

Abstract
Background: Fatty acid oxidation (FAO) disorders are a heterogeneous group of inborn errors in the transportation and oxidation of fatty acids. FAO disorders were thought to be very rare in the Chinese population. Newborn screening for FAO disorders beginning in 2002 in Taiwan may have increased the diagnosis of this group of diseases.
Materials and Methods: Till 2012, the National Taiwan University Hospital Newborn Screening Center screened more than 800,000 newborns for FAO disorders. Both patients diagnosed through screening and patients detected after clinical manifestations were included in this study.
Results: A total of 48 patients with FAO disorders were identified during the study period. The disorders included carnitine palmitoyltransferase I deficiency, carnitine acylcarnitine translocase deficiency, carnitine palmitoyltransferase II deficiency, very long-chain acyl-CoA dehydrogenase deficiency, medium-chain acyl-CoA dehydrogenase deficiency, multiple acyl-CoA dehydrogenase deficiency, short-chain defects, and carnitine uptake defect. Thirty-nine patients were diagnosed through newborn screening. Five false-negative newborn screening cases were noted during this period, and four patients who were not screened were diagnosed based on clinical manifestations. The ages of all patients ranged from 6 months to 22.9 years (mean age 6.6 years). Except for one case of postmortem diagnosis, there were no other mortalities.
Conclusions: The combined incidence of FAO disorders estimated by newborn screening in the Chinese population in Taiwan is 1 in 20,271 live births. Newborn screening also increases the awareness of FAO disorders and triggers clinical diagnoses of these diseases.
Introduction
Fatty acid oxidation (FAO) disorders are a heterogeneous group of inborn errors involving the transportation or oxidation of fatty acids, including the uptake and activation of fatty acids, the carnitine cycle, and the β-oxidation spiral (Roe and Ding 2001). Long-chain fatty acids, activated by being bound to their CoA esters in the cytosol, are shuttled across the barrier of the inner mitochondrial membrane by the carnitine cycle (Brivet et al. 1999). This cycle is composed of four steps mediated by a plasma membrane carnitine transporter, two carnitine palmitoyltransferases (CPT I and CPT II), and a carnitine-acylcarnitine translocase (CATC). In the mitochondria, β-oxidation of fatty acids occurs and requires a sequential action of very-long-chain acyl-CoA dehydrogenase (VLCAD), long-chain 3-hydroxy-acyl-CoA dehydrogenase (LCHAD), medium-chain acyl-CoA dehydrogenase (MCAD), and short-chain acyl-CoA dehydrogenase. Those dehydrogenases transfer electrons to ubiquinone via two flavoproteins—electron transfer flavoprotein (ETF) and ETF:ubiquinone oxidoreductase (ETF:QO). Impaired transfer of electrons affects multiple dehydrogenation reactions and is thus termed multiple acyl-CoA dehydrogenase (MAD) deficiency. FAO disorders can present with life-threatening symptoms, such as hypoketotic hypoglycemia, Reye-like syndrome in infants (Brivet et al. 1999; Baruteau et al. 2009; Spiekerkoetter 2010), acute encephalopathy (Gregersen et al. 2001; Spiekerkoetter 2010), cardiomyopathy (Bonnet et al. 1999; Spiekerkoetter et al. 2009a), myolysis (Spiekerkoetter et al. 2009a; van Adel and Tarnopolsky 2009), and liver dysfunction (Clayton 2003). Currently more than 15 distinct FAO disorders have been elucidated based on enzymatic and/or molecular analyses (Gregersen et al. 2008). Among them, MCAD deficiency is the most common disease in Caucasians of northern European origin (Roe and Ding 2001). Before newborn screening for these disorders, FAO disorders were characterized by high morbidity and mortality (Wanders et al. 1999) and required strict treatment measures. The mortality rate of FAO disorders was approximately 48% but could be higher in specific diseases such as VLCAD deficiency (60%) or CATC deficiency (92%) (Baruteau et al. 2012).
After the development of tandem mass spectrometry (MS/MS) analysis of acylcarnitines (Millington et al. 1990), the number of disorders that could be detected by newborn screening greatly increased (Huang et al. 2006), and most aminoacidopathies, FAO disorders, and organic acidurias became diagnosable. The combined incidence of FAO disorders reaches 1:9,000 in some countries, and half of the detected patients are affected by MCAD deficiency (Zytkovicz et al. 2001; Lindner et al. 2010). Early institution of high-glucose infusion, a low-fat diet, and avoidance of fasting can prevent the occurrence of metabolic decompensation (Baruteau et al. 2012). Therefore, MS/MS newborn screening (NBS) has been shown to improve the outcome of patients, and this screening is also cost effective (Pandor et al. 2004). Currently, different sets of FAO disorders are screened in different countries. The American College of Clinical Genetics proposed 29 core and 25 secondary conditions for screening, which include most of the FAO disorders (2006), while only 5 FAO disorders are included in screening in Germany (Lindner et al. 2011). In Taiwan, MS/MS was introduced in 2001 (Huang et al. 2006); however, only MCAD deficiency is recommended by the Bureau of Health Promotion, Department of Health, and all other FAO disorders can be screened only after the parents’ consent (Niu et al. 2010).
The incidence of FAO disorders in the Chinese population was thought to be very low, and no patients except those with carnitine uptake defects (CUD) had been diagnosed before the initiation of NBS. However, the under-diagnosis of FAO disorders is likely, and NBS provides an opportunity to learn their true incidences. Unfortunately, data from previously published NBS pilot studies were not comprehensive (Huang et al. 2006; Niu et al. 2010); for example, the high incidence of CUD in the Chinese population was only realized at a later time (Lee et al. 2010; Chen et al. 2013). In this report, we present single-center longitudinal data over a 10-year period for the diagnosis and treatment of FAO disorders from both NBS and clinical recognition.
Materials and Methods
MS/MS NBS was performed at the National Taiwan University Hospital (NTUH) Newborn Screening Center. Dried blood spot (DBS) samples were obtained from newborn babies 48 to 72 h after birth, and acylcarnitine profiles were analyzed by MS/MS NBS as described (Huang et al. 2006). The results of the screening were available 24–48 h after the DBS samples were received. Two cutoff values were set for each metabolite (Table 1). Newborns with an initial screening value exceeding the diagnostic cutoff were requested for a confirmatory test at our hospital. Newborns with an initial screening value not exceeding the diagnostic cutoff but equal to or exceeding the screening cutoff (the inconclusive cases) were requested for a second screening. If the second screening results were still abnormal, the babies were requested for a confirmatory test. The targeted FAO disorders were carnitine palmitoyltransferase I (CPT I) deficiency, carnitine palmitoyltransferase II (CPT II) deficiency, carnitine acylcarnitine translocase (CACT) deficiency, very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency, long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency, medium-chain acyl-CoA dehydrogenase (MCAD) deficiency, short-chain defects, multiple acyl-CoA dehydrogenase (MAD) deficiency, and carnitine uptake defect (CUD). The confirmatory test includes enzyme assays and/or mutation analyses by accredited clinical laboratories when available (information provided upon request). Immediately after receiving a positive confirmation test, the patients were put on treatment. Short chain defects are generally benign conditions (Gallant et al. 2012), so babies positive for them were not put on treatment. The treatment and follow up of CUD patients have been described previously (Lee et al. 2010; Chen et al. 2013). In brief, the CUD patients were all given carnitine supplements, and none of them experienced metabolic decompensation or cardiac problems after treatment. For other FAO disorders, a low-fat diet (equal to or less than 30% of energy from fat) was the main treatment; for long-chain FAO disorders, this could be combined with a medium-chain triglyceride (MCT) supplement, and for secondary carnitine deficiency, this could be combined with a carnitine supplement.
Table 1.
Cutoff values for individual acylcarnitines
| Concentration (μM) | Screening cutoff | Diagnostic cutoff* |
|---|---|---|
| C0 | ≤ 6.44 | |
| C03 | ≥ 4.74 | ≥ 8.8 |
| C03DC | ≥ 0.42 | |
| C04 | ≥ 0.79 | |
| C04DC | ≥ 0.6 | |
| C05 | ≥ 0.4 | ≥ 1.1 |
| C05DC | ≥ 0.23 | ≥ 0.23 |
| C05OH | ≥ 0.56 | ≥ 2.2 |
| C06 | ≥ 0.3 | |
| C08 | ≥ 0.28 | ≥ 1.1 |
| C08:1 | ≥ 0.56 | |
| C10 | ≥ 0.26 | |
| C10:1 | ≥ 0.25 | |
| C12 | ≥ 0.73 | |
| C12:1 | ≥ 0.3 | |
| C14 | ≥ 0.52 | |
| C14:1 | ≥ 0.52 | |
| C14OH | ≥ 0.18 | |
| C16 | ≥ 6.4 | ≥ 8.8 |
| C16:1 | ≥ 0.61 | |
| C16OH | ≥ 0.18 | |
| C18 | ≥ 1.78 | |
| C18:1 | ≥ 2.75 | ≥ 3.3 |
| C18:1OH | ≥ 0.16 | |
| C0/(C16+C18) | ≥ 50 |
*Diagnostic cutoff was set for selected acylcarnitines
Between January 1, 2003, and December 31, 2012, 790,569 newborns were screened by the NTUH Newborn Screening Center. Cutoff values were not changed during the study period except for CUD diagnosis. Approximately 80,000 newborns screened before 2003 were excluded from the study because the cutoff values were not set at that time. Both NBS-confirmed cases and patients diagnosed through clinical manifestations were included in the study. Data analyses included screening results, clinical manifestations, genotypes, treatments, and outcomes. Because differential diagnoses of short-chain defects were not completed, and those patients did not need treatment, we did not include them in the outcome analysis.
Results
Incidences Determined from NBS
During the study period, 39 newborns were identified by screening to have FAO disorders, with an incidence of 1 in 20,271 live births or 4.53 cases per 100,000 live births (Table 2). In addition, we also identified 12 newborns whose mothers had CUD (the maternal CUD cases); those newborns only had transient carnitine deficiency and were not included in the current study. Among all FAO disorders, CUD was the most prevalent disease, with an incidence of 1 in 35,935 or 2.78 cases per 100,000 live births.
Table 2.
Case numbers and incidences of FAO defects in Taiwan
| Disease | Case number | Incidence (per 100 000 newborns) |
|---|---|---|
| CPT I deficiency | 1 | 0.13 |
| CPT II/CACT deficiency | 2 | 0.25 |
| VLCHAD deficiency | 2 | 0.25 |
| LCHAD deficiency | 0 | 0.00 |
| MCAD deficiency | 3 | 0.38 |
| Short-chain defects | 8 | 1.01 |
| MAD deficiency | 1 | 0.13 |
| CUD | 22 | 2.78 |
| Total | 39 | 4.93 |
Initial Clinical Manifestations
Two patients were symptomatic at the time of diagnosis through NBS. One patient with VLCAD deficiency presented with hypoglycemia shortly after birth and was found to have cardiomegaly at 10 days of age. The other patient had CACT deficiency that presented with hyperammonemia and hypoglycemia after birth. Others patients were asymptomatic at the time that screening results were obtained.
The False-Negative Cases
Five known false-negative cases that received screening from either our screening center or other centers were noted and listed in Tables 2 and 3. FN-1 was found to be affected by CUD after her younger brother was diagnosed. In her first DBS test, her free carnitine level was low, but low free carnitine levels were not reported in the initial pilot MS/MS screening. FN-2 was referred for hypotonia, an elevated serum creatine kinase (CK) level, and hyperammonemia after a fever episode at 1 year of age. She had experienced two previous uneventful episodes of acute bronchiolitis at the ages of 3 and 5 months. Her DBS acylcarnitine profile was typical of MAD deficiency but was not picked up by the screening algorithm (Table 3), and ETFDH gene p.A84T/p.F128S mutations were later confirmed (Er et al. 2011). FN-3 was referred for hepatomegaly and liver failure at 6 months of age; CPT II deficiency was suspected and confirmed by mutation analysis, which found two CPT2 gene mutations, p.F383Y and p.G601fsX621. FN-4 was diagnosed because her younger brother was found to have MAD deficiency. Data from her first screening was not available because it was performed by another screening center. She had no symptoms until the end of this study.
Table 3.
Initial screening results of acylcarnitine levels in the four false-negative cases
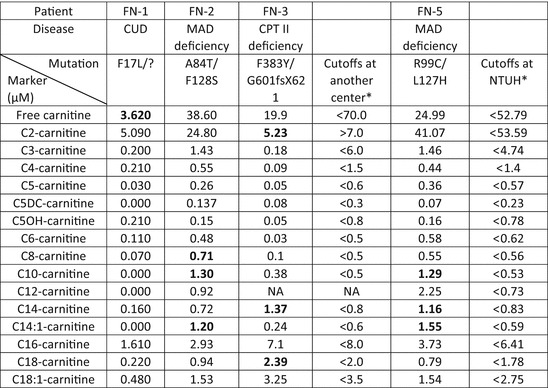
Bold characters indicate abnormal data
*FN-1, FN-2, and FN-3 were screened by another newborn screening center, and FN-5 was screened by the National Taiwan University Hospital (NTUH), so two different sets of cutoff values were presented
Recent studies suggest a high incidence of MAD deficiency in South China. Therefore, we retrospectively evaluated our newborn screening data using the Region 4 post-analytic tool (Marquardt et al. 2012). One newborn (FN-5) was found to have a high MAD deficiency score, and the diagnosis was later confirmed by mutation analysis.
The Clinical Cases
Four cases who did not receive NBS for FAO disorders came to our attention. Case 1 was a 20-year-old female patient who suffered from intermittent abdominal pain since the age of 15 years (Table 4). Chronic pancreatitis was suspected by magnetic resonance imaging (MRI) results, but a laparoscope examination at 19 years of age did not confirm pancreatitis; the laparoscopy showed many tiny stones in the gall bladder and resulted in a cystectomy. A diagnosis of CPT I deficiency was finally made when she was 20 years old. Case 3 was a 2.5-year-old female patient who presented with recurrent hypoglycemia, elevated liver enzymes, and hepatomegaly. She was put on a carnitine supplement, but still experienced three more attacks with consciousness disturbance after gastroenteritis. The final diagnosis made was CPT II deficiency when she was 3 years old. Case 9 died of poor activity and jaundice at 1 year of age. Her younger brother was later found to have VLCAD deficiency by NBS. Case 17 presented with proximal muscle weakness, dyspnea, and rhabdomyolysis at the age of 17 years. His DBS acylcarnitine profile was typical of MAD deficiency, and ETFDH gene p.A84T homozygous mutations were later found.
Table 4.
List of all patients except those with short-chain defect or CUD
| Case No. | Defective enzyme | Clinical presentation | Specific enzyme activity* | Genotype** | Diet fat intake |
|---|---|---|---|---|---|
| 1 | CPT I | At age of 15 years Recurrent acute pancreatitis |
0.080 (Normal > 0.42) |
c.1367C>T (p.S456L)/c.1433C>T (p.A478V) | 30% |
| 2 | CPT I | NBS | 0.110 (Normal 0.36 – 0.88) |
c.727C>T (p.L91P)/c.497G>T (p.S166I) | 30% |
| 3 | CPT II | At age of 3 years Hypoglycemia, hepatitis, hepatomegaly |
0.5 (Normal 15.8 ± 4.2) |
NA | 30% |
| 4 (FN-3) |
CPT II | At age of 6 months Hypoglycemia, metabolic acidosis, impaired liver function, hepatomegaly |
0.02 (Normal 0.4 – 1.85) |
c.1148 T>A (p.F383Y)/ c.1803delC (p.G601fsx621) | 25% |
| 5 | CPT-II? | NBS | 0.65 (Normal 0.4 – 1.85) |
c.1102 G>A (p.V368I) He | 30% |
| 6 | CACT | NBS (hyperammonemia, metabolic acidosis) |
NA | NA | NA |
| 7 | VLCAD | NBS | NA | c.1273 G>A (p.A425T)/c.1349 G>A (p.R450H) | 40% |
| 8 | VLCAD | NBS (hypoglycemia, cardiac hypertrophy) |
NA | c.1751+1 G>A/c.961 A>G (p.N321D) | <30% |
| 9 | VLCAD | Elevated liver enzymes, hyperammonemia, expired | NA | NA | NA |
| 10 | MCAD | NBS | NA | c.446_449delTGAC (p.T150RfsX4)/c.580A>G (p.N194D) | <30% |
| 11 | MCAD | NBS | NA | c.580A>G (p.N194D)/c.580A>G (p.N194D) | NA |
| 12 | MCAD | NBS | NA | NA | NA |
| 13 (FN-2) |
MAD | At age of 12 months Hypotonia, elevated liver enzymes, hyperammonemia |
Fibroblast assay: elevated C6-C12, C14, and C16-carnitines, very low C2 carnitine | c.250G>A (p.A84T)/c.383T>C (p.F128S) | <30% |
| 14 | MAD | NBS | Fibroblast assay: elevated C6-C16-carnitines, very low C2 carnitine | c. 295C>G (p.R99G)/? | Normal |
| 15 (FN-4) |
MAD | At age of 2.9 years Sibling screening |
NA | c. 295C>G (p.R99G)/? | Normal |
| 16 (FN-5) |
MAD | NBS | NA | c.295C>T (p.R99C)/c.380T>A (p.L127H) | NA |
| 17 | MAD | At age of 17 years Weakness and rhadomyolysis |
NA | c.250G>A (p.A84T)/c.250G>A (p.A84T) | <30% |
NA data not available, FN false-negative cases, NBS detected by newborn screening
*Specific enzyme activity: nmol/min/mg protein for CPT I and CPT II activity; fibroblast beta-oxidation study by acyl-3H-carnitine for MAD deficiency; normal ranges in parenthesis
**Genotype: CPT1A gene for CPT I deficiency; CPT2 gene for CPT II deficiency; ACADM gene for MCAD deficiency; ACADVL gene for VLCAD deficiency; ETFDH gene for MADD (Bold characters indicate novel mutations; ? indicates mutation not found)
Treatments and Courses
At the end of this study period, the mean age of all patients was 6.6 years (6 months to 22.9 years). A low fat diet was prescribed for 10 of the 12 (83%) patients with known dietary histories, and concomitant carnitine was prescribed for 3 (27%) patients. During the study period, no patients needed intensive care or prolonged hospitalization.
For the NBS cases, occasionally patients with CPT I, CPT II, VLCAD, or MAD deficiencies were put on oral or intravenous calorie supplementation because of concurrent illness. The two VLCAD deficiency patients experienced mild metabolic instability during follow up. Rhadomyolysis occurred in Case 7 (VLCAD deficiency) when she was infected by a Noro virus at the age of 2.5 years. One episode of elevation of CK (from 188 IU/L to 1838 IU/L) occurred in Case 8 (VLCAD deficiency) when he contracted an upper respiratory tract infection at the age of 3 months. None of these events were repeated.
Among the false-negative and clinically diagnosed patients, the girl affected by CPT I deficiency (Case 1) had poor compliance to the diet, and she continued to suffer from intermittent abdominal pain and muscle pain after stringent exercise. Case 3 (CPT II deficiency) was put on a low-fat diet, but she could not tolerate the MCT supplement and had approximately one episode of rhadomyolysis per year. Case 4 (FN-3, CPT II deficiency) started a low-fat diet, MCT supplement, and carnitine supplement after diagnosis. Although she received an intravenous calorie supply two times during concurrent illness, there was no metabolic decompensation. Case 13 (FN-2, MAD deficiency) was put on riboflavin, carnitine, and a low-fat, low-protein diet, but she had facial eczema and frequently needed hospitalization for fear of metabolic decompensation. Case 17 was put on riboflavin and a low-fat, low-protein diet, and he recovered from respiratory failure 1 month after the attack.
Discussion
Incidence of the FAO Disorders in the Chinese Population
Our NBS program disclosed an incidence of FAO disorders as 1 in 20,271. Han et al. have screened for FAO disorders by MS/MS NBS analysis for patients suspected to have inborn errors and found only a few cases (Han et al. 2007). Therefore, underdiagnosis before NBS was a serious issue. The incidence of FAO disorders in the Chinese population could be even higher if we included the false-negative cases. For example, CUD can only be detected by NBS in half of affected newborns because of carnitine transportation through the placenta from the mother (Wilcken et al. 2001; Lahjouji et al. 2004). CUD disease severity decreased in patients detected by NBS. Most of the mothers affected by CUD were asymptomatic (Chen et al. 2013). The carrier rate of the more severe CUD mutation in the Southern Chinese population, p.R254X, is approximately 1 in 125 (Tang et al. 2002). This mutation represented 50% of all mutations in clinically detected cases of CUD but represented only 30% of all mutations in NBS-detected cases (Lee et al. 2010), suggesting a decrease in the severity of the disease in the latter group of patients.
The Occurrences of False-Negative Cases in NBS
The occurrence of false-negative cases in NBS can be either technically or physiologically related. The acylcarnitine profile for MAD deficiency is composed of mild but multiple elevations of a group of acylcarnitines. Screening for MAD deficiency by analyzing the level of a single acylcarnitine results in low sensitivity. The detection rate of MAD deficiency may be improved by applying more sophisticated algorithms weighing several acylcarnitines together. Using an improved interpretive tool (Marquardt et al. 2012), we retrospectively detected one patient and confirmed the increased sensitivity of this tool. This is especially important because the incidence of the riboflavin-responsive form of MAD deficiency is high in Taiwan (Liang et al. 2009; Lan et al. 2010) and Southern China (Wang et al. 2011) owing to a high incidence of the p.A84T mutation in the ETFDH gene (a carrier rate of 1 in 74), and the treatment with carnitine and riboflavin is easy and effective.
Patients with FAO disorders may have normal acylcarnitines at birth. As we have discussed in the previous section, newborns with CUD can be missed by NBS because of normal free carnitine levels at birth (Wilcken et al. 2001; Lahjouji et al. 2004). A looser cut-off value can increase the sensitivity of the tests, but will also increase the rate of false-positives. Repeat screening for an inconclusive NBS result can also be dangerous. In many FAO disorders (Illsinger et al. 2008), especially VLCAD deficiency (Ficicioglu et al. 2010; Spiekerkoetter et al. 2010b; Sahai et al. 2011), repeat tests can have normal results in FAO patients, resulting in a false-negative screening. To solve these diagnostic issues, we have developed second-tier molecular tests for diseases in which common mutations occur (Wang et al. 2013).
Treat or Not
NBS detects significantly more patients with MCAD and short-chain acyl-CoA dehydrogenase (SCAD) deficiency than are detected by previous clinical manifestations (Wilcken et al. 2003; Gallant et al. 2012). Patients with MCAD deficiency can present with acute decompensation followed by a poor outcome, and, therefore, early diagnosis and treatment are important. However, some MCAD deficiency cases detected by NBS revealed novel mutations with no known clinical meaning. It may be prudent to follow such MCAD patients to avoid fasting during concurrent illnesses, but for SCAD deficiency the disease has been classified as a benign condition not needing treatment (Gallant et al. 2012).
In CUD, it can be difficult to predict the phenotype of asymptomatic patients detected by NBS; most of the mothers with CUD are also asymptomatic. However, we have encountered two mothers with CUD that were symptomatic: one died suddenly and one had undiagnosed cardiomegaly (Chen et al. 2013). Although phenotype prediction can be difficult (Rose et al. 2012), treatment for CUD is simple and safe. Therefore, it is prudent to maintain normal blood free carnitine levels for all patients with CUD (El-Hattab et al. 2010).
Clinical manifestations of VLCAD deficiency also vary. Patients with null mutations or other severe mutations (Andresen et al. 1996) should be immediately placed on a low-fat diet with supplemental calories provided by MCT supplementation (Solis and Singh 2002; Spiekerkoetter et al. 2009b). For patients with missense mutations or asymptomatic mothers with VLCAD deficiency detected through a false-positive NBS in their babies (McGoey and Marble 2011), a looser diet may be applicable (Spiekerkoetter et al. 2009b), but phenotype prediction for novel mutations may not be possible.
The role of low-fat diets in FAO disorders depends on the type and severity of the disease. For asymptomatic patients with long-chain defects, a normal diet may be tolerable. Intake of MCT oil prior to exercise can be effective to prevent myopathic symptoms (Spiekerkoetter et al. 2010a). L-carnitine supplementation in patients with long-chain defects can be dangerous (Spiekerkoetter et al. 2010a); however, we found secondary carnitine deficiency in 27% of our cases, and L-carnitine was prescribed to these patients to maintain a normal free carnitine level.
Conclusion
FAO disorders are not rare in Taiwan and the combined incidence is higher than 1 in 20,271 after NBS. CUD and MAD deficiency are the most common FAO disorders in Taiwan. NBS also increased the awareness of FAO disorders resulting in an increase in clinical diagnoses after the initiation of NBS. Early diagnosis and early initiation of treatment have led to a more favorable outcome for patients with FAO disorders as demonstrated in the current study.
Acknowledgment
We thank Dr. Nicola Longo from the University of Utah for performing the mutation analysis for patients of CPT I deficiency and CPT II deficiency. This study was partially support by grants (DOH94-HP-2203, DOH95-HP-2207) from the Department of Health, Taiwan.
Abbreviations
- CACT
Carnitine acylcarnitine translocase
- CPT I
Carnitine palmitoyltransferase 1
- CPT II
Carnitine palmitoyltransferase 2
- CUD
Systemic carnitine uptake defect
- FAO
Fatty acid oxidation
- LCHAD/mTFP
Long-chain 3-hydroxy acyl-CoA dehydrogenase/mitochondrial trifunctional protein
- MAD deficiency/GA II
Multiple acyl-CoA dehydrogenase deficiency/glutaric aciduria type II (synonym)
- MCAD
Medium-chain acyl-CoA dehydrogenase
- MS/MS
Tandem mass spectrometry
- NBS
Newborn screening
- SCAD
Short-chain acyl-CoA dehydrogenase
- VLCAD
Very-long-chain acyl-CoA dehydrogenase
Footnotes
Competing interests: None declared
Contributor Information
Wuh-Liang Hwu, Email: hwuwlntu@ntu.edu.tw.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- American College of Medical Genetics Newborn Screening Expert Group (2006) Newborn screening: toward a uniform screening panel and system--executive summary. Pediatrics 117(5 Pt 2): S296–307 [DOI] [PubMed]
- Andresen BS, Bross P, Vianey-Saban C, et al. Cloning and characterization of human very-long-chain acyl-CoA dehydrogenase cDNA, chromosomal assignment of the gene and identification in four patients of nine different mutations within the VLCAD gene. Hum Mol Genet. 1996;5(4):461–472. doi: 10.1093/hmg/5.4.461. [DOI] [PubMed] [Google Scholar]
- Baruteau J, Levade T, Redonnet-Vernhet I, Mesli S, Bloom MC, Broue P. Hypoketotic hypoglycemia with myolysis and hypoparathyroidism: an unusual association in medium chain acyl-CoA desydrogenase deficiency (MCADD) J Pediatr Endocrinol Metab. 2009;22(12):1175–1177. doi: 10.1515/JPEM.2009.22.12.1175. [DOI] [PubMed] [Google Scholar]
- Baruteau J, Sachs P, Broue P, et al (2012) Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: a French pediatric study of 187 patients. J Inherit Metab Dis 2012 Oct 3 [Epub ahead of print] [DOI] [PubMed]
- Bonnet D, Martin D, De Pascale L, et al. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation. 1999;100(22):2248–2253. doi: 10.1161/01.CIR.100.22.2248. [DOI] [PubMed] [Google Scholar]
- Brivet M, Boutron A, Slama A, et al. Defects in activation and transport of fatty acids. J Inherit Metab Dis. 1999;22(4):428–441. doi: 10.1023/A:1005552106301. [DOI] [PubMed] [Google Scholar]
- Chen YC, Chien YH, Chen PW, et al. Carnitine uptake defect (primary carnitine deficiency): risk in genotype-phenotype correlation. Hum Mutat. 2013;34(4):655. doi: 10.1002/humu.22286. [DOI] [PubMed] [Google Scholar]
- Clayton PT. Diagnosis of inherited disorders of liver metabolism. J Inherit Metab Dis. 2003;26(2–3):135–146. doi: 10.1023/A:1024429032116. [DOI] [PubMed] [Google Scholar]
- El-Hattab AW, Li FY, Shen J, et al. Maternal systemic primary carnitine deficiency uncovered by newborn screening: clinical, biochemical, and molecular aspects. Genet Med. 2010;12(1):19–24. doi: 10.1097/GIM.0b013e3181c5e6f7. [DOI] [PubMed] [Google Scholar]
- Er TK, Chen CC, Liu YY, et al. Computational analysis of a novel mutation in ETFDH gene highlights its long-range effects on the FAD-binding motif. BMC Struct Biol. 2011;11:43. doi: 10.1186/1472-6807-11-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficicioglu C, Coughlin CR, 2nd, Bennett MJ, Yudkoff M. Very long-chain acyl-CoA dehydrogenase deficiency in a patient with normal newborn screening by tandem mass spectrometry. J Pediatr. 2010;156(3):492–494. doi: 10.1016/j.jpeds.2009.10.031. [DOI] [PubMed] [Google Scholar]
- Gallant NM, Leydiker K, Tang H, et al. Biochemical, molecular, and clinical characteristics of children with short chain acyl-CoA dehydrogenase deficiency detected by newborn screening in California. Mol Genet Metab. 2012;106(1):55–61. doi: 10.1016/j.ymgme.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Gregersen N, Andresen BS, Corydon MJ, et al. Mutation analysis in mitochondrial fatty acid oxidation defects: exemplified by acyl-CoA dehydrogenase deficiencies, with special focus on genotype-phenotype relationship. Hum Mutat. 2001;18(3):169–189. doi: 10.1002/humu.1174. [DOI] [PubMed] [Google Scholar]
- Gregersen N, Andresen BS, Pedersen CB, Olsen RK, Corydon TJ, Bross P. Mitochondrial fatty acid oxidation defects– remaining challenges. J Inherit Metab Dis. 2008;31(5):643–657. doi: 10.1007/s10545-008-0990-y. [DOI] [PubMed] [Google Scholar]
- Han LS, Ye J, Qiu WJ, Gao XL, Wang Y, Gu XF. Selective screening for inborn errors of metabolism on clinical patients using tandem mass spectrometry in China: a four-year report. J Inherit Metab Dis. 2007;30(4):507–514. doi: 10.1007/s10545-007-0543-9. [DOI] [PubMed] [Google Scholar]
- Huang HP, Chu KL, Chien YH, et al. Tandem mass neonatal screening in Taiwan–report from one center. J Formos Med Assoc. 2006;105(11):882–886. doi: 10.1016/S0929-6646(09)60173-X. [DOI] [PubMed] [Google Scholar]
- Illsinger S, Lucke T, Peter M, et al. Carnitine-palmitoyltransferase 2 deficiency: novel mutations and relevance of newborn screening. Am J Med Genet A. 2008;146A(22):2925–2928. doi: 10.1002/ajmg.a.32545. [DOI] [PubMed] [Google Scholar]
- Lahjouji K, Elimrani I, Lafond J, Leduc L, Qureshi IA, Mitchell GA. L-Carnitine transport in human placental brush-border membranes is mediated by the sodium-dependent organic cation transporter OCTN2. Am J Physiol Cell Physiol. 2004;287(2):C263–C269. doi: 10.1152/ajpcell.00333.2003. [DOI] [PubMed] [Google Scholar]
- Lan MY, Fu MH, Liu YF, et al. High frequency of ETFDH c.250G>A mutation in Taiwanese patients with late-onset lipid storage myopathy. Clin Genet. 2010;78(6):565–569. doi: 10.1111/j.1399-0004.2010.01421.x. [DOI] [PubMed] [Google Scholar]
- Lee NC, Tang NL, Chien YH, et al. Diagnoses of newborns and mothers with carnitine uptake defects through newborn screening. Mol Genet Metab. 2010;100(1):46–50. doi: 10.1016/j.ymgme.2009.12.015. [DOI] [PubMed] [Google Scholar]
- Liang WC, Ohkuma A, Hayashi YK, et al. ETFDH mutations, CoQ10 levels, and respiratory chain activities in patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Neuromuscul Disord. 2009;19(3):212–216. doi: 10.1016/j.nmd.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner M, Hoffmann GF, Matern D. Newborn screening for disorders of fatty-acid oxidation: experience and recommendations from an expert meeting. J Inherit Metab Dis. 2010;33(5):521–526. doi: 10.1007/s10545-010-9076-8. [DOI] [PubMed] [Google Scholar]
- Lindner M, Gramer G, Haege G, et al. Efficacy and outcome of expanded newborn screening for metabolic diseases–report of 10 years from South-West Germany. Orphanet J Rare Dis. 2011;6:44. doi: 10.1186/1750-1172-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt G, Currier R, McHugh DM, et al. Enhanced interpretation of newborn screening results without analyte cutoff values. Genet Med. 2012;14(7):648–655. doi: 10.1038/gim.2012.2. [DOI] [PubMed] [Google Scholar]
- McGoey RR, Marble M. Positive newborn screen in a normal infant of a mother with asymptomatic very long-chain Acyl-CoA dehydrogenase deficiency. J Pediatr. 2011;158(6):1031–1032. doi: 10.1016/j.jpeds.2011.01.063. [DOI] [PubMed] [Google Scholar]
- Millington DS, Kodo N, Norwood DL, Roe CR. Tandem mass spectrometry: a new method for acylcarnitine profiling with potential for neonatal screening for inborn errors of metabolism. J Inherit Metab Dis. 1990;13(3):321–324. doi: 10.1007/BF01799385. [DOI] [PubMed] [Google Scholar]
- Niu DM, Chien YH, Chiang CC, et al. Nationwide survey of extended newborn screening by tandem mass spectrometry in Taiwan. J Inherit Metab Dis. 2010;33(Suppl 2):S295–S305. doi: 10.1007/s10545-010-9129-z. [DOI] [PubMed] [Google Scholar]
- Pandor A, Eastham J, Beverley C, Chilcott J, Paisley S (2004) Clinical effectiveness and cost-effectiveness of neonatal screening for inborn errors of metabolism using tandem mass spectrometry: a systematic review. Health Technol Assess 8(12): iii, 1–121 [DOI] [PubMed]
- Roe CR, Ding J. Mitochondrial fatty acid oxidation disorders. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The metabolic and molecular bases of inherited disease New York. New York: McGraw-Hill; 2001. pp. 2297–2326. [Google Scholar]
- Rose EC, di San Filippo CA, Ndukwe Erlingsson UC, Ardon O, Pasquali M, Longo N. Genotype-phenotype correlation in primary carnitine deficiency. Hum Mutat. 2012;33(1):118–123. doi: 10.1002/humu.21607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai I, Bailey JC, Eaton RB, Zytkovicz T, Harris DJ. A near-miss: very long chain acyl-CoA dehydrogenase deficiency with normal primary markers in the initial well-timed newborn screening specimen. J Pediatr. 2011;158(1):172. doi: 10.1016/j.jpeds.2010.09.026. [DOI] [PubMed] [Google Scholar]
- Solis JO, Singh RH. Management of fatty acid oxidation disorders: a survey of current treatment strategies. J Am Diet Assoc. 2002;102(12):1800–1803. doi: 10.1016/S0002-8223(02)90386-X. [DOI] [PubMed] [Google Scholar]
- Spiekerkoetter U. Mitochondrial fatty acid oxidation disorders: clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J Inherit Metab Dis. 2010;33(5):527–532. doi: 10.1007/s10545-010-9090-x. [DOI] [PubMed] [Google Scholar]
- Spiekerkoetter U, Lindner M, Santer R, et al. Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis. 2009;32(4):488–497. doi: 10.1007/s10545-009-1125-9. [DOI] [PubMed] [Google Scholar]
- Spiekerkoetter U, Lindner M, Santer R, et al. Treatment recommendations in long-chain fatty acid oxidation defects: consensus from a workshop. J Inherit Metab Dis. 2009;32(4):498–505. doi: 10.1007/s10545-009-1126-8. [DOI] [PubMed] [Google Scholar]
- Spiekerkoetter U, Bastin J, Gillingham M, Morris A, Wijburg F, Wilcken B. Current issues regarding treatment of mitochondrial fatty acid oxidation disorders. J Inherit Metab Dis. 2010;33(5):555–561. doi: 10.1007/s10545-010-9188-1. [DOI] [PubMed] [Google Scholar]
- Spiekerkoetter U, Haussmann U, Mueller M, et al. Tandem mass spectrometry screening for very long-chain acyl-CoA dehydrogenase deficiency: the value of second-tier enzyme testing. J Pediatr. 2010;157(4):668–673. doi: 10.1016/j.jpeds.2010.04.063. [DOI] [PubMed] [Google Scholar]
- Tang NL, Hwu WL, Chan RT, Law LK, Fung LM, Zhang WM. A founder mutation (R254X) of SLC22A5 (OCTN2) in Chinese primary carnitine deficiency patients. Hum Mutat. 2002;20(3):232. doi: 10.1002/humu.9053. [DOI] [PubMed] [Google Scholar]
- van Adel BA, Tarnopolsky MA. Metabolic myopathies: update 2009. J Clin Neuromuscul Dis. 2009;10(3):97–121. doi: 10.1097/CND.0b013e3181903126. [DOI] [PubMed] [Google Scholar]
- Wanders RJ, Vreken P, den Boer ME, Wijburg FA, van Gennip AH, Ijlst L. Disorders of mitochondrial fatty acyl-CoA beta-oxidation. J Inherit Metab Dis. 1999;22(4):442–487. doi: 10.1023/A:1005504223140. [DOI] [PubMed] [Google Scholar]
- Wang ZQ, Chen XJ, Murong SX, Wang N, Wu ZY. Molecular analysis of 51 unrelated pedigrees with late-onset multiple acyl-CoA dehydrogenation deficiency (MADD) in southern China confirmed the most common ETFDH mutation and high carrier frequency of c.250G>A. J Mol Med (Berl) 2011;89(6):569–576. doi: 10.1007/s00109-011-0725-7. [DOI] [PubMed] [Google Scholar]
- Wang L-Y, Chen N-I, Chen P-W, et al. Newborn screening for citrin deficiency and carnitine uptake defect using second-tier molecular tests. BMC Med Genet. 2013;14:24. doi: 10.1186/1471-2350-14-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcken B, Wiley V, Sim KG, Carpenter K. Carnitine transporter defect diagnosed by newborn screening with electrospray tandem mass spectrometry. J Pediatr. 2001;138(4):581–584. doi: 10.1067/mpd.2001.111813. [DOI] [PubMed] [Google Scholar]
- Wilcken B, Wiley V, Hammond J, Carpenter K. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N Engl J Med. 2003;348(23):2304–2312. doi: 10.1056/NEJMoa025225. [DOI] [PubMed] [Google Scholar]
- Zytkovicz TH, Fitzgerald EF, Marsden D, et al. Tandem mass spectrometric analysis for amino, organic, and fatty acid disorders in newborn dried blood spots: a two-year summary from the New England Newborn Screening Program. Clin Chem. 2001;47(11):1945–1955. [PubMed] [Google Scholar]