

Abstract
α-Mannosidosis, OMIM #248500, is an autosomal recessive lysosomal storage disease caused by acidic α-mannosidase deficiency. Treatment options include bone marrow transplantation (BMT) and, possibly in the future, enzyme replacement therapy. Brain magnetic resonance spectroscopy (MRS) enables non-invasive monitoring of cerebral treatment effect. Accumulated cerebral mannose-containing oligosaccharides were demonstrated by MRS in a patient who at age 2 years and 11 months received a BMT from a haploidentical non-carrier sibling. The cerebral mannose-containing oligosaccharides had disappeared as early as 9½ months after BMT. MRS furthermore demonstrated the persistent treatment effect at regular intervals up to 5½ years after BMT. MRS is a non-invasive tool that can demonstrate the effect of BMT treatment. Likewise, MRS may be used to demonstrate the cerebral effect of other potential treatments such as enzyme replacement therapy.
Introduction
α-Mannosidosis, OMIM #248500, is an autosomal recessive lysosomal storage disease caused by acidic α-mannosidase deficiency. Patients accumulate mannose-rich oligosaccharides within the lysosomes. Three forms of the disease are described, though the phenotypes of the patients represent a continuum with vast clinical variation: There is mild adult onset (type 1) at one end of the spectrum, at the other end a severe form (type 3), and finally an intermediate, moderate form (type 2) (Chester et al. 1982; Malm and Nilssen 2008).
Findings on neuroimaging include periventricular white matter changes, enlarged Virchow-Robin spaces, enlarged suprasellar cistern, narrow foramen magnum, and small posterior fossa (Ara et al. 1999; Dietemann et al. 1990; Niemann et al. 1996; Gutschalk et al. 2004).
Results of magnetic resonance spectroscopy (MRS) have not previously been reported in severe α-mannosidosis. In a report of three patients with adult-onset α-mannosidosis, MRS was normal (Gutschalk et al. 2004). Avenarius et al. (2011) performed MRS on three patients with moderate type 2 α-mannosidosis, one of whom had received a bone marrow transplantation 6 years prior to MRS; they found an abnormally broad peak representing oligosaccharides in the untreated patients, and this peak was not present in the patient, who had undergone bone marrow transplantation.
Feline models of α-mannosidosis showed abnormal apparent diffusion coefficients, hyperintensities in white matter on T2-weighted magnetic resonance images (Vite et al. 2001, 2008), and MRS showed a broad signal in the 3.4–4.3 ppm region thought to represent mannose-rich oligosaccharides (Magnitsky et al. 2010).
The mannose-rich oligosaccharides accumulating in α-mannosidosis contain mostly two to four mannose groups, but also branched or linear variants with up to nine mannose groups are found (Van Halbeek et al. 1980; Strecker et al. 1976; Abraham et al. 1986; Malm and Nilssen 2008). In vivo human MRS does not allow for individual characterization of the mannose-rich oligosaccharides, but it may complement urine analysis and provide in vivo measurements of the oligosaccharides in the brain where the composition and amounts may not necessarily reflect those found in urine. Hård et al. showed that the relative amounts of the different mannose-rich oligosaccharides differ in urine and brain (Hård et al. 1991) in a feline model of α-mannosidosis.
The purpose of this report is to demonstrate abnormal mannose-rich oligosaccharides by in vivo cerebral MRS in a patient with type 2 α-mannosidosis in the severe end of the spectrum, and to show the immediate and long-term effect of allogenous bone marrow transplantation (BMT) on presence of brain mannose-rich oligosaccharides and on normal cerebral metabolites seen on MRS in the same individual patient. Similarly, Takahashi et al. demonstrated the effect of BMT in patients with mucopolysaccharidoses using MRS (Takahashi et al. 2001).
Patient
The patient was a girl with severe type 2 α-mannosidosis in the severe end of the spectrum with early presenting signs of skeletal and CNS disease from about 5 months of age. She was compound heterozygous for c.2248C>T and c.2426T>C in MAN2B1. At age 2 years and 11 months, she received a BMT from a haploidentical non-carrier sibling. Psychomotor development was judged 6 to 9 months delayed at age 2½ years. Six months after BMT, biochemical analyses showed normalized excretion of urine oligosaccharides and normal leucocyte α-mannosidase activity, and both remained normal hereafter. Psychomotor development judged at age 6 years remained delayed with motor skills 2 years delayed and language skills about 1 year delayed.
Methods
The patient was evaluated by magnetic resonance imaging and spectroscopy, in general anaesthesia. Examinations were performed 8½ months before bone marrow transplantation, and 9½ months, 22 months, 3½ years, and 5½ years after. Brain magnetic resonance imaging used standard T1-, T2-, and diffusion-weighted imaging protocols. The last examination was supplemented by fluid attenuated inversion recovery (FLAIR). The short echo time magnetic resonance spectra were sampled from three volumes of interest located in the occipital grey matter, parieto-occipital white matter, and centrum semiovale, using methods as suggested by Elberling et al. (2003). The first three examinations were performed on a 1.5 Tesla Siemens Vision scanner (STEAM, TE 20 ms, TR 3000 ms, 86 acquisitions), the remaining on a 3 Tesla Siemens Trio scanner (PRESS, TE 30 ms, TR 3000 ms, 80 acquisitions). Magnetic resonance spectra were analysed by the manufacturers’ post-processing software for the purpose of spectral display. Post-processing of the spectra included eddy-current correction, zero filling, minimal Gaussian apodization (time constant 512 ms), fast Fourier transform, phase correction, and filtration of the residual water signal, but no corrections of the baseline was employed. The fully automated and user-input-independent quantitative post-processing analysis LCModel (Provencher 1993) was used for quantitation of cerebral metabolites. The mannose-containing oligosaccharide resonance complex (MC) was quantified using the LCModel basis-set with an additional simulated basis accounting for the broad resonance detected at baseline. Due to the broad nature of the MC, absence of MC was defined by the following three requirements: (1) the ratio MC/Cr gave a value similar to values obtained in normal controls, when MC was included in the basis-set; (2) excluding MC from the basis-set gave fitting results with normal baselines and residuals; (3) the broad hump between 3.5 and 3.9 ppm was undetectable by visual inspection. Routinely used neurometabolic markers of disease development included N-acetylaspartate (NAA), myo-inositol (mI), choline containing compounds (Cho), and glutamine+glutamate (Glx). They were calculated as ratios of metabolites related to total creatine (Cr) and compared to the normal controls used in the clinic for the relevant scanners. Values outside ± 2 SD were considered abnormal.
Results
Brain magnetic resonance imaging showed mild, diffuse hyperintense signals on T2-weighted images and increased apparent diffusion coefficients in white matter near the lateral horns in locations not overlapping with MRS volumes of interest. The apparent diffusion coefficient was normal in normal-appearing tissue. The imaging findings were stable throughout the observation period. FLAIR images obtained only from the last examination showed discrete signal abnormalities, also only at the lateral horns.
Brain MRS prior to bone marrow transplantation showed abnormally broad resonances in the 3.5–3.9 ppm region. It was clearly visible in the baseline spectrum from centrum semiovale and parieto-occipital white matter, but in the baseline spectrum from occipital grey matter, it was at the detection limit only. The broad resonance complex could be identified as a combination of mannose-rich oligosaccharides. Quantitation of the individual mannose-containing oligosaccharides was not attempted, because the information in the detected broad resonance was insufficiently detailed. The abnormal pretreatment MC signal however was quite prominent compared to findings done at the first follow-up 9½ months after BMT, when it had disappeared (Fig. 1). All the following MRS studies demonstrated continued absence of the MC in the spectra (Fig. 1).
Fig. 1.
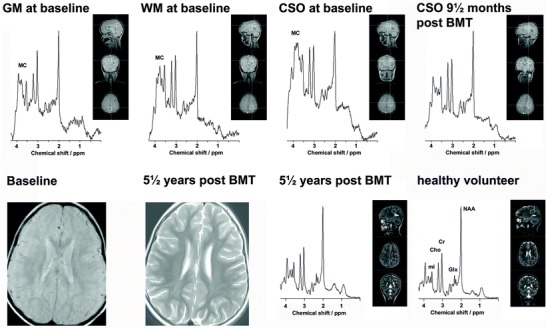
The short echo time magnetic resonance spectra (MRS) from grey matter (GM), white matter (WM), and centrum semiovale (CSO) at baseline 8½ months before bone marrow transplantation and from CSO 9½ months after bone marrow transplantation demonstrate disappearance of the mannose-containing oligosaccharide resonance complex (MC) after treatment (MRS from GM and WM also had normalized, not shown). These four spectra were sampled at 1.5 T using the pulse sequence STEAM. Brain magnetic resonance imaging showed mild, diffuse hyperintense signals on T2-weighted images without temporal evolution; images shown here are MRI at baseline (1.5 T) and MRI 5½ years after BMT (3 T). The persistent absence of the mannose-containing oligosaccharide resonance complex (MC) is illustrated by MRS from WM in the patient 5½ years after BMT in comparison to MRS from a healthy volunteer measured using identical conditions. These two spectra were sampled at 3 T using the pulse sequence PRESS
Analysis of the NAA/Cr, Cho/Cr, mI/Cr, and Glx/Cr ratios in grey and white matter measured during the five examinations showed normal values compared to normal controls with a few exceptions, Cho/Cr in grey matter and mI/Cr in white matter: In the grey matter, Cho/Cr was 30% decreased at the fourth examination and 25% decreased at the fifth examination. In the white matter, the mI/Cr ratio was 40% increased at baseline, at the second examination it was 40% increased, at the third it was 30% increased, at the fourth it was 35% increased, and finally it was 20% increased indicating a tendency towards normalization of mI/Cr over time.
Discussion
Information on the natural course of early presenting type 2 α-mannosidosis is limited. As our patient progressed relatively quickly, BMT was offered and this successfully stopped further progression. The metabolic benefit of the intervention was demonstrated by brain MRS that showed the disappearance of MC within 9½ months after the BMT. Future studies of BMT treated α-mannosidosis patients should include earlier follow-up to find the earliest time for intracerebral removal of MC.
This study has for the first time demonstrated the effect of BMT in the same individual. Avenarius et al. (2011) demonstrated MC in two untreated patients and absence of MC in a BMT treated patient, but this patient may not have had increased MC in the brain before BMT. Gutschalk et al. (2004) reported three untreated cases, all without MC. Including this study, a total of six untreated patients with α-mannosidosis have been studied, and only half showed elevated MC. The frequency and correlation to symptoms of elevated cerebral MC in untreated patients remains to be studied in a larger population.
Five examinations were carried out during a period of 6 years, monitoring the effect of BMT on neurometabolism in the patient presented here. The last two examinations showed a mild reduction of Cho/Cr ratio compared to normal grey matter; the significance of this is unknown, but it may reflect a minor abnormality of the composition of grey and white matter in the volume of interest.
All examinations showed elevated mI/Cr ratio compared to normal white matter. Over time, mI/Cr ratio moved towards normal values. Likewise, data from Gutschalk et al. (2004) showed a non-significant tendency towards elevated mI/Cr ratio. Elevated mI/Cr ratio in white matter was detected in MRI normal-appearing tissue and may be a result of gliosis or demyelination reflecting MRI invisible damage that has occurred before treatment became effective. Another possibility that cannot be excluded as part of the reason for the elevated mI/Cr ratio is residual underlying MC, too little to be recognized by in vivo MRS, but sufficient to affect mI/Cr.
Preclinical animal studies have shown efficacy of enzyme replacement therapy in mice and guinea pigs, including data to suggest that the enzyme crosses the blood-brain barrier (Blanz et al. 2008; Roces et al. 2004; Crawley et al. 2006). Whether the enzyme replacement therapy passes the blood-brain barrier in humans, however, remains to be demonstrated. Another possible future avenue of treatment may be gene therapy. Such developments of less-invasive treatments hopefully will replace BMT in the future. We have demonstrated that MRS may be a biomarker of disease activity after BMT. This opens the possibility that in vivo cerebral studies could be carried out in future studies of treatment alternatives to BMT in α-mannosidosis.
Conclusion
Brain MRS has detected MC at baseline and demonstrated its disappearance after successful BMT in a patient with α-mannosidosis. Future studies with evaluation of new treatment protocols including enzyme replacement therapies should include brain MRS to monitor the intracerebral efficiency of the treatments.
Electronic Supplementary Material
1H MR spectroscopy of gray and white matter in carbon monoxide poisoning (PDF 189 kb)
Fatal encephalopathy after an isolated overdose of cocaine (PDF 110 kb)
Footnotes
Electronic supplementary material: The online version of this chapter (doi:10.1007/8904_2013_221) contains supplementary material, which is available to authorized users.
Contributor Information
Else R. Danielsen, Email: else.rubaek.danielsen@regionh.dk
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Abraham D, Daniel P, Dell A, et al. Structural analysis of the major urinary oligosaccharides in feline alpha-mannosidosis. Biochem J. 1986;233(3):899–904. doi: 10.1042/bj2330899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ara JR, Mayayo E, Marzo ME, et al. Neurological impairment in alpha-mannosidosis: a longitudinal clinical and MRI study of a brother and sister. Childs Nerv Syst. 1999;15(8):369–371. doi: 10.1007/s003810050416. [DOI] [PubMed] [Google Scholar]
- Avenarius DF, Svendsen JS, Malm D. Proton nuclear magnetic resonance spectroscopic detection of oligomannosidic n glycans in alpha-mannosidosis: a method of monitoring treatment. J Inherit Metab Dis. 2011;34(5):1023–1027. doi: 10.1007/s10545-011-9331-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanz J, Stroobants S, Lüllmann-Rauch R, et al. Reversal of peripheral and central neural storage and ataxia after recombinant enzyme replacement therapy in alpha-mannosidosis mice. Hum Mol Genet. 2008;17:3437–3445. doi: 10.1093/hmg/ddn237. [DOI] [PubMed] [Google Scholar]
- Chester MA, Lundblad A, Öckerman PA, et al. Mannosidosis. In: Durand P, O'Brian J, et al., editors. Genetic errors of glycoprotein metabolism. Milan, Italy: Edi-Ermes; 1982. pp. 89–120. [Google Scholar]
- Crawley AC, King B, Berg T, et al. Enzyme replacement therapy in alpha-mannosidosis guinea-pigs. Mol Genet Metab. 2006;89(1–2):48–57. doi: 10.1016/j.ymgme.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Dietemann JL, Filippi de la Palavesa MM, Tranchant C, et al. MR findings in mannosidosis. Neuroradiology. 1990;32(6):485–487. doi: 10.1007/BF02426460. [DOI] [PubMed] [Google Scholar]
- Elberling TV, Danielsen ER, Rasmussen AK, et al. Reduced myo-inositol and total choline measured with cerebral MRS in acute thyrotoxic Graves’ disease. Neurology. 2003;60(1):142–145. doi: 10.1212/01.WNL.0000038911.07643.BF. [DOI] [PubMed] [Google Scholar]
- Gutschalk A, Harting I, Cantz M, et al. Adult alpha-mannosidosis: clinical progression in the absence of demyelination. Neurology. 2004;63:1744–1746. doi: 10.1212/01.WNL.0000143057.25471.4F. [DOI] [PubMed] [Google Scholar]
- Hård K, Mekking A, Kamerling JP, et al. Different oligosaccharides accumulate in the brain and urine of a cat with alpha-mannosidosis: structure determination of five brain-derived and seventeen urinary oligosaccharides. Glycoconj J. 1991;8(1):17–28. doi: 10.1007/BF00731639. [DOI] [PubMed] [Google Scholar]
- Magnitsky S, Vite CH, Delikatny EJ, et al. Magnetic resonance spectroscopy of the occipital cortex and the cerebellar vermis distinguishes individual cats affected with alpha-mannosidosis from normal cats. NMR Biomed. 2010;23(1):74–79. doi: 10.1002/nbm.1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm D, Nilssen Ø. Alpha-mannosidosis. Orphanet J Rare Dis. 2008;3:21. doi: 10.1186/1750-1172-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann S, Beck M, Seidel G, et al. Neurology of adult alpha-mannosidosis. J Neurol Neurosurg Psychiatry. 1996;61(1):116–117. doi: 10.1136/jnnp.61.1.116-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med. 1993;30(6):672–679. doi: 10.1002/mrm.1910300604. [DOI] [PubMed] [Google Scholar]
- Roces DP, Lüllmann-Rauch R, Peng J, et al. Efficacy of enzyme replacement therapy in alpha-mannosidosis mice: a preclinical animal study. Hum Mol Genet. 2004;13(18):1979–1988. doi: 10.1093/hmg/ddh220. [DOI] [PubMed] [Google Scholar]
- Strecker G, Fournet B, Bouquelet S, et al. Chemistry of urinary mannosides excreted in mannosidosis. 1976;58(5):579–586. doi: 10.1016/s0300-9084(76)80227-1. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Sukegawa K, Aoki M, et al. Evaluation of accumulated mucopolysaccharides in the brain of patients with mucopolysaccharidoses by 1H-magnetic resonance spectroscopy before and after bone marrow transplantation. Pediatr Res. 2001;49(3):349–355. doi: 10.1203/00006450-200103000-00008. [DOI] [PubMed] [Google Scholar]
- Van Halbeek H, Dorland L, Veldink GA, et al. A 500 MHz 1H NMR study of urinary oligosaccharides from patients with mannosidosis. FEBS Lett. 1980;121(1):71–77. doi: 10.1016/0014-5793(80)81269-5. [DOI] [PubMed] [Google Scholar]
- Vite CH, Magnitsky S, Aleman D, et al. Apparent diffusion coefficient reveals gray and white matter disease, and T2 mapping detects white matter disease in the brain in feline alpha-mannosidosis. AJNR Am J Neuroradiol. 2008;29(2):308–313. doi: 10.3174/ajnr.A0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vite CH, McGowan JC, Braund KG, et al. Histopathology, electrodiagnostic testing, and magnetic resonance imaging show significant peripheral and central nervous system myelin abnormalities in the cat model of alpha-mannosidosis. J Neuropathol Exp Neurol. 2001;60(8):817–828. doi: 10.1093/jnen/60.8.817. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1H MR spectroscopy of gray and white matter in carbon monoxide poisoning (PDF 189 kb)
Fatal encephalopathy after an isolated overdose of cocaine (PDF 110 kb)