hearing loss being suspected from an extremely early age. Interestingly, this review also indicated that there was little genotype/phenotype correlation with patients in that study surviving until middle age with null mutations. Thus, it cannot be inferred that the null mutation found in this patient was the major determinant of the severity of presentation.
Distinguishing whether the developmental delay was secondary to the seizures or the underlying mannosidosis is impossible, though it seems that once seizures are noted in β-mannosidosis, a loss of skills does ensue (Cooper et al. 1991; Cherian 2004). This lack of consistent clinical features means that there is increased reliance on screening tests diagnostically and would support the use of urinary oligosaccharides as part of initial investigations of potential metabolic disorders. The first unique feature of this case is the onset of seizures in the perinatal period. Previously the earliest manifestation of any neurological impairment in a patient with β-mannosidosis was the dysphagia seen at 8 weeks, in one of the Turkish siblings described by Dorland (Dorland et al. 1988). The earliest persistent seizure activity documented was in the Saudi siblings described by Cherian at 6 months of age, however in both of these patients the seizures were initially isolated and associated with temperatures, unlike our patient, and only became persistent later (Cherian 2004). The historical patient, who appears to be most similar to our patient, was the girl described by Cooper et al. (1991). She also presented with sudden onset of persistent generalised tonic-clonic seizures which were also difficult to control. However, the age of onset of the seizures was at 9 months of age and was preceded by a history of developmental delay. Including this case, seizures have occurred in 4 cases out of the 21 reported (Cooper et al. 1991; Cherian 2004) making seizures a relatively common presentation. As may be expected, seizures seem to be associated with a more severe phenotype, with all four cases presenting at less than 1 year of age and all of them having marked developmental delay. However, it is to be noted that EEG abnormalities are not restricted to those with clinical seizure activity (Wijburg et al. 1992).
The other unique feature in this case is the communicating hydrocephalus, a feature that has not been described before. While cerebral atrophy has been described (Labauge et al. 2009), there have been no documented cases of hydrocephalus. Given the scarcity and heterogeneity of the disease, it is hard to be certain that the hydrocephalus is directly attributable to the mannosidosis, but it is the most likely explanation. Indeed in storage disorders it is occasionally hard to differentiate between communicating hydrocephalus and cerebral atrophy (Manara et al. 2011) though the progressive macrocephaly would be indicative of the former.
The pathogenesis underlying the hydrocephalus, however, remains unclear. There have been a number of different mechanisms suggested for the development of hydrocephalus in storage disorders. This has been best described in the mucopolysaccharidoses (MPS) where a number of different theories have been put forward for their development. These include venous hypertension secondary to skull base abnormalities (Vedolin et al. 2007); however, there was no obvious crowding of the posterior fossa structures or bony craniovertebral junction anomaly in our patient. Other potential mechanisms include the deposition of storage material in the leptomeninges (Matheus et al. 2004) with a subsequent decrease in CSF absorption. The loculation of fluid in the extra-axial cystic lesions suggests the trapping of fluid in the arachnoid space (see Fig. 2). This feature of arachnoid loculation, including the development of hydrocephalus, is a recognised feature of MPS and has been attributed to the deposition of glycosaminoglycans in the subarachnoid space. In this case, there was no enlargement of the perivascular spaces, which is an additional feature, described in MPS. The possibility of alternative pathological mechanisms cannot be discounted, however, since both the type and degree of substrate deposition differ between disorders giving rise to the potential triggering of different pathological cascades.
Fig. 2.
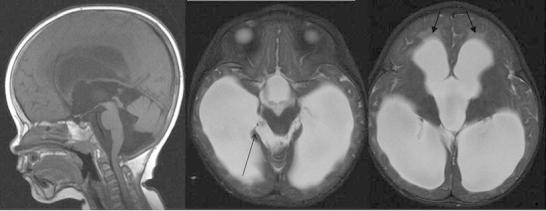
Second brain MRI performed at 23 months. While there is some coexistent cerebral volume loss, the sagittal T1W and axial T2W images show macrocrania, dilatation of the lateral, third and fourth ventricles with transependymal oedema (closed arrowheads) and some bulging of the inferior recesses of the third ventricle in keeping with extraventricular hydrocephalus. The pons and medulla are displaced posteriorly, the inferior cerebellar vermis superiorly and there are new extra-axial cystic lesions within the ambient cisterns, larger on the right (open arrowhead) in keeping with loculated fluid within the arachnoid space
In summary, this case would indicate that β-mannosidosis although phenotypically heterogeneous in presentation is not necessarily a milder disease in humans than in other animals and is a potential cause of neonatal seizures. Children with β-mannosidosis should also be closely observed as they do have the potential to develop hydrocephalus.
Footnotes
Competing interests: None declared
Contributor Information
A Broomfield, Email: alexander.broomfield@gosh.nhs.uk.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Abbitt B, Jones MZ, Kasari TR, Storts RW, Templeton JW, Holland PS, Castenson PE. Beta-mannosidosis in twelve Salers calves. J Am Vet Med Assoc. 1991;198:109–113. [PubMed] [Google Scholar]
- Bedilu R, Nummy KA, Cooper A, Wevers R, Smeitink J, Kleijer WJ, Friderici KH. Variable clinical presentation of lysosomal beta-mannosidosis in patients with null mutations. Mol Genet Metab. 2002;77:282–290. doi: 10.1016/S1096-7192(02)00172-5. [DOI] [PubMed] [Google Scholar]
- Cherian MP. Beta-mannosidase deficiency in two mentally retarded girls with intractable seizures. Ann Saudi Med. 2004;24:393–395. doi: 10.5144/0256-4947.2004.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A, Sardharwalla IB, Roberts MM. Human beta-mannosidase deficiency. N Engl J Med. 1986;315:1231. doi: 10.1056/NEJM198608143150722. [DOI] [PubMed] [Google Scholar]
- Cooper A, Hatton C, Thornley M, Sardharwalla IB. Human β-mannosidase deficiency: biochemical findings in plasma, fibroblasts, white cells and urine. J Inher Metab Dis. 1988;1:17–29. doi: 10.1007/BF01800054. [DOI] [PubMed] [Google Scholar]
- Cooper A, Wraith JE, Savage WJ, Thornley M, Noronha MJ. Beta-mannosidase deficiency in a female infant with epileptic encephalopathy. J Inherit Metab Dis. 1991;14:18–22. doi: 10.1007/BF01804383. [DOI] [PubMed] [Google Scholar]
- Dorland L, Duran M, Hoefnagels FE, et al. Beta-mannosidosis in two brothers with hearing loss. J Inherit Metab Dis. 1988;11(Suppl 2):255–258. doi: 10.1007/BF01804251. [DOI] [PubMed] [Google Scholar]
- Gort L, Duque J, Fabeiro JM, Zulaica A, Coll MJ, Chabás A. Molecular analysis in two beta-mannosidosis patients: description of a new adult case. Mol Genet Metab. 2006;89:398–400. doi: 10.1016/j.ymgme.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Jones MZ, Cunningham JG, Dade AW, et al. Caprine beta-mannosidosis: clinical and pathological features. J Neuropathol Exp Neurol. 1983;42:268–285. doi: 10.1097/00005072-198305000-00005. [DOI] [PubMed] [Google Scholar]
- Jones MZ, Rathke EJ, Gage DA, Costello CE, Murakami K, Ohta M, Matsuura F. Oligosaccharides accumulated in the bovine beta-mannosidosis kidney. J Inherit Metab Dis. 1992;15:57–67. doi: 10.1007/BF01800344. [DOI] [PubMed] [Google Scholar]
- Labauge P, Renard D, Castelnovo G, Sabourdy F, de Champfleur N, Levade T. Beta-mannosidosis: a new cause of spinocerebellar ataxia. Clin Neurol Neurosurg. 2009;111:109–110. doi: 10.1016/j.clineuro.2008.09.007. [DOI] [PubMed] [Google Scholar]
- Manara R, Priante E, Grimaldi M, et al. Brain and spine MRI features of Hunter disease: frequency, natural evolution and response to therapy. J Inherit Metab Dis. 2011;34:763–780. doi: 10.1007/s10545-011-9317-5. [DOI] [PubMed] [Google Scholar]
- Matheus MG, Castillo M, Smith JK, Armao D, Towle D, Muenzer J. Brain MRI findings in patients with mucopolysaccharidosis types I and II and mild clinical presentation. Neuroradiology. 2004;46:666–672. doi: 10.1007/s00234-004-1215-1. [DOI] [PubMed] [Google Scholar]
- Poorthuis BJ, Wevers RA, Kleijer WJ, et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999;105:151–156. doi: 10.1007/s004399900075. [DOI] [PubMed] [Google Scholar]
- Poupetova H, Ledvinova J, Berna L, Dvoráková L, Kozich V, Elleder M. The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis. 2010;33:387–396. doi: 10.1007/s10545-010-9093-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GH, et al. Disorders of glycoprotein degradation: a-mannosidosis, b-mannosidosis, fucosidosis, and sialidosis. In: Scriver CR, Beudet AL, Valle D, et al., editors. The Metabolic and molcular basis of inherited disease. 8. New York: McGraw-Hill; 2001. pp. 3507–3535. [Google Scholar]
- Vedolin L, Schwartz IV, Komlos M, et al. Brain MRI in mucopolysaccharidosis: effect of aging and correlation with biochemical findings. Neurology. 2007;69:917–924. doi: 10.1212/01.wnl.0000269782.80107.fe. [DOI] [PubMed] [Google Scholar]
- Wenger DA, Sujansky E, Fennessey PV, Thompson JN. Human beta-mannosidase deficiency. N Engl J Med. 1986;315:1201–1205. doi: 10.1056/NEJM198611063151906. [DOI] [PubMed] [Google Scholar]
- Wijburg H, de Jong J, Wevers R, Bakkeren J, Bakkeren J, Trijbels F, Sengers R. Beta-mannosidosis and ethanolaminuria in a female patient. Eur J Pediatr. 1992;151:311. doi: 10.1007/BF02072238. [DOI] [PubMed] [Google Scholar]
- Zhu M, Lovell KL, Patterson JS, Saunders TL, Hughes ED, Friderici KH. Beta-mannosidosis mice: a model for the human lysosomal storage disease. Hum Mol Genet. 2006;15:493–500. doi: 10.1093/hmg/ddi465. [DOI] [PubMed] [Google Scholar]