

Abstract
Objective: Limited evidence is available about the early cardiac manifestation of Fabry disease (FD) in children. We aimed to evaluate cardiac involvement in children with FD by analysing serial structural and electrocardiographic changes.
Methods: The data were acquired from 22 children with FD [11 males; median age 9.8 (ranging 2.5–16) years]. Seven patients (5 males) were on enzyme replacement therapy (ERT) with Agalasidase alpha. Echocardiography, ECG and 24-h ECG monitoring recordings were acquired during routine annual clinical controls. ECG data were compared to a group of age-and gender-matched controls.
Results: At baseline, ECG and ECHO parameters of left ventricular mass were similar in both males and females. Three boys (all were on ERT) developed left ventricular hypertrophy (LVH) during two-year follow-up. The progression to LVH was accompanied by the appearance of frequent ventricular premature beats in two cases and supraventricular premature beats (SPBs) with T wave inversion in one case. T wave inversion and SPBs were detected in two younger relatives of a patient with LVH, in the absence of detectable LVH. Seven out of 22 patients had T wave abnormalities. Five of them were males (p = 0.03) all carrying the N215S mutation (p = 0.03). At baseline, median PR intervals were prolonged in FD subjects compared to controls [143 (122–177) vs. 122 (82–165) ms; p < 0.0001].
Conclusions: Cardiac complications of FD become apparent in childhood as subtle changes with slow but detectable progression over time, with males more frequently affected than females. Progression of LVH was apparent in three children despite ERT.
Introduction
Anderson–Fabry disease (FD) (OMIM Number 301500) is an X-linked inherited lysosomal storage disease caused by a defect in the gene encoding lysosomal enzyme α-galactosidase A (Desnick et al. 2003). The resulting absence or severe deficiency of enzymatic activity leads to intracellular storage of glycosphingolipids, in particular globotriaosylceramide (Gb3). This accumulation can be detected in different organs and tissues.
The storage within the heart affects all type of cells including cardiomyocytes, conduction system tissue, vascular endothelial and smooth muscle cells and valvular fibroblasts (Brady and Schiffmann 2000). Cardiac involvement represents the major cause of premature morbidity and mortality in adult patients with FD (Cybulla et al. 2007; Patel et al. 2011). Left ventricular hypertrophy (LVH) is the most frequent and well-recognised cardiac manifestation in adults (Linhart et al. 2000; Kampmann et al. 2002; Sachdev et al. 2002; Elliott et al. 2011). Other frequent complications and changes in adult patients with FD include atrial fibrillation, non-sustained ventricular tachycardia, QRS broadening, shortening of PR interval or atrio-ventricular blocks (AVBs) and sinus node dysfunction (Mehta et al. 1977; Pochis et al. 1994; Shah et al. 2005). Several patients may develop mild to moderate valve insufficiencies, dilatation of the proximal aorta, arterial hypertension and coronary artery disease (Linhart et al. 2000; Kampmann et al. 2002; Nagueh 2003; Kleinert et al. 2006; Elliott et al. 2006; Barbey et al. 2010).
Symptoms of the disease typically start in childhood and subtle organ changes can be detected early (Ries et al. 2003; Ramaswami et al. 2006; Tøndel et al. 2008). However, life-threatening complications appear almost exclusively in adult patients. Severe arrhythmias, LVH and cardiac failure in children were not reported. Very little is known about cardiac changes in children. Only Kampmann et al. demonstrated that cardiac manifestations in FD are detectable already in paediatric population as well (Kampmann et al. 2008b).
Therefore, the aim of our study is to evaluate early onset cardiac manifestations in children with FD by analysing serial changes on electrocardiography (ECG), 24-h ECG monitoring (Holter ECG) and echocardiography.
Methods
Study Population
We performed a retrospective single-site observational analysis of available electrocardiographic (standard ECG and Holter ECG data) and echocardiographic recordings from 22 paediatric patients with FD acquired during systematic annual visits. All children were under the care of Uma Ramaswami (UR) in a tertiary centre in the Paediatric Metabolic Unit, Addenbrooke’s Hospital, Cambridge, UK between the years 2005 and 2011. Patients were included to the clinical care program for patients with FD having either a newly established diagnosis of FD or being identified as affected relatives of patients with already known FD. All patients included to the study had the disease confirmed by the identification of the underlying mutation within the α-galactosidase A gene. Clinical, ECG, Holter ECG and echocardiography recordings were acquired during routine clinical visits performed on annual basis. Clinical examination was performed in all children by a single clinician (UR). Anonymised data provided by UR were compiled and processed by an independent observer during his short stay in Addenbrooke’s Hospital. The research passport was obtained specifically to review link-anonymised data. No data were exported outside Addenbrooke’s Hospital. The protocol of analysis was approved by the local institutional ethics board.
Seven patients (32%) received enzyme replacement therapy (ERT) based on fulfilling UK National Guidelines for Treatment of FD. All subjects were treated with Agalasidase alpha (Shire Human Genetic Therapies, Cambridge, MA) at the dose of 0.2 mg/kg delivered biweekly as a 40-min intravenous infusion.
Baseline ECG parameters were compared to a control population of age- and gender-matched healthy children without evidence of cardiovascular and metabolic diseases. The healthy controls were recruited prospectively from children investigated in a preventive program of the Department of Paediatrics and Adolescent Medicine, First Faculty of Medicine, Charles University in Prague, Czech Republic. The anonymised data were acquired exclusively for the purpose of this study and analysed by the same observer as the data from FD patients.
Echocardiography
Echocardiography was done at the annual review for FD patients as per the National UK guidelines.
Complete echocardiographic examinations were performed according to the recommendations of American Society of Echocardiography (Douglas et al. 2011). For the purpose of the study, we calculated left ventricular (LV) mass by using the Devereux modified formula (Devereux et al. 1986). LV mass was then indexed to body height (in metres squared to 2.7). LVH was defined as LV mass > 95th percentile for the population of the given age (Khoury et al. 2009).
ECG Data and Holter Monitoring
Conventional 12-channel resting ECG analysis included measurement of following parameters: heart rhythm; heart rate (HR); conduction intervals: PR, QRS, QT/QTc; heart axis; T wave polarity in V5 and V6 and voltage or voltage/duration markers of LVH: Sokolow–Lyon voltage, Cornell voltage, Cornell index. QTc was calculated according to Bazzet’s formula. Sokolow–Lyon voltage was defined as sum of voltages of S in V1 and R in V5 or V6 (whichever was higher). The sum of voltages of S in V3 and R in aVL constituted Cornell voltage. Cornell index was calculated as the product of Cornell voltage multiplied by QRS duration. The measurements were performed on digitised paper tracings which are acquired as standard recordings at 25 mm/s paper speed with the amplitude of 1 mV/mm. Holter ECG monitoring was used to obtain following parameters: heart rhythm, HR (maximal, minimal and mean), occurrence and type of arrhythmias. Special concern was given about pauses and bradycardia episodes, conduction blocks; supraventricular (SPBs) and ventricular premature beats (VPBs) and both supraventricular and ventricular tachyarrhythmias. Frequently, both SPBs and VPBs were defined as occurrence of more than 10 beats per 24 h.
Statistical Analysis
Statistical analysis was carried out using Statistica 6.1 (StatSoft, Inc., Tulsa, OK, USA) statistical package. Continuous variables were expressed as means with standard deviations after testing for normality of distribution (Shapiro Wilk’s test) and compared by the two-tailed t-test for independent samples. Non-normally distributed variables were expressed as medians with range and compared by Mann–Whitney U test. Categorical variables were expressed as percentages and compared by χ2-test. A P-value < 0.05 was considered significant.
Results
Study Population Characteristics
The data were obtained from 22 children with FD from 16 families [11 males; baseline mean age 9.6 ± 4.6 years; median 9.8 (ranging 2.5–16.0) years]. The most frequently represented mutation was N215S type in 7 (32%) cases. Seven (32%) patients [5 males, mean age 8.6 ± 3.0; median 7.8 (ranging 4.3–14.3) years] were on ERT (Agalasidase alpha in all cases) during the entire study period, none of them carrying the N215S mutation. Baseline clinical and demographic data are shown in Table 1. At baseline, no significant differences between males and females were detected. The list of mutations, occurrence of symptoms and signs of FD, treatment and summary of abnormal findings is shown in Table 2.
Table 1.
Baseline clinical and demographical characteristics
| FD patients (total) (n = 22) |
Males (n = 11) |
Females (n = 11) |
|
|---|---|---|---|
| Male | 11 (50%) | – | – |
| Age at baseline (years) | 9.8 (2.5–16) | 7 (4–14) | 9.5 (2.5–16.5) |
| Enzyme activity < 2 nmol/mg per hour |
12 (55%) | 9 (82%) | 3 (27%) |
| Typical symptoms of FD | 19 (86%) | 11 (100%) | 9 (82%) |
| ERT anytime | 7 (32%) | 5 (45.5%) | 2 (18%) |
| ERT baseline | 4 (18%) | 3 (27%) | 1 (9%) |
FD Fabry disease, ERT enzyme replacement therapy with Agalasidase alpha
Value expressed as: No (%) or median (range)
Table 2.
List of mutation and case profiles
| Mutation type | Patient # (n = 22) |
Family # (n = 16) |
Baseline age (Y/M) |
Gender | Typical symptoms of FD | ERT | Detected abnormality |
|---|---|---|---|---|---|---|---|
| Gln107X | 1 | 1 | 4/0 | Male | Yes | No | No |
| N215S | 2 | 2 | 13/6 | Male | Yes | No | T wave inversion |
| 3 | 2 | 14/4 | Male | Yes | No | No | |
| 4 | 3 | 15/4 | Female | Yes | No | T wave inversion | |
| 5 | 4 | 14/1 | Female | Yes | No | Prolonged PR interval | |
| 6 | 5 | 4/5 | Male | Yes | No | T wave inversion | |
| 7 | 6 | 4/10 | Male | Yes | No | T wave inversion | |
| 8 | 7 | 15/1 | Female | Yes | No | T wave inversion | |
| N298H | 9 | 8 | 10/1 | Female | Yes | No | No |
| A143T | 10 | 9 | 7/10 | Male | Yes | Yes | LVH + T wave inversion + SPBs |
| 11 | 9 | 6/5 | Male | Yes | Yes | T wave inversion | |
| p.N33D | 12 | 10 | 9/6 | Male | Yes | No | No |
| c.950T>C | 13 | 11 | 6/9 | Male | Yes | Yes | LVH + VPBs |
| c.802-3_802-2delCA | 14 | 12 | 11/6 | Female | Yes | No | No |
| R301X | 15 | 13 | 16/0 | Female | No | No | No |
| 16 | 13 | 7/7 | Female | Yes | No | No | |
| R49S | 17 | 14 | 14/4 | Male | Yes | Yes | LVH + VPBs + sinus bradycardia |
| 18 | 14 | 10/9 | Female | No | No | SPBs | |
| P2005T | 19 | 15 | 7/0 | Male | Yes | Yes | No |
| W209X | 20 | 16 | 10/5 | Female | Yes | Yes | No |
| 21 | 16 | 7/5 | Female | Yes | No | SPBs | |
| 22 | 16 | 2/6 | Female | No | No | No |
ERT enzyme replacement therapy with Agalasidase alpha, LVH left ventricular hypertrophy, SPBs supraventricular beats, VPBs ventricular beats, Y year, M month
The serial ECG, Holter ECG and echocardiography data were available in 22, 20 and 16 subjects at baseline, first and second annual control, respectively. At baseline, both ECG and ECHO parameters were comparable between FD patients and controls and no gender differences in both ECG and ECHO parameters of LVH were detected in patients with FD, as shown in Table 3.
Table 3.
ECHO and ECG parameters of left ventricular hypertrophy at baseline
| FD patients (total) (n = 22) | FD patients males (n = 11) | FD patients females (n = 11) | ECG controls (total) (n = 44) | ECG controls males (n = 22) | ECG controls females (n = 22) | |
|---|---|---|---|---|---|---|
| ECG | ||||||
| Sokolow–Lyon voltage (mV) | 2.7 (1.1–4.8) | 2.8 (1.9–4.6) | 2.7 (1.1–4.8) | 2.9 (0.6–5.8) | 2.9 (1.2–5.8) | 2.8 (0.6–3.9) |
| Cornel voltage (mV) | 1.1 (0.3–3.0) | 1.8 (0.9–2.7) | 0.9 (0.3–3.0) | 1.6 (0.4–2.3) | 1.7 (0.8–2.3) | 1.6 (0.4–2.1) |
| Cornel index (mV * ms) | 888 (267–2808) | 1484 (753–2808) | 781 (267–2808) | 1050 (320–2070) | 1123 (523–2070) | 985 (320–1946) |
| Echocardiography | ||||||
| LV mass index (g/h2.7) | 36 (24–46) | 41 (36–45) | 35 (24–46) | – | – | – |
LV left ventricle, FD Fabry disease
Value expressed as: median (range)
The control group for baseline ECG comparison consisted of 44 children [22 males, mean age 9.1 ± 1.2 years; median 9.1 (ranging 5.2–10.9) years] of comparable age and gender.
Progression of Left Ventricular Hypertrophy
Figure 1 shows the proportion of patients with LV mass above 75th percentile and LVH at baseline and during follow-up. The number of patients with LV mass in the upper percentiles at baseline (> 75th percentile at baseline) progressively increased during the follow-up. Whilst no patient met the criteria of LVH at baseline, one developed LVH after the first year (patient #13) and two additional patients (#10 and 17) after the second year of follow-up. The characteristics of these patients are shown in Table 4. All three patients were males with very low or undetectable levels of enzyme activity, and all were symptomatic and receiving ERT with Agalasidase alpha already at baseline. All three had mutations in the α-galactosidase A gene known to be associated with the classical phenotype of FD. In these three patients, at least two baseline LVH parameters on ECG (Sokolow–Lyon voltage, Cornel voltage or Cornel index) were within the upper quartile of the values measured in FD male population. In contrast, their PR intervals were within interquartile range in male subgroup. Patient #10 manifested frequent SPBs before detection of LVH. In this patient, the repolarisation changes appeared at the same time as LVH. The remaining two patients demonstrated frequent VPBs detected within 1 year after the appearance of LVH. The last patient (#17) had asymptomatic sinus bradycardia.
Fig. 1.
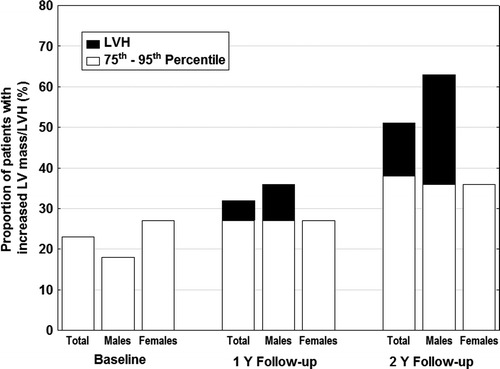
Proportion of patients with left ventricular mass ≥ 75th percentile and those with left ventricular hypertrophy at baseline and during follow-up. LV mass ≥ 75th percentile – white parts of the column. LVH – black parts of the column. LV left ventricle, LVH left ventricular hypertrophy, Y year
Table 4.
Profiles of three FD male patients with progression of left ventricular mass to left ventricular hypertrophy
| Patient # | Mutation | ERT since baseline | Typical clinical signs of FD | Detected (year of follow-up) |
Enzyme activity (nmol/mg per hour) | Baseline Sokolow– Lyon voltage (mV) | Baseline Cornel voltage (mV) | Baseline Cornel index (mV * ms) | Changes in Holter ECG | Repolarisation changes |
|---|---|---|---|---|---|---|---|---|---|---|
| 10 | A143T | Yes | Yes | 3rd | UD | 2.3 | 2.7 | 2808 | SPBs in 2nd year | Yes |
| 13 | c.950T > C | Yes | Yes | 2nd | <0.5 | 3.7 | 3.0 | 2467 | VPBs in 3rd year | No |
| 17 | R49S | Yes | Yes | 3rd | <0.5 | 3.9 | 2.2 | 1936 | Sinus bradycardia + VPBs in 3rd year | No |
FD Fabry disease, M male, ERT enzyme replacement therapy with Agalasidase alpha, UD undetectable, SPBs supraventricular premature beats, VPBs ventricular premature beats
12-Leads ECG Parameters
As shown in Table 5, PR intervals at baseline were prolonged in patients with FD as compared to controls. Moreover, PR intervals after the first and second year of follow-up were significantly shorter in boys than in girls. This significance was lost when one female outlier was excluded. The outlier (patient # 5) is a symptomatic 14-year-old girl carrying the N215S mutation not receiving ERT, with a reduced enzyme activity. She progressively exceeded the normal value ranges of PR at the second year of follow-up, Fig. 2.
Table 5.
ECG parameters at baseline and during follow-up
| FD patients (total) (n = 22) |
FD patients males (n = 11) |
FD patients females (n = 11) |
ECG controls (total) (n = 44) |
ECG controls males (n = 22) |
ECG controls females (n = 22) |
|
|---|---|---|---|---|---|---|
| HR (bpm) | 77 (63–120) | 80 (65–120) | 78 (63–112) | 85 (64–118) | 82 (66–114) | 92 (64–118) |
| 1 Y follow-up | 77 (59–112) | 83 (69–112) | 77 (59–112) | – | – | – |
| 2 Y follow-up | 79 (59–104) | 86 (68–89) | 81 (59–104) | – | – | – |
| PR (ms) | 143 (122–177)* | 145 (122–166) | 143 (122–177) | 122 (82–165) | 121 (105–155) | 133 (82–165) |
| 1 Y follow-up | 144 (127–185) | 135 (127–148) | 145 (127–185)*** | – | – | – |
| 2 Y follow-up | 144 (118–186) | 138 (118–151) | 146 (118–186)*** | – | – | – |
| QRS (ms) | 79 (67–104) | 81 (70–104) | 79 (67–103) | 81 (52–95) | 82 (61–93) | 77 (52–95) |
| 1 Y follow-up | 82 (68–89) | 86 (69–89) | 80 (68–87) | – | – | – |
| 2 Y follow-up | 86 (67–91) | 89 (68–91) | 86 (67–91) | – | – | – |
| QT (ms) | 355 (288–408) | 345 (302–408) | 356 (288–408) | 330 (295–405) | 354 (312–405) | 312 (295–340) |
| 1 Y follow-up | 357 (305–397) | 349 (307–371) | 357 (305–397) | – | – | – |
| 2 Y follow-up | 333 (315–390) | 329 (315–365) | 333 (315–390) | – | – | – |
| QTc (ms) | 393 (372–459) | 395 (384–459) | 393 (372–459) | 412 (362–444) | 391 (362–433) | 422 (368–444) |
| 1 Y follow-up | 393 (368–446) | 405 (374–446) | 393 (368–446) | – | – | – |
| 2 Y follow-up | 391 (368–426) | 394 (380–426) | 391 (368–426) | – | – | – |
| T wave inversion | 4 (18%)* | 4 (36%) | 0 (0%)** | 0 (0%) | 0 (0%) | 0 (0%) |
| New onset at 1 Y | 1 (4.5%) | 1 (9%) | 0 (0%) | – | – | – |
| New onset at 2 Y | 2 (9%) | 0 (0%) | 2 (18%) | – | – | – |
HR heart rate, PR, QRS, QT, QTc conduction intervals on ECG, FD Fabry disease, Y year
Value expressed as: No (%) or median (range)
*p < 0.05 (compare FD vs. controls)
**p < 0.05 (compare males vs. females)
***p < 0.05 (compare males vs. females), see the text – the difference is lost when one female outlier is excluded from analysis
Fig. 2.
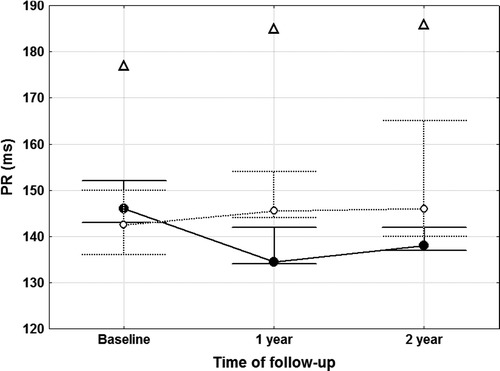
PR intervals at baseline and during follow-up in paediatric patients with Fabry disease. For explanation, see the text. Male Fabry patients – closed circles, solid line. Female Fabry patients – open circles, dotted line. Open triangle – outlier female patient. Points and whiskers represent medians and quartile ranges values, respectively
Mean HR, median QRS, median QT and QTc intervals in FD patients were not different from healthy control group and did not change over time.
The T wave inversion indicating repolarisation changes in lateral leads were present in four (18%) patients with FD at baseline. In contrast, such finding was not found in the control group. The number of patients with T wave inversion increased over time. At the end of 2-year follow-up, seven FD patients (32%) had T wave abnormalities. Of note, five of the seven patients with T wave abnormalities were males (p = 0.03 vs. females) and five had the N215S mutation (p = 0.03 vs. other mutations). Only one patient with isolated T wave inversion was on ERT with Agalasidase alpha. Of note, one of the boys with T wave inversion (patient #11) was the brother (aged 6 years at baseline) of a patient #10 with manifest LVH and T wave abnormality.
Holter ECG Parameters
No serious arrhythmias were detected on Holter ECG monitoring in any of the study subgroups, neither at baseline nor during follow-up. Only minor changes were present with increasing manifestation rate, Table 6. We detected only SPBs and VPBs which were considered clinically benign. Of note, two asymptomatic girls presented with frequent SPBs. The first patient (aged 11 years at baseline with mildly reduced enzyme activity) is the younger sister of patient # 17 with LVH. The second patient (# 21) was a girl (aged 7 years at baseline with mildly reduced enzyme activity). Her two sisters had manifest classical symptoms of FD without cardiac involvement.
Table 6.
Holter ECG parameters at baseline and during follow-up
| FD patients (total) (n = 22) |
FD males (n = 11) |
FD females (n = 11) |
|
|---|---|---|---|
| Mean HR (bpm) | 85 (72–109) | 89 (78–98) | 82 (72–109) |
| 1 Y follow-up | 84 (70–102) | 87 (75–96) | 83 (70–102) |
| 2 Y follow-up | 80 (71–103) | 89 (79–103) | 79 (71–88) |
| Min HR (bpm) | 59 (50–77) | 63 (50–77) | 58 (53–70) |
| 1 Y follow-up | 59 (48–72) | 60 (48–72) | 58 (49–71) |
| 2 Y follow-up | 60 (47–75) | 62 (54–75) | 58 (47–65) |
| Max HR (bpm) | 143 (114–188) | 159 (114–188) | 140 (118–175) |
| 1 Y follow-up | 141 (118–179) | 149 (131–163) | 136 (118–179) |
| 2 Y follow-up | 141 (120–163) | 153 (131–163) | 138 (120–143) |
| Supraventricular premature beats | 8 (36.5%) | 3 (27%) | 5 (45%) |
| New onset in 1 Y | 2 (9%) | 0 (0%) | 2 (18%) |
| New onset in 2 Y | 2 (9%) | 1 (9%) | 1 (9%) |
| Ventricular premature beats | 11 (50%) | 7 (64%) | 4 (36%) |
| New onset in 1 Y | 2 (9%) | 0 (0%) | 2 (18%) |
| New onset in 2 Y | 0 (0%) | 0 (0%) | 0 (0%) |
| Bradycardia episodes | 1 (4.5%) | 0 (0%) | 1 (9%) |
| New onset in 1 Y | 1 (4.5%) | 1 (9%) | 0 (0%) |
| New onset in 2 Y | 0 (0%) | 0 (0%) | 0 (0%) |
| Sinus arrest | 2 (9%) | 1 (9%) | 1 (9%) |
| New onset in 1 Y | 2 (9%) | 1 (9%) | 1 (9%) |
| New onset in 2 Y | 1 (4.5%) | 1 (9%) | 0 (0%) |
| AVB first degree | 1 (4.5%) | 0 (0%) | 1 (9%) |
| New onset in 1 Y | 0 (0%) | 0 (0%) | 0 (0%) |
| New onset in 2 Y | 1 (4.5%) | 0 (0%) | 1 (9%) |
| AVB second degree (type 1) | 4 (18%) | 2 (18%) | 2 (18%) |
| New onset in 1 Y | 0 (0%) | 0 (0%) | 0 (0%) |
| New onset in 2 Y | 1 (4.5%) | 1 (9%) | 0 (0%) |
| Junctional beats | 9 (40%) | 4 (36%) | 5 (45%) |
| New onset in 1 Y | 2 (9%) | 1 (9%) | 1 (9%) |
| New onset in 2 Y | 0 (0%) | 0 (0%) | 0 (0%) |
| Ventricular escaped | 1 (4.5%) | 0 (0%) | 1 (9%) |
| New onset in 1 Y | 0 (0%) | 0 (0%) | 0 (0%) |
| New onset in 2 Y | 0 (0%) | 0 (0%) | 0 (0%) |
HR heart rate, AVB atrio-ventricular block, FD Fabry disease, Y year
Value expressed as: No (%) or mean ± SD; median (range)
There were no clinically important brady-arrhythmias including serious sinus arrests or AVB of advanced degree observed in this cohort of patients. Only two patients demonstrated intermittent first-degree AVB (both female) and five patients presented with second-degree AVB type 1 during study period (3 boys). The incidence of all minor changes was not gender dependent.
Discussion
The major finding of our study is the early detection of subtle cardiac changes in children with FD. Although no patient met criteria for LVH at baseline, three males progressed to LVH during the follow-up. The progression towards LVH was accompanied with the appearance of abnormalities on ECG and Holter ECG (i.e. premature beats and T wave inversions). The progression of disease occurred in some cases despite established ERT with Agalasidase alpha. Some ECG abnormalities were detectable in younger siblings of these rapidly progression patients. In addition, we found a longer PR interval as compared to healthy gender- and age-matched controls in both males and females. Interestingly, detected conduction abnormalities were separated from occurrence of LVH in time.
Left Ventricular Mass and Left Ventricular Hypertrophy
LVH is the most frequent cardiac manifestation of FD, and thereby FD represents an important differential diagnosis in patients with unexplained hypertrophic cardiomyopathy (Kampmann et al. 2002; Sachdev et al. 2002; Monserrat et al. 2007; Elliott et al. 2011). In contrast to other hypertrophic cardiomyopathies, FD-associated hypertrophy of significant degree seldom develops in young patients (Kampmann et al. 2008a). In agreement with previous data (Kampmann et al. 2008b), we have found only mildly increased LV mass with its slight progression over time, more so in boys than in girls with FD. However, three boys had fulfilled the definition of LVH during the follow-up. Progression of LVH was apparent in three children on ERT, and therefore the timing of ERT in children requires attention. Moreover, the manifestation of LVH was accompanied with other ECG and Holter ECG abnormalities in all three cases: T wave inversion in the first case, frequent VPBs in the second case and frequent VPBs with sinus bradycardia in the third case. Some other abnormalities on Holter ECG (frequent SPBs) preceded the LVH by one year in first case (patient # 10) emphasising the importance of annual follow-up. As LVH seems to respond to the therapy only before it reaches a significant degree (Weidemann et al. 2009), its early detection appears to be of paramount importance since its appearance advocates early enzyme replacement initiation. The association of LVH with described ECG abnormalities underlines the significant role of at least annual ECG and Holter ECG monitoring.
T Wave Inversion
In our cohort, only one patient presented both T wave inversion and LVH. One younger brother to the patient with LVH manifested T wave inversion without detectable LVH. The ECG analysis of our patients shows relatively frequent changes in T wave polarity with progressive increase in incidence of these abnormalities. Our data is in concordance with previous literature in adult patients with FD, where the inversion of T wave was described as a frequent finding in combination with or without manifest LVH in adult patients with FD (Mehta et al. 1977; Linhart et al. 2002). We have found that the T wave inversion was predominantly expressed in male patients and those carrying N215S mutation. The progressively increasing polarity changes within lateral leads should be considered as abnormal and may reflect the impact of myocardial storage before the onset of manifest LVH.
It should be mentioned that T wave morphology, particularly in precordial leads, is progressively changing throughout childhood even in healthy subjects. The most stable pattern (upright polarity) of T wave in childhood is in leads V5–V6. Opposite to this T wave inversion is physiological in leads V1–V3 in children and normalises in relatively prolonged period during late childhood or early adolescence (Dickinson 2005). For this reason in our study, T wave polarity was evaluated in lateral leads only.
ECG LV Mass Indexes
The ECG analysis of our cohort has shown that at least two parameters of LVH (Sokolow–Lyon voltage, Cornel voltage or Cornel index) were within the upper quartile of all values measured in total FD male population at baseline. On the other hand, in two patients with LVH one out of three parameters was not symmetrically elevated. In addition, differences in QRS voltage and LVH indices between male and female patients were minimal.
The ECG voltage parameters in our population were lower than previously reported by Kampmann (Kampmann et al. 2008). We hypothesise that this difference might be due to the wide variation in QRS amplitudes seen in normal children and adolescents (Kampmann et al. 2002; Rijnbeek et al. 2008b) or may be attributable to ERT administration in our cohort. ERT has been shown to reduce LV mass (Ries et al. 2006) and also in potentially preventing its onset (Weidemann et al. 2005; Mehta et al. 2009; Ramaswami et al. 2012). The likely influence of ERT is supported by our finding of lower LV mass in our cohort in comparison to previously reported data by Kampmann (Kampmann et al. 2008b).
24-h ECG Monitors
In patients with LVH, frequent VPBs and SPBs were noticed to appear before the onset of LVH. One patient with frequent SPBs was the younger sister of the patient with manifest LVH. Therefore, we hypothesise that premature beats may be considered as a marker of disease progression. Considering the limited number of patients, we have no clear evidence whether all patients with frequent SPBs will develop LVH. Nevertheless, the simultaneous existence of LVH and premature beats emerges as a potentially clinically important observation of our study.
The comparison of Holter ECGs obtained in Fabry patients with previously published cohorts of healthy children aged 7–16 years (Southall et al. 1981; Scott et al. 1980; Dickinson and Scott 1984) revealed that most of the abnormalities may be present in otherwise healthy children as well. As in healthy children, our Fabry patients had SPBs or VPBs in the majority as isolated beats with uniform morphology and usually with low frequency per 24 h. The HR variations in our study are within previously described normal ranges. Bradycardia and junctional rhythm episodes, sino-atrial blocks, AVB of first degree and pauses, including incidental and AVB of second degree Mobitz type I, have been detected also in all healthy groups particularly during sleep.
Our data have not shown any serious rhythm disturbances in paediatric FD patients. This is in contrast with findings described in adults with FD who present with high incidence of paroxysmal atrial fibrillation, non-sustained ventricular tachycardia, AVB and sinus node dysfunction (Shah et al. 2005; Frustaci and Chimenti 2007).
Conduction Intervals
Short PR intervals and QRS broadening have been shown as frequent findings in adult patients with FD on ECG (Mehta et al. 1977; Pochis et al. 1994). Shortening of PR interval occurs by rapid AV conduction and is often observed in early stage of FD (Mehta et al. 1977). Invasive studies excluded the presence of accessory bypass pathways causing PR interval changes and confirmed that PR shortening is result of accelerated atrio-ventricular nodal conduction (Jastrzebski et al. 2006; Aryana et al. 2008) or by shortening of atrial conduction characterised by shorter P wave duration (Namdar et al. 2011). The PR interval prolongs during aging in normal paediatric population (Rijnbeek et al. 2001; O`Connor et al. 2008). Significant increase of PR duraton with age was described also in the adult population of FD (O'Mahony et al. 2011). Our data showed an unexpected observation in children and adolescents. Patients with FD had baseline PR interval prolonged compared to healthy population. On comparison of genders, in females the PR interval prolonged over time. One female patient (case # 5) had PR interval length above the 98th percentile of PR interval for a given age derived from the normal population (Rijnbeek et al. 2001). When this outlier is excluded from the analysis, the PR intervals appear comparable in both genders. In addition, this girl manifested the slowest HR in both first year and second year of follow-up. An important aspect is that conduction abnormalities were separated from occurrence of LVH. This temporal separation implies more likely different mechanisms of both changes.
Our finding of longer PR intervals among patients with FD is, in part, in agreement with the observation that short PR is less frequent than originally reported (Namdar et al. 2010). Moreover, FD is known to be associated not only with short PR interval, but also with significant increases in PR interval duration with age in adults with FD and with high rates of AV conduction impairments, occurring particularly in older patients (O'Mahony et al. 2011). Broadening of QRS interval along with decreasing heart rate was described in adult patients with FD (Mehta et al. 1977; Pochis et al. 1994). In our study, the QRS and QTc interval duration as well as HR were found within the ranges of normal values for paediatric patients (Rijnbeek et al. 2001).
N215S Mutation
To our knowledge, we are reporting about the largest cohort of N215S paediatric patients. The N215S mutation in the alpha-galctosidase A gene is known to be associated with late-onset cardiac variant of the disease. In our cohort, it was the most frequent mutation and the patients presented mostly only subtle ECG changes including prolongation of PR interval or T wave abnormalities. N215S as a cardiac variant has been described previously (Eng et al. 1993b; Sachdev et al. 2002). No patient in our cohort who progressed to LVH had the N215S mutation. The absence of detection of LVH in N215S patients confirms the typically delayed onset of symptoms in this variant (Eng et al. 1993b). The late onset of symptoms was shown to be associated with the presence of significant residual enzymatic activity in patients with N215S mutation. This activity seems to prevent the accumulation of ceramide trihexoside storage in organs with a high cellular renewal or with lesser α-galactosidase A substrate turnover. This may explain why other symptoms of the disease (renal, neurological, gastrointestinal or cutaneous) do not develop early or are significantly attenuated (Eng et al. 1993a, b; Desnick et al. 2003). Subjects with N215S mutation develop clinically relevant symptoms (including LVH, electrophysiological abnormalities and heart failure symptoms) relatively late (usually after the third decade of age). Although the authors are aware of anecdotal unpublished cases of FD patients with N215S mutation who have a more severe phenotype (unpublished; personal communication), these appear very rare. Longer-term follow-up of patients is essential to understand the natural history and disease progression.
Limitations
We recognise several limitations of our study including the relatively limited number of patients and the retrospective character of the analysis. However, this is a single centre study with regular follow-up by one experienced clinician. The follow-up was based on pre-specified National Guidelines and the data collection was satisfactory with only few data missing. Although our patients are not covering the entire infancy age distribution, it should be underlined that our comparisons between genders are relevant since males and females were of comparable age and the control group was age matched. The potential impact of ERT is unfortunately unavoidable since ERT is part of the current standards of care in symptomatic children with FD and based on National Guidelines for Treatment of FD. In our cohort, five boys and two girls (total 32 %) were receiving ERT with Agalasidase alpha. This study addressed cardiac parameter changes in both ERT and non-ERT patients. ERT may certainly influence conduction abnormalities, repolarisation changes, LVH and arrhythmias progression rate. Our data therefore reflect in part a natural history of the disease in the current era with the availability of ERT and also the natural history of the untreated disease. Of note, patients with LVH progressed despite receiving ERT. This however does not allow any conclusions about the ERT efficacy. On the other hand, since the heart appears to be relatively unresponsive to ERT-induced lysosomal clearance (Thurberg et al. 2009), it may reflect the need of early initiation before the development of significant LVH. The subtle ECG changes noticed in these children are often overlooked and may prove to be important early indicators for initiating early ERT. However, to confirm this observation, a substantially larger cohort of patients should be studied.
Conclusion
Early changes in ECG/Holter ECG and ECHO related to cardiac involvement caused by FD can be demonstrated already in children of both genders. The progression of disease to LVH was more apparent in FD males. Interestingly, some changes on ECG and Holter ECG preceded LVH development and were noted also in the siblings of these rapidly progressing individuals. Although clinically relevant arrhythmias were not found in this cohort of children with FD, our findings justify regular sequential annual follow-up in all children (both males and females) with FD including a complex cardiac evaluation.
Acknowledgements
We thank the metabolic nurses and the cardiology technicians at Addenbrooke’s Hospital for their help with collating data and performing cardiac ECHO and Holter monitoring.
Abbreviations
- AVB
AV block
- ECHO
Echocardiography
- ERT
Enzyme replacement therapy
- FD
Fabry disease
- Holter ECG
24-h ECG monitoring
- HR
Heart rate
- LV
Left ventricle
- LVH
Left ventricular hypertrophy
- M
Month
- SPBs
Supraventricular premature beats
- UR
Uma Ramaswami
- VPBs
Ventricular premature beats
- Y
Year
Synopsis
Early changes in ECG/Holter ECG and echocardiography related to cardiac involvement caused by Fabry disease can be observed in children, both males and females.
Details of the Contributions of Individual Authors
Stepan Havranek: analysis and interpretation of data; drafting of manuscript
Ales Linhart: analysis and interpretation of data; revising article critically for important intellectual content
Zuzana Urbanova: analysis of data; revising article critically for important intellectual content
Uma Ramaswami: Developing concept, design of study, patient follow-up, data collection; revising article critically for important intellectual content
Guarantor Author
Dr. Uma Ramaswami MD, FRCPCH.
Funding
Supported by Program for research development in Charles University: PRVOUK-P35/LF1/5.
The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors.
Details of Ethics Approval
The study was approved by Norfolk Research Ethics Committee. Reference number: 11/H0310/4.
Disclosure
None declared.
Footnotes
Competing interests: None declared
Contributor Information
Uma Ramaswami, Email: uma.ramaswami@cmft.nhs.uk.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Aryana A, Fifer MA, Ruskin JN, Mela T. Short PR interval in the absence of preexcitation: a characteristic finding in a patient with Fabry disease. Pacing Clin Electrophysiol. 2008;31:782–783. doi: 10.1111/j.1540-8159.2008.01088.x. [DOI] [PubMed] [Google Scholar]
- Barbey F, Qanadli SD, Juli C, et al. Aortic remodelling in Fabry disease. Eur Heart J. 2010;31:347–353. doi: 10.1093/eurheartj/ehp426. [DOI] [PubMed] [Google Scholar]
- Brady RO, Schiffmann R. Clinical features of and recent advances in therapy for Fabry disease. JAMA. 2000;248:2771–2775. doi: 10.1001/jama.284.21.2771. [DOI] [PubMed] [Google Scholar]
- Cybulla M, Walter K, Neumann HP, et al. Fabry disease: demographic data since introduction of enzyme replacement therapy. Dtsch Med Wochenschr. 2007;132:1505–1509. doi: 10.1055/s-2007-982060. [DOI] [PubMed] [Google Scholar]
- Desnick RJ, Brady R, Barranger J, et al. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003;138:338–346. doi: 10.7326/0003-4819-138-4-200302180-00014. [DOI] [PubMed] [Google Scholar]
- Devereux RB, Alonso DR, Lutas EM, et al. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol. 1986;57:450–458. doi: 10.1016/0002-9149(86)90771-X. [DOI] [PubMed] [Google Scholar]
- Dickinson DF. The normal ECG in childhood and adolescence. Heart. 2005;91:1626–1630. doi: 10.1136/hrt.2004.057307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson DF, Scott O. Ambulatory electrocardiographic monitoring in 100 healthy teenage boys. Br Heart J. 1984;51:179–183. doi: 10.1136/hrt.51.2.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas PS, Garcia MJ, Haines DE, et al. ACCF/ASE/AHA/ASNC/HFSA/HRS/SCAI/SCCM/SCCT/SCMR 2011 appropriate use criteria for echocardiography. J Am Soc Echocardiogr. 2011;24:229–267. doi: 10.1016/j.echo.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Elliott PM, Kindler H, Shah JS, et al. Coronary microvascular dysfunction in male patients with Anderson-Fabry disease and the effect of treatment with alpha galactosidase A. Heart. 2006;92:357–360. doi: 10.1136/hrt.2004.054015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott P, Baker R, Pasquale F, et al. Prevalence of Anderson-Fabry disease in patients with hypertrophic cardiomyopathy: the European Anderson-Fabry Disease Survey. Heart. 2011;97:1957–1960. doi: 10.1136/heartjnl-2011-300364. [DOI] [PubMed] [Google Scholar]
- Eng CM, Niehaus DJ, Desnick RJ. Molecular analysis of classical and variant phenotypes. Pediatr Res. 1993;33:128A. [Google Scholar]
- Eng CM, Resnick-Silverman LA, Niehaus DJ, Astrin KH, Desnick RJ. Nature and frequency of mutations in the α-galactosidase A gene that cause Fabry disease. Am J Hum Genet. 1993;53:1186–1197. [PMC free article] [PubMed] [Google Scholar]
- Frustaci A, Chimenti C. Cryptogenic ventricular arrhythmias and sudden death by Fabry disease: prominent infiltration of cardiac conduction tissue. Circulation. 2007;116:350–351. doi: 10.1161/CIRCULATIONAHA.107.723387. [DOI] [PubMed] [Google Scholar]
- Jastrzebski M, Bacior B, Dimitrow PP, Kawecka-Jaszcz K. Electrophysiological study in a patient with Fabry disease and a short PQ interval. Europace. 2006;8:1045–1047. doi: 10.1093/europace/eul121. [DOI] [PubMed] [Google Scholar]
- Kampmann C, Baehner F, Whybra C, et al. Cardiac manifestations of Anderson-Fabry disease in heterozygous females. J Am Coll Cardiol. 2002;40:1668–1674. doi: 10.1016/S0735-1097(02)02380-X. [DOI] [PubMed] [Google Scholar]
- Kampmann C, Linhart A, Baehner F, et al. Onset and progression of the Anderson-Fabry disease related cardiomyopathy. Int J Cardiol. 2008;130:367–373. doi: 10.1016/j.ijcard.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Kampmann C, Wiethoff CM, Whybra C, Baehner FA, Mengel E, Beck M. Cardiac manifestations of Anderson-Fabry disease in children and adolescents. Acta Paediatr. 2008;97:463–469. doi: 10.1111/j.1651-2227.2008.00700.x. [DOI] [PubMed] [Google Scholar]
- Khoury PR, Mitsnefes M, Daniels SR, Kimball TR. Age-specific reference intervals for indexed left ventricular mass in children. J Am Soc Echocardiogr. 2009;22:709–714. doi: 10.1016/j.echo.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Kleinert J, Dehout F, Schwarting A, et al. Prevalence of uncontrolled hypertension in patients with Fabry disease. Am J Hypertens. 2006;19:782–787. doi: 10.1016/j.amjhyper.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Linhart A, Palecek T, Bultas J, et al. New insights in cardiac structural changes in patients with Fabry’s disease. Am Heart J. 2000;139:1101–1108. doi: 10.1067/mhj.2000.105105. [DOI] [PubMed] [Google Scholar]
- Linhart A, Magage S, Palecek T, Bultas J. Cardiac involvment in Fabry disease. Acta Paediatr Suppl. 2002;91:15–20. doi: 10.1111/j.1651-2227.2002.tb03104.x. [DOI] [PubMed] [Google Scholar]
- Mehta J, Tuna N, Moller JH, Desnick RJ. Electrocardiographic and vectorcardiographic abnormalities in Fabry’s disease. Am Heart J. 1977;93:699–705. doi: 10.1016/S0002-8703(77)80064-1. [DOI] [PubMed] [Google Scholar]
- Mehta A, Beck M, Elliott P, et al. Enzyme replacement therapy with agalsidase alfa in patients with Fabry’s disease: an analysis of registry data. Lancet. 2009;374:1986–1996. doi: 10.1016/S0140-6736(09)61493-8. [DOI] [PubMed] [Google Scholar]
- Monserrat L, Gimeno-Blanes JR, Marin F, et al. Prevalence of Fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2007;50:2399–2403. doi: 10.1016/j.jacc.2007.06.062. [DOI] [PubMed] [Google Scholar]
- Nagueh SF. Fabry disease. Heart. 2003;89:819–820. doi: 10.1136/heart.89.8.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namdar M, Kampmann C, Steffel J, et al. PQ interval in patients with Fabry disease. Am J Cardiol. 2010;105:753–756. doi: 10.1016/j.amjcard.2009.10.056. [DOI] [PubMed] [Google Scholar]
- Namdar M, Steffel J, Vidovic M, et al. Electrocardiographic changes in early recognition of Fabry disease. Heart. 2011;97:485–490. doi: 10.1136/hrt.2010.211789. [DOI] [PubMed] [Google Scholar]
- O`Connor M, McDaniel N, Brady W. The pediatric electrocardiogram Part I. Am J Emerg Med. 2008;26:221–228. doi: 10.1016/j.ajem.2007.08.003. [DOI] [PubMed] [Google Scholar]
- O'Mahony C, Coats C, Cardona M, et al. Incidence and predictors of anti-bradycardia pacing in patients with Anderson-Fabry disease. Europace. 2011;13:1781–1788. doi: 10.1093/europace/eur267. [DOI] [PubMed] [Google Scholar]
- Patel MR, Cecchi F, Cizmarik M, et al. Cardiovascular events in patients with Fabry disease natural history data from the Fabry registry. J Am Coll Cardiol. 2011;57:1093–1099. doi: 10.1016/j.jacc.2010.11.018. [DOI] [PubMed] [Google Scholar]
- Pochis WT, Litzow JT, King BG, Kenny D. Electrophysiologic findings in Fabry’s disease with a short PR interval. Am J Cardiol. 1994;74:203–204. doi: 10.1016/0002-9149(94)90106-6. [DOI] [PubMed] [Google Scholar]
- Ramaswami U, Whybra C, Parini R, et al. Clinical manifestations of Fabry disease in children: data from the Fabry Outcome Survey. Acta Paediatr. 2006;95:86–92. doi: 10.1080/08035250500275022. [DOI] [PubMed] [Google Scholar]
- Ramaswami U, Parini R, Pintos-Morell G, Kalkum G, Kampmann C, Beck M. Fabry disease in children and response to enzyme replacement therapy: results from the Fabry Outcome Survey. Clin Genet. 2012;81:485–490. doi: 10.1111/j.1399-0004.2011.01671.x. [DOI] [PubMed] [Google Scholar]
- Ries M, Ramaswami U, Parini R, et al. The early clinical phenotype of Fabry disease: a study on 35 European children and adolescents. Eur J Pediatr. 2003;162:767–772. doi: 10.1007/s00431-003-1299-3. [DOI] [PubMed] [Google Scholar]
- Ries M, Clarke JTR, Whybra C, et al. Enzyme-replacement therapy with agalsidase alfa in children with Fabry disease. Pediatrics. 2006;118:924–932. doi: 10.1542/peds.2005-2895. [DOI] [PubMed] [Google Scholar]
- Rijnbeek PR, Witsenburg M, Schrama E, Hess J, Kors JA. New normal limits for the paediatric electrocardiogram. Eur Heart J. 2001;22:702–711. doi: 10.1053/euhj.2000.2399. [DOI] [PubMed] [Google Scholar]
- Rijnbeek PR, Herpen G, Kapusta L, Harkel DJ, Witsenburg M, Kors JA. Electrocardiographic criteria for left ventricular hypertrophy in children. Pediatr Cardiol. 2008;29:923–928. doi: 10.1007/s00246-008-9235-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdev B, Takenaka T, Teraguchi H, et al. Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation. 2002;105:1407–1411. doi: 10.1161/01.CIR.0000012626.81324.38. [DOI] [PubMed] [Google Scholar]
- Scott O, Williams GJ, Fiddler GI. Results of 24 hour ambulatory monitoring of electrocardiogram in 131 healthy boys aged 10 to 13 years. Br Heart J. 1980;44:304–308. doi: 10.1136/hrt.44.3.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah JS, Hughes DA, Sachdev B, et al. Prevalence and clinical significance of cardiac arrhythmia in Anderson-Fabry disease. Am J Cardiol. 2005;96:842–846. doi: 10.1016/j.amjcard.2005.05.033. [DOI] [PubMed] [Google Scholar]
- Southall DP, Johnston F, Shinebourne EA, Johnston PGB. 24-hour electrocardiographic study of heart rate and rhythm patterns in population of healthy children. Br Heart J. 1981;45:281–291. doi: 10.1136/hrt.45.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurberg BL, Fallon JT, Mitchell R, Aretz T, Gordon RE, O'Callaghan MW. Cardiac microvascular pathology in Fabry disease: evaluation of endomyocardial biopsies before and after enzyme replacement therapy. Circulation. 2009;119:2561–2567. doi: 10.1161/CIRCULATIONAHA.108.841494. [DOI] [PubMed] [Google Scholar]
- Tøndel C, Bostad L, Hirth A, Svarstad E. Renal biopsy findings in children and adolescents with Fabry disease and minimal albuminuria. Am J Kidney Dis. 2008;51:767–776. doi: 10.1053/j.ajkd.2007.12.032. [DOI] [PubMed] [Google Scholar]
- Weidemann F, Breunig F, Beer M, et al. The variation of morphological and functional cardiac manifestation in Fabry disease: potential implications for the time course of the disease. Eur Heart J. 2005;26:1221–1227. doi: 10.1093/eurheartj/ehi143. [DOI] [PubMed] [Google Scholar]
- Weidemann F, Niemann M, Breunig F, et al. Long-term effects of enzyme replacement therapy on Fabry cardiomyopathy: evidence for a better outcome with early treatment. Circulation. 2009;119:524–529. doi: 10.1161/CIRCULATIONAHA.108.794529. [DOI] [PubMed] [Google Scholar]