

Abstract
Lysosomal storage disorders (LSDs) are a very heterogeneous group of hereditary disorders. The diagnostic process usually involves complex sampling, processing, testing, and validation procedures, performed by specialized laboratories only, which causes great limitations in reaching a diagnosis for patients affected by these diseases.
There are few studies about LSDs in Colombia. The diagnostic limitations often make medical practitioners disregard the possibility of these disorders while diagnosing their patients. The current study documents the results of a 7-year screening in high-risk patients, aimed to detect LSDs using dried blood spots (DBS) collected on filter paper, with a micromethodology that facilitates diagnosis even with a large number of samples.
The activities of α-galactosidase A, α glucosidase, α-l-iduronidase, arylsulfatase B, β-galactosidase, β-glucosidase, total hexosaminidase, iduronate sulfatase, and chitotriosidase were analyzed in high-risk patients for lysosomal disease. The catalytic activity was evaluated with fluorometric micromethods using artificial substrates marked with 4-methylumbelliferone.
The reference values for a control population were established for the enzymes listed above, and 242 patients were found to have an enzyme deficiency, guiding to the following diagnoses: Fabry disease (n = 31), Pompe disease (n = 16), Hurler Syndrome (n = 15), Maroteaux-Lamy Syndrome (n = 34), GM1 Gangliosidosis (n = 10), Morquio B (n = 1), Gaucher disease (n = 101), Sandhoff disease (n = 1), Mucolipidosis (n = 2), and Hunter Syndrome (n = 31). In conclusion, this protocol provides a comprehensive diagnostic approach which could be carried out in Colombia and made it available to medical services spread around the country, enabling the identification of a large number of patients affected by LSDs, which could potentially benefit from the therapeutic tools already available for many of these diseases.
Introduction
Lysosomal storage disorders (LSDs) include approximately 50 different diseases with a combined incidence of 1:1,500 to 1:7,000 births (Wraith 2002; Staretz-Chacham et al. 2009). Each of these diseases occurs due to a deficiency in the enzymes, activating proteins or transport proteins involved in the catabolism of macromolecules that takes place in the lysosomes, causing substrate storage and progressive cell damage (Gieselmann 1995). The majority of these genetic anomalies is inherited in an autosomal recessive manner, with the exceptions of Fabry, Hunter, and Danon diseases, which are linked to the X chromosome (Staretz-Chacham et al. 2009).
The clinical findings surrounding these pathologies demonstrate multisystemic and progressive difficulties with a broad spectrum of manifestations, including skeletal abnormalities, visceromegalies, and, often, central nervous system dysfunction, ranging from behavior problems to profound mental retardation (Vellodi 2005; Scriver et al. 2001). The phenotypic expression of these diseases is so diverse and heterogeneous that their definitive identification and diagnosis must be made through specialized clinical and laboratory studies (Wenger et al. 2002)
The laboratory diagnostic approach, directed at high-risk populations, first uses protocols typically based on the detection of metabolites that are excreted in the urine after accumulating in tissues as a result of enzymatic deficiencies. Lysosomal storage disorders usually involve an accumulation of three groups of compounds: sphingolipids, mucopolysaccharides, and oligosaccharides, molecules that may be detected in affected individuals but do not offer specific diagnostic clues (Futerman et al. 2004; Filocamo and Morrone 2011). Next, these preliminary tests were followed by specific enzymatic studies – which involved plasma, leukocytes, or cultured fibroblasts samples – aimed to provide definitive diagnosis (Civallero et al. 2006).
In Colombia, and in the majority of Latin American countries, laboratory diagnosis of these metabolic disorders is not a routine practice in health services, due to the complexity of the process and also because these disorders have a very low incidence and are not among the health priorities. According to the European Union, a rare disease is classified as having a prevalence of fewer than five cases for every 10,000 inhabitants (Stolk et al. 2006). Since a complex infrastructure is required to perform the diagnostic tests for LSDs, the protocols for screening and diagnosis are primarily developed by few specialized centers. This situation becomes a limitation for the investigation of a large number of patients, either because they do not have direct access to these centers or because sending liquid samples to these laboratories (plasma, whole blood, or urine) requires strict storage conditions and short transportation times. These circumstances can either cause underdiagnosis or late diagnosis, leading to a delay in starting the treatment and, in some cases, to the death of affected individuals without the benefit of a diagnosis.
Recently, a new methodology to facilitate patient access to this type of screening has been developed in Latin America and implemented worldwide. It consists in the enzymatic analysis of dried blood spots (DBS) collected on filter paper (Fig. 1) (Chamoles et al. 2001a, 2001b; Niizawa et al. 2005; Civallero et al. 2006; Müller et al. 2010). This methodology facilitates the packing and shipment of samples because it does not require strict refrigeration and eases the collection of samples for analysis at reference testing centers, allowing the study of high-risk populations without regard to their geographical location. Given that in the recent years a number of treatments are available to treat some of these diseases, diagnosis becomes relevant. This point is especially important in the case of enzyme replacement therapy for disorders such as Fabry, Gaucher, Pompe, and Mucopolysaccharidosis type I, II, and VI for which an early diagnosis could dramatically improve the outcome of the disease (Brady 2006).
Fig. 1.
Final results of the application of enzymatic studies for dried blood collected on filter paper
The protocols for LSDs screening are usually made with a punch of 3.2 mm diameter (1/8 in.). This is considered the “universal punch” and is the most widely used because its blood volume (3.2 ul) has been reported with the greater exactitude (Reuser et al. 2011). In Colombia, there are no government screening programs that include these kinds of diseases, so all the initiatives to study them are from private universities, which leads to the development of sustainable projects based on donations and self-reliance. In this context, we propose the use of a 1.2 mm punch with the aim of diminishing the sample, materials, and reagents quantities used and in that way, reduce costs even in a 40 %, making the project viable and sustainable in time.
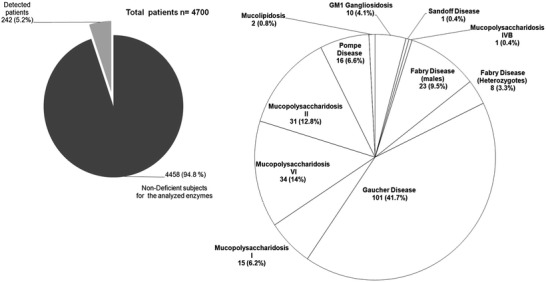
The results presented here correspond to seven years of selective screening for LSDs in Colombia. This study involves the enzymatic evaluation of DBS samples from 4,700 patients, with ages ranging from 4 months to 73 years. This study also involved the analysis of control samples to obtain reference values for α-galactosidase A, α-glucosidase, α-l-iduronidase, arylsulfatase B, β-galactosidase, β-glucosidase, total hexosaminidase, iduronate sulfatase, and chitotriosidase using modified protocols for a punch of filter paper with a diameter of 1.2 mm. These enzymatic analyses of DBS resulted in the identification of 242 individuals affected by LSDs, an unprecedented accounting in Colombia. The techniques reported here have facilitated the diagnostic approach and rapid incorporation of affected individuals into available treatment programs, which helped to change the clinical trajectory of these pathologies and to improve the quality of life for these patients and their families.
Materials and Methods
Sample Collection for the Controls and Patients in the Study
A total of 4,700 blood samples were collected on filter paper for analysis. These samples were taken from patients, aged between 4 months and 73 years, who had clinical findings related to disorders affecting glycosaminoglycan and sphingolipid metabolism. The samples were subjected to different enzymatic analyses according to clinical criteria and informed consent was obtained. Blood samples were obtained via intravenous or capillary puncture, depending on the age of the person being evaluated and were collected directly on grade 903 filter paper (Schleicher and Schuell, Whatman®) provided by GE Healthcare Life Sciences (Piscataway, NJ, USA). The specimens were kept at room temperature for 8–12 h to assure complete drying and then stored at 4 °C in self-sealing plastic bags to prevent deterioration from humidity. The samples were received from different regions in Colombia, did not undergo refrigeration, and had transport times ranging from 2 to 7 days.
To establish reference values for each enzyme tested in this study, a total of 2,520 healthy subjects were analyzed during 7 years of study (approximately 360 subjects per year, 30 per month), following the same sampling procedure applied to patients. Control subjects are distributed in two groups: the first one consisted of 250 completely healthy newborns (10%), who were <36 days old (range between 4–36 days). These samples came as part of the extended newborn screening program offered by our laboratory.
The second group consisted of 2,270 control subjects, from 3 months to 88 years, distributed as follows: 1,463 samples were from control subjects (58%) >12 years old. These samples were taken from students and employees of Universidad de los Andes, and their relatives (all of them healthy subjects). Another 605 samples (24%) were from subjects ranging from 1.5 to 11.5 years old, who were in routine growth and nutritional control in pediatric services. Finally, 202 samples (8%) corresponded to subjects clinically defined who were between 3 months and 1.3 years old. These subjects did not show clinical findings related with inborn errors of metabolism.
Enzyme Activity Assay
DBS analysis employed substrates marked with 4-methylumbelliferone (4-MU) provided by Sigma (St. Louis, MO, USA), with the exception of 4-methylumbelliferyl-α-l-iduronide (enzyme: α-l-iduronidase), which was provided by Toronto Research Chemicals (Toronto, Canada), and 4-methylumbelliferyl-iduronate 2-sulfate (enzyme: iduronate sulfatase), which was provided by Moscerdam Substrates (Rotterdam, the Netherlands). All solutions were prepared using reagents with a high level of purity.
Analytical assays on DBS for β-galactosidase, total hexosaminidase, α-galactosidase A, α-L-iduronidase, β-glucosidase, and chitotriosidase were based on the protocols proposed by Chamoles et al. (2001a, 2001b, 2002) and Civallero et al. (2006). The analysis of α-glucosidase was performed according to a standardized methodology described by Kallwass et al. (2007) and Li et al. (2004). The analysis of arylsulfatase B was performed using a standardized methodology based on the analytical principle of the leukocyte analysis proposed by Shapira et al. (1989) and Civallero et al. (2006). The analysis of iduronate sulfatase was performed using an adaptation of the methods of Voznyi et al. (2001) and Civallero et al. (2006).
The aforementioned methodologies were modified to implement the use of 1.2 mm punches (~ 0.52 μl of blood) and the corresponding volumes of substrate and buffers (Table 1). To determine the approximate blood volume in the 1.2 mm punch, we calculate and average quantity according to what Reuser et al. (2011), Daitx et al. (2012), and Rodrigues et al. (2009) reported before (see Supplementary Fig. 1).
Table 1.
Analytical conditions for the lysosomal enzymes under study
| Enzyme | Reaction buffer | Substrate (Concentration in reaction buffer) | Incubation time/T° | Stop buffer |
|---|---|---|---|---|
| Arylsulfatase B | 40 μl of 0.5 M acetate buffer 0.5 M, pH 6.0 with 10 mM barium acetate | 4-Methylumbelliferyl Sulfate 5.5 mM | 24 h/37°C | 150 μl 0.17 M glycine: carbonate, pH 10 |
| α-Galactosidase A | 20 μl of 0.15 M citrate phosphate buffer, pH 4.4 with 10 μl of 0.25 M N-acetil-d-galactosamine | 4-Methylumbelliferyl α-d-Galactoside 5 mM | 20 h/37°C | 150 μl 0.17 M glycine: carbonate, pH 10 |
| α-I-Iduronidase | 20 μl of 0.2 M sodium formiate buffer, pH 2.8 with 10 μl of 3.1 mM D-saccharic acid 1,4-lactone monohydrate | 4-Methylumbelliferyl α-l-Iduronide 2 mM | 20 h/37°C | 200 μl 0.17 M glycine: carbonate, pH 10 |
| α-Glucosidase (AαG) a | Total AαG determination: 40 μl of 0.04 M acetate buffer, pH 3.8. : Residual AαG: 40 μl of 0.04 M acetate buffer, pH 3.8 with 40 μl of 40 μM acarbose. Neutral isoforms: 40 μl of 0.04 M acetate buffer, pH 7.0 | 4-Methylumbelliferyl α-d-Glucopyranoside 2.8 mM | 24 h/37°C | 200 μl 0.17 M glycine: carbonate, pH 10 |
| β-Galactosidase | 40 μl of 0.1 M citrate phosphate buffer, pH 4.4 with 20 μl of 0.15 M NaCl | 4-Methylumbelliferyl β-d-Galactoside 0.8 mM | 3 h/37°C | 200 μl 0.17 M glycine: carbonate, pH 10 |
| β-Glucosidase | 60 μl of 0.3 M citrate phosphate buffer, pH 5.0 with sodium 1% taurodeoxycholate and conduritol-β-epoxide 0.5 mMb | 4-Methylumbelliferyl β-d-Glucoside 20 mM | 24 h/37°C | 150 μl 0.17 M glycine: carbonate, pH 10 |
| Total Hexosaminidase | 60 μl of 0.01 M citrate phosphate buffer, pH 4.4 | 4-Methylumbelliferyl 2-Acetamide-2 deoxy- β-d-Glucopyranoside 3 mM | 1 h/ 37 °C | 120 μl 0.17 M glycine: carbonate, pH 10 |
| Iduronate Sulfatase | Pre-Incubation: Place each 1.2 mm punch in 25 μl of BSA 0.2 % Incubation 1: Add 16 μl of 0.1 M of acetate buffer, pH 5.0 with lead acetate 10 mM and the substrate to 1.25 mM. Incubation 2: Add 25 μl of 0.2 M of citrate phosphate buffer, pH 4.5 with 0.02 % sodium azide and auxiliary enzyme |
Incubation 1: 4-Methylumbelliferyl α-Iduronate-2-Sulfate 1.25 mM Incubation 2: Enzyme α-I-Iduronidase 3.4 mg/ml in distilled water |
Pre-incubation: 20 min/room T° Incubation 1: 24 h/37 °C Incubation 2: 24 h/37 °C |
200 μl 0.17 M glycine: carbonate, pH 10 (Added at the end of Incubation 2) |
| Chitotriosidase | 30 μl of 0.2 M acetate buffer, pH 5.5 with 10 mM barium acetate | 4-Methylumbelliferyl- β-d-N,N′,N″ Triacetylchitotrioside 0.25 mM | 20 min/37°C | 150 μl 0.17 M glycine: carbonate, pH 10 |
aFor each reaction mixture, the experimental assay requires a 1.2 mm punch of filter paper. An evaluation of AαG in the presence of the inhibiting agent acarbose permits the elimination of maltase glucoamylase activity, that can degrade the substrate, but it is not related with Pompe disease. Evaluation at pH 7.0 determines the activity of neutral isoforms (Glucosidase II and α-Glucosidase C). The quotient obtained from dividing this value over activity in the presence of acarbose offers a discrimination criterion for individuals affected by Pompe disease and healthy controls
bSee Olivova et al. 2008
A volume of substrate, equal to that used in the samples, was included to test unspecific substrate degradation. This volume was incubated with the samples in a separate Eppendorf tube for each protocol. At the end of the incubation, the stop buffer (glycine:carbonate buffer 0.17 M, pH 10) was added to samples and blanks. Then, the incubated substrate was added to the blank wells to evaluate unspecific substrate degradation in the reaction buffer. The difference between sample and blank fluorescence values allowed differentiating the real enzymatic activity from the unspecific degradation.
Assays were carried out by combining the punches of filter paper and the reaction solutions in black, 96-well polypropylene microplates. Evaporation was prevented using aluminum foil thermo-sealing sheets provided by Corning (Lowell, USA). The elution (5 min at room temperature) and incubation processes were performed with the orbital agitation of samples (200 rpm for elution and 120 rpm for incubation), using a Titramax 1000 plate vibrator and agitator and a Unimax 1010 incubator/agitator from the Heidolph group (Schwabach, Germany). A Spectramax M2 (Molecular Devices Corp.) was used as a fluorescence reader (excitation 360 nm, emission 460 nm), and the results were compared with a 10 points 4-MU calibration curve. Enzymatic activity was expressed as nanomoles of hydrolyzed substrate per milliliter of blood per hour (nmol/ml/h).
As quality control measures for the procedure, every referred patient was evaluated for betagalactosidase and total hexosaminidase as control enzymes, besides the requested enzyme (Supplementary Fig. 2). To avoid enzyme activity loss due to sample storage, the maximum processing time between sample taking and processing was 30 days. All samples were processed by triplicate to establish an intra assay coefficient of variance and in each plate, besides the group of patients, a control sample and a previously detected patient were included to see the feasibility of the assay. It is important to note that these control samples were not the same in each plate but they were evaluated several times to determine inter-assay variation. Additionally, since 2010 we are involved in an external quality control that includes sample exchange between research groups from Argentina, Brazil, and Mexico (Burin et al. 2011).
Deficient activity values were found in the screening assays for the DBS for the different enzymes under study and were confirmed in leukocyte assays performed in accordance with the protocols designed by Shapira et al. (1989), Voznyi et al. (2001), and Kallwas et al. (2007) (see Supplementary Table). Protein quantities were determined according to Lowry et al. (1951) and BCA (ThermoScientific) assays.
Statistical Analysis
An inter- and intra-assay coefficient of variance (CV) was determined as the average value taken from all the enzymes analyzed. Descriptive statistics were performed using the IBM SPSS Statistic 19 software provided by SPSS Inc. (Special Package for the Social Sciences, Chicago, USA). A nonparametric Shapiro-Wilk test was used to evaluate the distribution of control groups and a Mann-Whitney test was used to determine the difference between the populations under study. The cutoff between the enzymatic activities of controls and patients was established using ROC (Receiver Operating Characteristics) curves with the same program.
Results and Discussion
Enzymatic analysis of the control population samples on filter paper allowed establishing reference values for the Colombian population for nine enzymes that are directly related to hereditary disorders involving lysosomal metabolism. Samples from a total of 4,700 patients, whose clinical results were indicative of an LSD, were received during the study period (2005–2011) from all over the nation.
This high-risk screening led to the detection of 242 patients (5.2 %) affected by different LSDs. We found 242 with enzyme deficiencies, leading to the following diagnoses: Fabry disease (n = 31), Pompe disease (n = 16), Hurler Syndrome (n = 15), Maroteaux-Lamy Syndrome (n = 34), GM1 Gangliosidosis (n = 10), Morquio B (n = 1), Gaucher disease (n = 101), Sandhoff disease (n = 1), Mucolipidosis (n = 2), and Hunter Syndrome (n = 31). The enzymatic activities obtained showed significant differences (p < 0.001) in the catalytic agents measured with respect to the control group (Table 2).
Table 2.
Results of enzymatic activity on controlaand patient DBS
| Enzyme | n | Activity (nmol/ml/h) Average DS Range | CV1 | CV2 | Pb | Cutoffc |
|---|---|---|---|---|---|---|
| Arylsulfatase B | ||||||
|
Mucopolysaccharidosis type VI
(age range: 1.5–17 years) |
34 | 0.0–2.06 M: 1.2 DS:0.67 |
6.4 | 11.47 | <0.0001 | 2.7 |
| Controls (<36 days) | 21 | 4.0–31.5 M: 10.1 DS: 5.9 |
||||
| Controls (3 months–59 years) | 625 | 2.9–43.2 M: 9.2 DS: 5.6 |
||||
| α-Galactosidase A | ||||||
|
Fabry disease – men
(age range: 10–50.1 years) |
23 | 0.0 –0.3 M: 0.08 DS: 0.09 |
8.2 | 10.7 | <0.0001 | 1.15 |
|
Fabry disease
d
- heterozygotes
(age range: 28–67 years) |
8 | 0.21–3.34 M: 1.0 DS: 1.0 |
||||
| Controls (<36 days) | 80 | 2.5–21.9 M: 11.3 DS: 5.2 |
||||
| Controls (3 months–88 years) | 2337 | 2.0–21.8 M: 7.4 DS: 3.5 |
||||
| α-Iduronidase | ||||||
|
Mucopolysaccharidosis type I
(age range: 8 months–27.7 years) |
15 | 0.0–0.65 M: 0.25 DS: 0.23 |
5.2 | 11.3 | <0.0001 | 1.1 |
| Normal control (<36 days) | 48 | 3.0–19.6 M: 9.9 DS: 3.6 |
||||
| Normal controls (3 months–59 years) | 1585 | 1.5–20.1 M: 9.5 DS: 3.8 |
||||
| β-Galactosidase | ||||||
|
Gangliosidosis GM 1
(age range: 7 months–4 years) |
10 | 0.1–2.3 M: 1.4 DS: 0.7 |
5.3 | 13.4 | <0.01 | 11.4 |
|
Mucopolysaccharidosis type IVB
(age: 4 years) |
1 | 2.1 | ||||
| Controls (3 months–88 years) | 2354 | 19–99 M: 47 DS: 16 |
||||
| Controls (<36 days) | 166 | 28–164 M: 71 DS: 26 |
||||
| β-Glucosidase | ||||||
|
Gaucher disease
(age range: 4 months–72.8 years) |
101 | 0.39–5.3 M:3.6 DS:1.14 |
15 | 14.7 | <0.0001 | 5.6 |
| Controls (6 months–63 years) | 715 | 5.9–16.8 M:9.6 DS: 2.6 |
||||
| Total Hexosaminidase | ||||||
|
Sandoff disease
(age: 1 years) |
1 | 42.1 | 4.1 | 10.2 | <0.0001 | 111.4 |
| Normal controls (3 months–88 years) | 1820 | 180.8–750.2 M: 396.3 DS:120.8 |
||||
| Controls (<36 days) | 114 | 284.3–884.1 M: 527.2 DS: 130.2 |
||||
|
Overexpression of total
Hexosaminidase activity** |
32 | 856.4–4882.8 M: 1538.3 DS: 913.1 |
||||
| Iduronate Sulfatase | ||||||
|
Mucopolysaccharidosis type II
(age range: 1.1–35 years) |
31 | 0.0 –1.86 M: 0.86 DS: 0.43 |
8.0 | 11.2 | <0.0001 | 6.3 |
| Controls (<36 days) | 11 | 19.1–44.2 M:32.8 DS: 9.3 |
||||
| Controls (3 months–59 years) | 210 | 10.7–45.2 M: 24.8 DS: 7.3 |
||||
| Chitotriosidase | ||||||
|
Patients affected by Gaucher disease
and elevated chitotriosidase |
86 | 120.9–3479.8 M:1108.9 DS: 789.9 |
9.4 | 12.4 | <0.0001 | 96.7 |
|
Patients affected by Gaucher disease
without elevated chitotriosidase* |
15 | 0.0–80.8 M: 41.3 DS: 34.8 |
||||
|
Patients affected by other lysosomal
disorders*** |
74 | 20.3 – 1649.6 M: 229.1 DS:231.5 |
||||
| Controls (3 months–78 years) | 518 | 2.3 –96.1 M:36.3 DS:23.2 |
aAccording to the Shapiro-Wilk test, all control groups analyzed failed to show a normal distribution (Sig. <0.05 with 95% confidence)
bCited values correspond to the results of the non-parametric Mann-Whitney test comparing activity values of individuals affected by enzyme deficiency and healthy controls
cObtained through ROC (Receiver Operating Characteristics) analysis, based on a comparison between results for patients and control individuals (99 % confidence, 100 % sensitivity, and 100 % specificity). Both sensitivity and specificity values refer to the certainty offered by each cutoff for the enzyme analyzed. The ROC analysis was made with the complete dataset
dResults correspond to obligated heterozygotes (mothers of Fabry patients) who at the time of this writing are still not under clinical follow-up
*All referred patients with clinical findings suggesting Gaucher disease were evaluated for chitotriosidase. Eight cases showed increased Chitotriosidase activities (range: 105–1650 nmol/ml/h) with normal β–glucosidase activity in leukocyte samples; subsequent studies indicated Niemmann Pick disease (data not shown)
**The values shown do not establish a cutoff for the overexpression. Some patients with a confirmed diagnosis of LSD (n = 32) showed increases of 1.5 to 6.5 times above the reference value. (range: 180.8–750.2 nmol/ml/h). This study is in progress
***Patients with other LSDs and increased chitotriosidase: Gangliosidosis GM1 (n = 3), Fabry disease (n = 3), MPS I (n=2), MPS II (n = 1), MPS VI (n=1), MPS VII (n = 1)
CV1: Intra assay variability coefficient. (%)
CV2: Inter assay variability coeffcient. (%) (n = 30)
Internal quality controls provided evidence that the analytical techniques used here are reproducible. To this end, samples were evaluated in triplicate and the time between sampling and processing was interior to 30 days. The assays demonstrated an average intra-assay coefficient of variance of 7.7 % for all the enzymes analyzed and an inter-assay variability coefficient of 11.9 % (n = 30). The highest values found for any the enzymes analyzed in this study were detected in the β-glucosidase assay, where the intra- and inter-assay coefficients showed values of 15 % and 14.7 %.
When working with DBS samples, the suggested storage temperature for the filter paper is − 20 °C. However, in accordance to our experience and previous studies (Gasparotto et al. 2009; de Castilhos et al. 2011; De Jesus et al. 2009), the decrease in enzymatic activity from DBS samples stored at 4 °C in 60 days is less than 6 %, which means that there is no significant difference in the sample storage at 4 °C or − 20 °C.
Upon following up on the condition of the patients remitted for β-glucosidase, all cases showed evidence of various levels of leukopenia, which can affect sample quality and has been reported to be a variant that might affect the analytical processes (Civallero et al. 2006; Jiménez et al. 2011) (Table 2). In the present study, DBS analyses of β-glucosidase detected 103 deficient patients and the leukocyte assay confirmed only 101 Gaucher patients, finding two false positives and suggesting a possible interference due to low leukocyte counts. The other enzymes analyzed did not have this problem, and showed a 100% correlation between DBS and leukocyte samples.
In regard to the interferences reported in the analysis of β-glucosidase, both the studies in DBS and the leukocytes included the use of conduritol-β-epoxide (CBE, 0.5 mM), which has been described as a potent inhibitor of β-glucosidase, and allowed us to discriminate the activity of other isoenzymes that could degrade the fluorogenic substrate (Olivova et al. 2008). Additionally, it is important to highlight that the described DBS protocols are not made for diagnostic purposes, they are designed to orient the diagnosis, but definitely this must be confirmed with the measurement of enzymatic activities in cellular extracts.
The different methodologies led to a significant differentiation of enzymatic activities between control subjects and patients with an enzymatic deficiency. However, the evaluation of α-glucosidase levels from whole blood samples presented a challenge, given that three enzyme forms unrelated to the disease showed catalytic activity on 4-MU-α-d-glucopyranoside. This finding was demonstrated in the present study, where total catalytic activity was not different between the healthy controls and the patients (p < 0.176). A modified technique was used based on the one reported by Kallwass et al. (2007) and Li et al. (2004), which permitted the discrimination of the activity of the different isoforms using acarbose as an inhibitor. This method achieved significant differences between the controls and the individuals affected by Pompe disease (see Table 3).
Table 3.
Results of DBS enzyme activity for α-glucosidase
| Healthy controls | |||
|---|---|---|---|
| Age | <36 days | 6 months–89 years | Affected by Pompe disease* |
| n | 209 | 1,574 | 16 |
| Range/Median/Standard deviationa | |||
|
α-Glucosidase Total Activity
1
(α-Glu)=A |
R: 5.3–70.6 M:18 DS: 7.6 |
R: 3.7–57.6 M:18.2 DS:7.7 |
R: 4.8–64.2 M:20.1 DS:16.1 |
|
Inhibited α-Glucosidase
2
(α-GluINH)=B |
R: 1.8–10.9 M:4.4 DS: 1.9 |
R: 1.8–10.7 M:4.9 DS:2.0 |
R: 0.3–4.6 M:1.5 DS:1.2 |
|
Neutral α-Glucosidase
3
(α-GluNeu)=C |
R: 12.1–96.5 M:36.3 DS: 13.6 |
R: 9.8–147.3 M:38.6 DS:14.6 |
R: 24.2–191.6 M:57.0 DS:42.9 |
|
Ratio
4,5
α-GluNeu/α-GlulNH |
R: 3.0–16.0 M:8.0 DS: 2.9 |
R: 1.4–16.0 M:9.5 DS:3.2 |
R: 19.0–114.2 M:46.6 DS:24.8 |
| % Inhibition 5 | R: 40.4–86.0 M:71.0 DS:9.8 |
R: 25.8–85.0 M:74.7 DS:8.4 |
R: 87.2–95.5 M:92.4 DS:2.4 |
1A comparative statistical analysis of α-Glu between healthy controls and affected patients did not reveal significant differences (Mann-Whitney test P<0.176, Sig. 0.05)
2The significant differences (Mann-Whitney test P<0.0001, Sig. 0.05) among different evaluation of α-GlulNH is performed using acarbose as an inhibitor. Comparative analysis demonstrates control groups and patients affected with Pompe disease
3α-GluNeu corresponds to an isoform of α-Glucosidase evaluated at pH 7.0. Statistical analysis shows significant differences between healthy controls and affected patients (Mann-Whitney test P <0.0001, Sig. 0.05)
4Ratio neutral / inhibited = C/B, % inhibition = (B*100)/A
5Enzymatic evaluations for α-Glu, α-GlulNH and α-GluNeu allowed the calculation of the relation between enzymatic isoforms and the percentage of inhibition generated by the presence of acarbose. Values found for individuals affected by Pompe disease were significantly different from control groups analyzed (Mann-Whitney test P <0.0001, Sig. 0.05)
aActivity in nmol/ml/h
*Molecular studies has been made for some of these patients (see Niño et al. 2012 and Supplementary Table)
Chitotriosidase analyses, which were originally intended to assist in the diagnosis of patients suspected to suffer from Gaucher disease, also allowed for the evaluation of other LSDs in many cases (n = 74). Patients with Niemann-Pick disease, Mucopolysaccharidosis, and Gangliosidosis, among others, showed variable levels of this biomarker (Table 2). While its detection was nonspecific, an increase in the levels of this biomarker could be of great use in the detection of these disorders. In relation to Gaucher patients, Table 2 shows two different groups classified according to their chitotriosidase activities (elevated and non elevated with respect to the reference values), indicating that, although useful, chitotriosidase cannot always be used as a tool for a diagnostic approach. In the present study, we found that 14.9 % of patients with Gaucher disease did not show a marked increase in this biomarker. Moreover, in a previous study that evaluated Colombian healthy subjects and patients with LSDs, 2.6 % of control subjects showed no activity for this enzyme (data not shown, article in preparation).
The present study also allowed the detection of one patient with Morquio IVB. In this case, the patient was referred because he showed dysmorphic features and short stature without mental commitment. The obtained results for arylsulfatase B, α-l-iduronidase, and iduronate sulfatase showed normal values. However, this patient showed a marked decrease in the enzymatic activity of one of the control enzymes (β-galactosidase). In order to support diagnosis, we made a quantification of glycosaminoglycans in urine samples, obtaining a value 1.5 times above the reference value. The electroforetic pattern fund in this sample showed excretion of keratan sulfate, which, in correlation with clinical symptoms, allowed the diagnosis of a mucopolysaccharidosis type IVB (Morquio B). We also find a patient with Sandhoff disease, whose deficiency was confirmed measuring total hexosaminidase activity and the percentage of hexosaminidase A in leukocytes (see Supplementary Table).
Another finding of great relevance was observed while evaluating β-galactosidase and total hexosaminidase in all samples as a quality control for blood extraction, drying, and sample transport. The total hexosaminidase analysis exhibited variable levels of increased enzymatic activity (n = 32, from 1.2 to 6.5 times above the reference value) with significant differences (p < 0.001) compared to the control groups. In conducting a follow-up with previously confirmed cases, increases in activity could be related to patients with Mucopolysaccharidosis, Gangliosidosis, Gaucher disease, Pompe disease, and Mucolipidosis. This result might suggest that for this enzymatic quantification, beyond the ability to detect Sandhoff disease, a nonspecific elevation might be a useful indicator of a lysosomal disease. This aspect is currently under investigation and could be of great assistance for the high-risk screening of these disorders (Table 2).
When comparing DBS and leukocyte samples, we could not demonstrate a quantitative exact correlation between both samples because of the sample’s nature. While the activity in DBS is calculated over an approximate blood volume in the punch, the leukocyte activity is normalized to the protein quantity measured in the sample, making the latter a more accurate measure. Nevertheless, qualitatively the correlation is very good.
We could not perform molecular studies for all the deficiencies found due to the costs that these would imply. However, in association with another research group, we were able to develop a molecular protocol for Pompe disease, and 11 volunteer patients were tested for a complete study (Niño et al. 2012). At this time, patients with Fabry disease and Hunter syndrome are under mutation analyses.
Some articles have described interferences due to fluorescence quenching caused by hemoglobin (Oermardien et al. 2011). Our protocol uses a 1.2 mm punch, which dilutes the quantity of hemoglobin in the reaction, considerably diminishing this kind of interference. These modifications also allow to make the sample processing easier, since there is no need to make a previous elution (the punch is directly placed in the 96-well plate) and does not need addition of other reagents or posterior centrifugation to get better lectures, as has been described by Oermardien et al. (2011). Additionally, a smaller punch allowed us to make retrospective studies, which is a very important fact given the difficulty of finding some of these patients after the first sampling. The use of 1.2 mm punches allows us to reanalyze them several times and orient to a diagnosis that can, in some cases, be different from the initial approach.
Given the little information available in the literature about the state of the art of these entities in our countries, it is difficult to implement early detection protocols for LSDs, leading to a late diagnosis and, in the worst situation, to the death of patients without a diagnosis. Given that physicians and health management entities are more interested in the studies when these are made in their own countries than when they take place outside, we think this study will generate more interest from the medical community toward lysosomal storage diseases.
Although working with 4-MU substrates is not new and several authors have reported similar assays before, in Colombia, Andean countries, and several Central American countries, there are no high-risk screening studies for lysosomal storage diseases that involve such a large number of patients with a representative number of affected subjects, over such a long period of time. In our country, the funding for research in these orphan entities and the investment from the government in these diseases is very low. With this study we want to present an alternative solution that would help achieve diagnosis for patients in Colombia and the neighboring countries.
Conclusions
The results of this study offer reference values for healthy controls and individuals affected by nine lysosomal enzymes, allowing for the diagnostic approach of eight lysosomal disorders.
To date in Colombia, Andean countries, and several Central American countries, there are no high-risk screening studies for lysosomal storage diseases that involve such a large number of patients with a representative number of affected subjects, over such a long period of time. In this regard, in the 10 years prior to the start of this project (1995–2004), our laboratory performed enzymatic evaluations on only 751 patients with clinical characteristics related to these disorders which resulted in 26 confirmed cases. This limitation was due to the aforementioned difficulties regarding the handling of liquid samples, which led us to believe that the implementation of dried blood spots collected on filter paper would increase the number of patients that could be tested in the upcoming years. Here we report our increased testing capacity that allows individuals with clinical results related to these disorders to be more easily studied regardless of their geographical location, and, in addition, permits the analysis of a high volume of samples.
The final results of this study offer a general view of LSDs in Colombia, disorders that were, until recently, considered an extreme rarity in the medical field. It is expected that these preliminary findings could stimulate state-sponsored health systems to systematically screen for these disorders and to support reference centers which could offer diagnostic testing, facilitating the detection of these disorders and enabling a timely treatment for many patients affected by LSDs.
Electronic Supplementary Material
Acknowledgments
The authors would like to express thanks to Dr. Barbara Zimmermann at the University of los Andes, Bogota, Colombia; Dr. Maira Burin and the laboratory of Inborn Errors of Metabolism, Medical Genetics Service of Hospital de Clinicas de Porto Alegre, Federal University of Rio Grande do Sul (UFRGS), Brazil; and Dr. Nestor Chamoles (deceased) and his research group at the Neurochemistry Laboratory, Buenos Aires, Argentina, for their guidance and support in the development of this study.
The author also wishes to thank the Faculty of Sciences and Department of Biological Sciences at the University of los Andes, Genzyme of Colombia, and Biomarin of Colombia for the support they provided for this project.
Synopsis
In this report, of a 7-year screening study, we demonstrate the usefulness of dried blood spots on filter paper for the high-risk screening of lysosomal storage disorders.
Footnotes
Electronic supplementary material: The online version of this chapter (doi:10.1007/8904_2013_229) contains supplementary material, which is available to authorized users.
Contributor Information
Alfredo Uribe, Email: jeuribe@uniandes.edu.co.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Beratis N, Aaron A, Hirschhorn K. Metachromatic leukodystrophy: detection in serum. J Pediatr. 1973;83(5):824–827. doi: 10.1016/S0022-3476(73)80377-4. [DOI] [PubMed] [Google Scholar]
- Brady RO. Enzyme replacement for lysosomal diseases. Annu Rev Med. 2006;57:283–296. doi: 10.1146/annurev.med.57.110104.115650. [DOI] [PubMed] [Google Scholar]
- Burin M, D’Almeida V, Carrillo J, Kallwas H, Schenone A, Uribe A (2011) Latin American external quality control: preliminary experience in analysis lysosomal enzymes from blood collected in filter paper. Oral presentation. Rev Gastroenterol Peru 31(Suppl 1):29
- Chamoles NA, Blanco MB, Gaggioli D. Fabry disease: enzymatic diagnosis in dried blood spots on filter paper. Clin Chim Acta. 2001;308:195–196. doi: 10.1016/S0009-8981(01)00478-8. [DOI] [PubMed] [Google Scholar]
- Chamoles NA, Blanco MB, Gaggioli D, Casentini C. Hurler-like phenotype: enzymatic diagnosis in dried blood spots on filter paper. Clin Chem. 2001;47:2098–2102. [PubMed] [Google Scholar]
- Chamoles NA, Blanco MB, Gaggioli D, Casentini C. Gaucher and Niemann-Pick diseases – Enzymatic diagnosis in dried blood spots on filter paper: retrospective diagnoses in newborn-screening cards. Clin Chim Acta. 2002;317:191–197. doi: 10.1016/S0009-8981(01)00798-7. [DOI] [PubMed] [Google Scholar]
- Chamoles NA, Niizawa G, Blanco M, Gaggioli D, Casentini C. Glycogen storage disease type II: enzymatic screening in dried blood spots on filter paper. Clin Chim Acta. 2004;347:97–102. doi: 10.1016/j.cccn.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Civallero G, Michelin K, de Mari J, Viapiana M, Burin M, Coelho JC, et al. Twelve different enzyme assays on dried-blood filter paper samples for detection of patients with selected inherited lysosomal storage diseases. Clin Chim Acta. 2006;372(1–2):98–102. doi: 10.1016/j.cca.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Daitx VV, Mezzalira J, Goldim MP, Coelho JC. Comparison between alpha-galactosidase A activity in blood samples collected on filter paper, leukocytes and plasma. Clin Biochem. 2012;45(15):1233–1238. doi: 10.1016/j.clinbiochem.2012.04.030. [DOI] [PubMed] [Google Scholar]
- de Castilhos CD, Mezzalira J, Goldim MP, Coelho JC. Influence of pre-analytical factors on α-galactosidase A, arylsulfatase B and α-glucosidase activities measured on dried blood spots on filter paper. Clin Biochem. 2011;44(10–11):922–926. doi: 10.1016/j.clinbiochem.2011.03.138. [DOI] [PubMed] [Google Scholar]
- De Jesus VR, Zhang XK, Keutzer J, Bodamer OA, Mühl A, Orsini JJ, Caggana M, Vogt RF, Hannon WH. Development and evaluation of quality control dried blood spot materials in newborn screening for lysosomal storage disorders. Clin Chem. 2009;55(1):158–164. doi: 10.1373/clinchem.2008.111864. [DOI] [PubMed] [Google Scholar]
- Filocamo M, Morrone A. Lysosomal storage disorders: molecular basis and laboratory testing. Hum Genomics. 2011;5(3):156–169. doi: 10.1186/1479-7364-5-3-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5:554–565. doi: 10.1038/nrm1423. [DOI] [PubMed] [Google Scholar]
- Gasparotto N, Tomanin R, Frigo AC, Niizawa G, Pasquini E, Blanco M, Donati MA, Keutzer J, Zacchello F, Scarpa M. Rapid diagnostic testing procedures for lysosomal storage disorders: alpha-glucosidase and beta-galactosidase assays on dried blood spots. Clin Chim Acta. 2009;402(1–2):38–41. doi: 10.1016/j.cca.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Gieselmann V. Lysosomal storage diseases. Biochim Biophys Acta. 1995;1270:103–136. doi: 10.1016/0925-4439(94)00075-2. [DOI] [PubMed] [Google Scholar]
- Jiménez LM, Bobillo J, Caro A, Duran P. β-galactosidase activity as an index of quality control of dried blood sample collected on paper. Revista Laboratorio Clínico. 2011;4(3):153–157. doi: 10.1016/j.labcli.2011.04.001. [DOI] [Google Scholar]
- Kallwass H, Carr C, Gerrein J, Titlow M, Pomponio R, Bali D, Dai J, Kishnani P, Skrinar A, Corzo D, Keutzer J. Rapid diagnosis of late-onset Pompe disease by fluorometric assay of alpha-glucosidase activities in dried blood spots. Mol Genet Metab. 2007;90(4):449–452. doi: 10.1016/j.ymgme.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Li Y, Scott CR, Chamoles NA, Ghavami A, Pinto BM, Turecek F, Gelb MH. Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clin Chem. 2004;50(10):1785–1796. doi: 10.1373/clinchem.2004.035907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193(1):265–275. [PubMed] [Google Scholar]
- Müller KB, Rodrigues MD, Pereira VG, Martins AM, D'Almeida V. Reference values for lysosomal enzymes activities using dried blood spots samples - a Brazilian experience. Diagn Pathol. 2010;29(5):65. doi: 10.1186/1746-1596-5-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niizawa G, Blanco MB, Levin C, Aranda C, Chamoles NA. Retrospective diagnosis of glycogen storage disease type II by use of newborn-screening card. Clin Chim Acta. 2005;359:205–206. doi: 10.1016/j.cccn.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Niño MY, Mateus HE, Fonseca DJ, Kroos MA, Ospina SY, Mejía JF, Uribe JA, Reuser AJ, Laissue P. Identification and functional characterization of GAA mutations in Colombian patients affected by Pompe disease. JIMD Rep. 2012;7:39–48. doi: 10.1007/8904_2012_138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oemardien LF, Boer AM, Ruijter GJ, van der Ploeg AT, de Klerk JB, Reuser AJ, Verheijen FW. Hemoglobin precipitation greatly improves 4-methylumbelliferone-based diagnostic assays for lysosomal storage diseases in dried blood spots. Mol Genet Metab. 2011;102(1):44–48. doi: 10.1016/j.ymgme.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Olivova P, Cullen E, Titlow M, Kallwass H, Barranger J, Zhang K, Keutzer J. An improved high-throughput dried blood spot screening method for Gaucher disease. Clin Chim Acta. 2008;398(1–2):163–164. doi: 10.1016/j.cca.2008.08.024. [DOI] [PubMed] [Google Scholar]
- Reuser AJ, Verheijen FW, Bali D, van Diggelen OP, Germain DP, Hwu WL, Lukacs Z, Mühl A, Olivova P, Piraud M, Wuyts B, Zhang K, Keutzer J. The use of dried blood spot samples in the diagnosis of lysosomal storage disorders–current status and perspectives. Mol Genet Metab. 2011;104(1–2):144–148. doi: 10.1016/j.ymgme.2011.07.014. [DOI] [PubMed] [Google Scholar]
- Rodrigues MD, de Oliveira AC, Müller KB, Martins AM, D'Almeida V. Chitotriosidase determination in plasma and in dried blood spots: a comparison using two different substrates in a microplate assay. Clin Chim Acta. 2009;406(1–2):86–88. doi: 10.1016/j.cca.2009.05.022. [DOI] [PubMed] [Google Scholar]
- Scriver CR, Sly WS, Childs B, Beaudet AR. The metabolic and molecular basis of inherited disease. 8. New York: McGraw-Hill; 2001. [Google Scholar]
- Shapira E, Blitzer MG, Miller JB, Africk DK. Biochemical genetics. A laboratory manual. New York: Oxford University Press; 1989. pp. 17–36. [Google Scholar]
- Singh J, Tavella D, Ferrante ND. Measurements of arylsulfatases A and B in human serum. J Pediatr. 1975;86(4):574–576. doi: 10.1016/S0022-3476(75)80152-1. [DOI] [PubMed] [Google Scholar]
- Staretz-Chacham O, Lang TC, LaMarca ME, Krasnewich D, Sidransky E. Lysosomal storage disorders in the newborn. Pediatrics. 2009;123:1191–1207. doi: 10.1542/peds.2008-0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolk P, Willeme JC, Leufkens GM. Rare essentials: drugs for rare diseases as essential medicines. Bull World Health Organ. 2006;84(9):745–751. doi: 10.2471/BLT.06.031518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellodi A. Lysosomal storage disorders. Br J Haematol. 2005;128:413–431. doi: 10.1111/j.1365-2141.2004.05293.x. [DOI] [PubMed] [Google Scholar]
- Voznyi YV, Keulemans J, Beyer EM, van Diggelen OP. A fluorogenic assay for the diagnosis of Hunter disease (MPS II) J Inherit Metab Dis. 2001;24:675–80. doi: 10.1023/A:1012763026526. [DOI] [PubMed] [Google Scholar]
- Wenger DA, Coppola S, Liu SL. Lysosomal storage disorders: diagnostic dilemmas and prospects for therapy. Genet Med. 2002;4(6):412–419. doi: 10.1097/00125817-200211000-00003. [DOI] [PubMed] [Google Scholar]
- Whiteman P. The Quantitative measurement of Alcian Blue glycosaminoglycan complexes. Biochem J. 1973;131:343–350. doi: 10.1042/bj1310343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wraith JE. Lysosomal disorders. Semin Neonatol. 2002;7:75–83. doi: 10.1053/siny.2001.0088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.