

Abstract
Wilson disease (WD) is a disorder of copper transport that can cause hepatic and neuropsychiatric symptoms. Because of its broad spectrum of clinical manifestations that can present in almost any decade of life, a high degree of clinical suspicion is needed for diagnosis. We present an exceptional family with three consecutive generations affected by WD. Autosomal recessive disorders are not typically present in consecutive generations, but this can occur, particularly when carrier frequencies are as high as in WD. This point is of critical importance in counseling families affected by WD. This case also highlights the importance of genetic testing in confirming the diagnosis of WD, particularly when there is a positive family history. To our knowledge, this is the first report of WD in three consecutive generations.
Introduction
Wilson disease (WD) is an autosomal recessive disorder of copper transport. The causative gene, ATP7B, was identified in 1993 and remains the only gene associated with this disorder (Bull et al. 1993; Petrukhin et al. 1993; Tanzi et al. 1993; Yamaguchi et al. 1993). ATP7B is required for efficient transport of copper from hepatocytes into the biliary system (Sato and Gitlin 1991). In the absence of ATP7B function, there is toxic accumulation of copper in various body tissues, resulting in a wide variety of symptoms, including acute and chronic hepatitis, liver failure, and neurologic dysfunction. The wide spectrum of clinical manifestations, which can present in almost any decade of life, can make diagnosis challenging. However, early and accurate diagnosis is critical as oral chelating agents such as penicillamine and trientine, or agents that reduce intestinal copper absorption such as zinc, are simple, effective, and can prevent the development of neurologic and hepatic symptoms (Roberts and Schilsky 2008). The diagnosis of WD has traditionally been made by identifying low serum ceruloplasmin levels and elevated urine and hepatic copper levels, but because the biochemical values can overlap with those of healthy controls, the sensitivity and specificity of these tests can be problematic. For this reason, direct sequencing of the ATP7B gene has been suggested to be a critical component of the diagnosis of WD (Bennett and Hahn 2011). Indeed the sensitivity of ATP7B sequencing is estimated to be greater than 95% (http://www.ncbi.nlm.nih.gov/books/NBK1512/). The utility of direct gene sequencing is even greater in cases with a positive family history, as presymptomatic patients can be easily identified without ambiguity or invasive liver biopsy, allowing therapy to begin and prevent symptoms from ever developing. The ease and effectiveness of treating presymptomatic patients has prompted proposals to include WD in newborn screening (Kroll et al. 2006; deWilde et al. 2008).
In this report, we describe an exceptional family with three consecutive generations affected by WD. Although this is not a typical pattern for autosomal recessive disorders, it can occur, particularly when the carrier frequency is as high as is seen in WD. In addition, this family highlights the importance of genetic testing in diagnosing Wilson disease, as gene testing was used to confirm the diagnosis in two asymptomatic individuals who will, because of that test, be maintained on effective, life-saving therapies.
Case Report
The proband (II-2, see Fig. 1) presented for management of WD after moving cross-country at 28 years of age. She was diagnosed at 12 years of age after undergoing a diagnostic workup because her mother (I-2) had died of WD. At that time, the proband had mild elevations in her liver function tests (AST/ALT = 57/110 IU/L) and an elevated urine copper (220 ug/24 h, normal range 15–60). Her ceruloplasmin was normal (26 mg/dL, normal range 21–53), and liver biopsy showed mild focal portal inflammation and occasional hepatocytes positive for copper staining. She was started on penicillamine and has remained asymptomatic, without hepatic or neurologic symptoms.
Fig. 1.
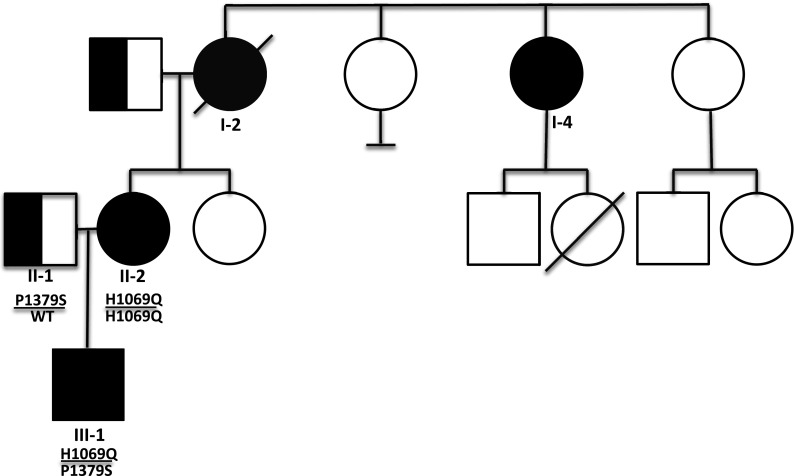
Individuals affected with Wilson disease are fully filled in with black; obligate carriers are indicated by half-filled symbols. ATP7B genotypes are also indicated below each individual, when known
Her mother (I-2) developed symptoms of liver failure, including ascites and esophageal varices, at age 39 years. At that time, she had an elevated urine copper (202 ug/24 h) and normal ceruloplasmin (24 mg/dL). Liver biopsy showed macronodular cirrhosis with marked hepatocellular copper accumulation. A slit lamp exam revealed Kayser-Fleischer rings. She was treated with penicillamine but died from complications of liver failure within 6 months of her presentation. Her three sisters and two daughters were screened for WD at that time. One of her sisters (I-4) had an elevated urine copper (464 ug/24 h) as well as hepatocellular copper accumulation on liver biopsy. She was 45 years old at that time. She developed signs of liver failure but responded to penicillamine. She is currently 63 years old and asymptomatic. The remaining family members’ screens were normal. Genetic testing was not performed as the gene for WD had not yet been identified.
To confirm II-2’s diagnosis, we analyzed the sequence of ATP7B. She was homozygous for p.H1069Q, the most common WD associated allele (Thomas et al. 1995). Her treatment was changed from penicillamine to zinc acetate because she was trying to get pregnant.
A year later, II-2 became pregnant. Her husband’s carrier testing showed that he is a heterozygote for a known pathogenic mutation (p.P1379S) in ATP7B (Cox et al. 2005). The family was appropriately counseled that there was a 50 % chance of their child being affected with WD. Prenatal testing was not pursued, but postnatal genetic testing showed that their newborn son (III-1) is a compound heterozygote for p.H1069Q/p.P1379S. He is currently 21 months of age, healthy, and developing normally. He has not yet been started on any dietary restriction and was started on low-dose zinc treatment at 16 months of age. At 1 year of age, prior to starting zinc treatment, he had a very mildly elevated AST with a normal ALT and ceruloplasmin (see Table 1).
Table 1.
Laboratory values for individuals in this family affected by Wilson disease
| AST (U/L) | ALT (U/L) | Urine copper (ug/24 h) | Ceruloplasmin (mg/dL) | |
|---|---|---|---|---|
| Normal range | 5–41 | 6–40 | 15–60 | 21–53 |
| I-2 | Unknown | Unknown | 202 | 24 |
| I-4 | Unknown | Unknown | 464 | Unknown |
| II-2 | 57 | 110 | 220 | 26 |
| III-1 | 45 | 22 | Not done | 30 |
In this exceptional family, there are four individuals in three consecutive generations affected with WD. To our knowledge, this has not been previously reported. There is no known consanguinity in this family.
Discussion
The availability of safe and effective treatment for WD highlights the importance of early and accurate diagnosis. However, there are reports of patients being misdiagnosed with WD and inappropriately treated, which has resulted in symptoms of copper deficiency (Kumar et al. 2003). When our proband (II-2) initially presented to us, she had never had any signs or symptoms of hepatic or neurologic dysfunction, and we did not initially have records of her childhood testing. Since WD does not usually present in consecutive generations, we wondered if her diagnosis was correct. Confirmatory genetic testing allowed unambiguous confirmation of her diagnosis and provided clear guidance that continued therapy for WD was indicated.
The presence of multiple affected generations is atypical for autosomal recessive disorders. In the absence of consanguinity, the chances of it occurring are related to the population carrier frequency. The prevalence of WD has been estimated to be 1/30,000, though it can be significantly higher in some populations (Dedoussis et al. 2005). This family highlights the fact that, with an estimated carrier frequency of 1/90 (Olivarez et al. 2001), WD is not rare. Given this carrier frequency, the chance of an individual with WD having an affected child is one half of one percent (½ × 1/90), and the chance of having an affected child and grandchild is ~0.003 % (½ × 1/90 × ½ × 1/90). There is one previous report of WD occurring in two generations, but consanguinity was present (Firneisz et al. 2001). Our case is, to our knowledge, the only report of WD occurring in three consecutive generations. It also underscores the utility of ATP7B sequence analysis in confirming a diagnosis of WD, particularly when there is a positive family history. As Table 1 indicates, all the individuals in this family with WD had normal ceruloplasmin levels. Although variants of uncertain significance remain a complication of ATP7B sequencing, the biochemical tests of WD can also be misleading (Bennett and Hahn 2011).
Abbreviations
- WD
Wilson disease
Grant Support
JB receives funding from NIH T32 training grant. KS receives funding from National Institute of Diabetes, Digestive and Kidney Diseases, Bristol Myers Squibb, Novartis, and Vertex Pharmaceuticals. PS and SH have no sources of funding to report.
Author Contributions
JB, KS, PS, and SH saw patients and contributed clinical information. JB and SH wrote the manuscript.
Disclosures
There are no conflicts of interest.
Take-Home Message
Providers must counsel their patients with Wilson disease carefully about recurrence risks since there can be vertical transmission of WD due to the fact that the carrier frequency is fairly high.
Footnotes
Competing interests: None declared
References
- Bennett J, Hahn SH. Clinical molecular diagnosis of Wilson disease. Semin Liver Dis. 2011;31(3):233–238. doi: 10.1055/s-0031-1286054. [DOI] [PubMed] [Google Scholar]
- Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5(4):327–337. doi: 10.1038/ng1293-327. [DOI] [PubMed] [Google Scholar]
- Cox DW, Prat L, Walshe JM, Heathcote J, Gaffney D. Twenty-four novel mutations in Wilson disease patients of predominantly European ancestry. Human Mutation. 2005;26(3):280. doi: 10.1002/humu.9358. [DOI] [PubMed] [Google Scholar]
- Dedoussis GV, Genschel J, Sialvera TE, et al. Wilson disease: high prevalence in a mountainous area of Crete. Ann Hum Genet. 2005;69(Pt 3):268–274. doi: 10.1046/J.1469-1809.2005.00171.x. [DOI] [PubMed] [Google Scholar]
- deWilde A, Sadilkova K, Sadilek M, Vasta V, Hahn SH. Tryptic peptide analysis of ceruloplasmin in dried blood spots using liquid chromatography-tandem mass spectrometry: application to newborn screening. Clin Chem. 2008;54(12):1961–1968. doi: 10.1373/clinchem.2008.111989. [DOI] [PubMed] [Google Scholar]
- Firneisz G, Szonyi L, Ferenci P, Gorog D, Nemes B, Szalay F. Wilson disease in two consecutive generations: an exceptional family. Am J Gastroenterol. 2001;96(7):2269–2271. doi: 10.1111/j.1572-0241.2001.03983.x. [DOI] [PubMed] [Google Scholar]
- Kroll CA, Ferber MJ, Dawson BD, et al. Retrospective determination of ceruloplasmin in newborn screening blood spots of patients with Wilson disease. Mol Genet Metab. 2006;89(1–2):134–138. doi: 10.1016/j.ymgme.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Kumar N, Gross JB, Jr, Ahlskog JE. Myelopathy due to copper deficiency. Neurology. 2003;61(2):273–274. doi: 10.1212/01.WNL.0000073542.02761.5F. [DOI] [PubMed] [Google Scholar]
- Olivarez L, Caggana M, Pass KA, Ferguson P, Brewer GJ. Estimate of the frequency of Wilson's disease in the US Caucasian population: a mutation analysis approach. Annals Human Genetics. 2001;65(Pt 5):459–463. doi: 10.1046/j.1469-1809.2001.6550459.x. [DOI] [PubMed] [Google Scholar]
- Petrukhin K, Fischer SG, Pirastu M, et al. Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nat Genet. 1993;5(4):338–343. doi: 10.1038/ng1293-338. [DOI] [PubMed] [Google Scholar]
- Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47(6):2089–2111. doi: 10.1002/hep.22261. [DOI] [PubMed] [Google Scholar]
- Sato M, Gitlin JD. Mechanisms of copper incorporation during the biosynthesis of human ceruloplasmin. J Biol Chem. 1991;266(8):5128–5134. [PubMed] [Google Scholar]
- Tanzi RE, Petrukhin K, Chernov I, et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993;5(4):344–350. doi: 10.1038/ng1293-344. [DOI] [PubMed] [Google Scholar]
- Thomas GR, Forbes JR, Roberts EA, Walshe JM, Cox DW. The Wilson disease gene: spectrum of mutations and their consequences. Nature Genetics. 1995;9(2):210–217. doi: 10.1038/ng0295-210. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Heiny ME, Gitlin JD. Isolation and characterization of a human liver cDNA as a candidate gene for Wilson disease. Biochem Biophys Res Commun. 1993;197(1):271–277. doi: 10.1006/bbrc.1993.2471. [DOI] [PubMed] [Google Scholar]