

Abstract
With over 1,000 nuclear genes that could potentially cause a mitochondrial disorder, the current diagnostic approach requires targeted molecular analysis, guided by a combination of clinical and biochemical features. However, the expanding molecular and clinical spectrum means that this approach does not always yield a result. Here we report the unusual clinical presentation of “Progressive External Ophthalmoplegia (PEO) plus” Leigh syndrome in three children from a consanguineous family where exome sequencing identified mutations in NDUFS8. NDUFS8 is a nuclear-encoded structural core protein of complex I, and mutations are expected to cause infantile onset and severe disease. Our patients had a later onset, milder and a clinically distinct phenotype, and this gene would not normally be considered in this context. Being untargeted to specific genes, whole exome analysis has the potential to re-write the phenotype and reveal an unexpected molecular aetiology, as illustrated by this family.
Introduction
Mitochondrial complex I (NADH ubiquinone oxidoreductase) is the largest enzyme of the mitochondrial respiratory chain and catalyzes the transfer of electrons from NADH through the respiratory chain (Distelmaier et al. 2009; Pagniez-Mammeri et al. 2012). It is composed of 37 nuclear-encoded and 7 mitochondrial-encoded subunits, based on a recent study, which showed that the NDUFA4 subunit is actually a subunit of complex IV (Balsa et al. 2012), and requires several additional proteins for its assembly. Mutations in many of these genes were reported in patients with different clinical presentations associated with isolated complex I deficiency, which is the most frequently encountered defect, causing ~25–35 % of all disorders of the mitochondrial energy metabolism (Distelmaier et al. 2009; Pagniez-Mammeri et al. 2012; Nouws et al. 2012).
Mutations in the mitochondrial subunit genes (MTND1-MTND6) of complex I cause maternally inherited disease with variable clinical presentations such as Leber’s hereditary optic neuropathy, mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes or Leigh syndrome. More frequent clinical presentations of nuclear gene defects are Leigh syndrome (25 %), neonatal cardiomyopathy with lactic acidosis, leukoencephalopathies or other undefined encephalomyopathies, which follow Mendelian inheritance. In general, mutations in assembly factor genes result in later onset and milder phenotypes compared to deficiencies of nuclear-encoded structural complex I subunit genes (Nouws et al. 2012). The number of nuclear mutations responsible for isolated complex I deficiency is increasing rapidly, which was recently facilitated by the new high-throughput technologies. However, thorough clinical reports on patients are still lacking (Haack et al. 2012; Calvo et al. 2010).
Here we report three children with a mild and variable clinical presentation due to a homozygous mutation in a nuclear complex I structural subunit gene, a totally unexpected finding detected through non-targeted whole exome sequencing.
Case Reports
Patient 1
The now 13-year-old index patient (Fig. 1a) is the first of three children of consanguineous Afghan parents (first cousins). Our index patient was born at term after uneventful pregnancy. He exhibited generalized muscular hypotonia from infancy with delayed motor milestones (sitting at 16 months, walking at 24 months, frequent falls). His speech development was delayed (first words at age 24 months). Facial hypotonia and mild ptosis were also noted.
Fig. 1.
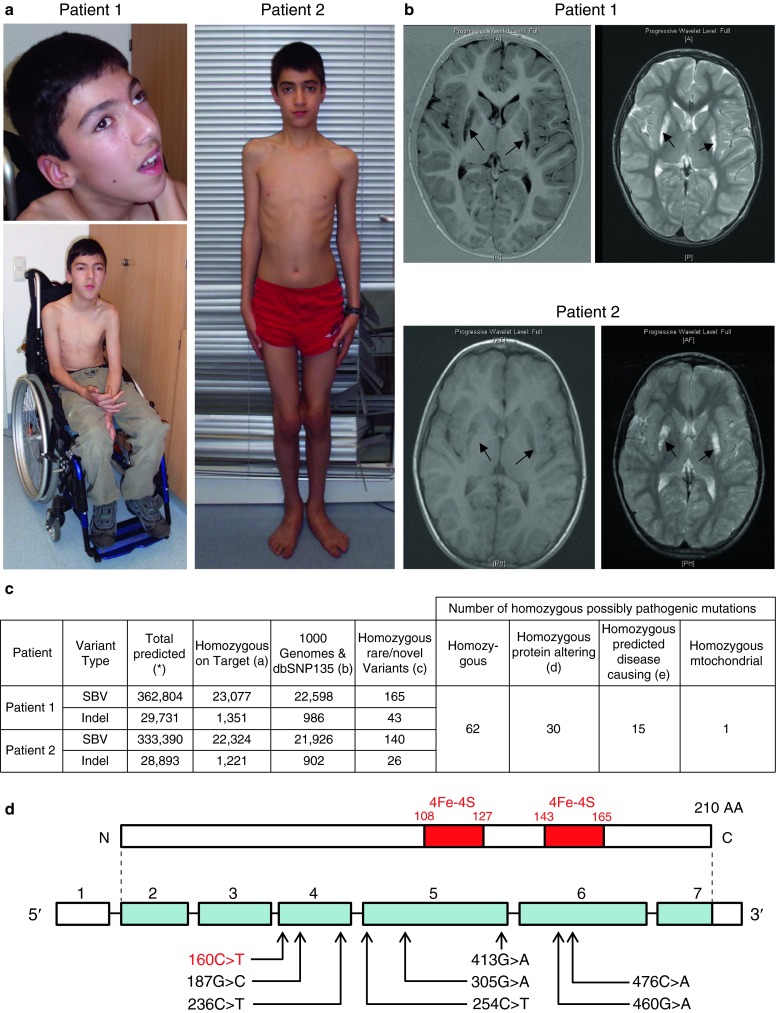
(a) The picture shows patient 1 (13 years) and patient 2 (12 years). Note external ophthalmoplegia on upgaze in patient 1. (b) Brain MRI of patient 1 at 9 years of age showed hyperintense lesions in the bilateral putamen and nucleus caudatus as well as in the bilateral frontal subcortical region on T2w sequences without enhancement after gadolinium. The lesions in the bilateral putamen and nucleus caudatus were hypointense on T1 images. There was a slight ventricular asymmetry, but the configuration of the cerebellum and brainstem were normal. Brain MRI of patient 2 at 8 years of age showed an abnormal symmetrical signal in the bilateral putamen (hypointense on T1 images, hyperintense on T2 images) but no other malformations of the brain structures. (c) Exome sequencing detected 62 novel homozygous changes shared in both patients; 30 were protein altering and 15 of these were predicted to be disease causing. Only one of these homozygous changes was in a gene encoding a known mitochondrial protein, NDUFS8, a complex I related gene. (d) Localization of the c.160C>T, pArg54Trp mutation in the NDUFS8 gene. 4Fe-4S refers to the iron-sulphur binding domains
From 5 years of age he suffered from a slowly progressive muscle weakness, and developed dysarthria and an ataxic gait. There was a marked worsening of his symptoms, especially of muscle strength and respiratory function, frequently caused by infections. Currently, at the age of 13 years he is able to walk only a few steps independently. He complains of pain after physical exercise and for longer distances he uses a wheelchair.
On clinical examination his height was on the 3rd percentile (144 cm) and weight 2 kg below the 3rd percentile (31.2 kg). He showed facial hypotonia, mild ptosis, external ophthalmoplegia (Fig. 1a) and had intermittent hypersalivation and difficulties in swallowing. He had atrophic muscles with thoracolumbal scoliosis and contractures; deep tendon reflexes were reduced. Further symptoms were dystonic posturing, decelerated movements and an ataxic gait. He had a reduced intelligence quotient (IQ) 61 (Snijders Oomen nonverbal Test for Intelligence), was not able to read or write apart from his own name and attends special schooling.
Laboratory measurements including creatine kinase (CK) were normal, but serum lactate was mildly elevated (23.5 mg/dl, normal<19.8 mg/dl). Echocardiography detected a borderline left ventricular dysfunction (fractional shortening 25.6 %); electrocardiogram was normal.
Cranial magnetic resonance imaging (MRI) at 5 years of age showed bilateral increased signals in the putamen. MRI at age 9 years showed increased signals in the bilateral putamen, nucleus caudatus and frontal subcortical region (Fig. 1b), consistent with mitochondrial encephalopathy (Leigh syndrome).
Substitution with creatine and riboflavin was initiated at 10 years of age, with no clearly demonstrable effect.
Patient 2
The younger brother of the index patient had a normal motor and mental development until 5 years of age, when he developed dysarthria. Severe myopia was diagnosed at 6 years of age, followed by slowly progressive muscle weakness leading to frequent falls. Currently, at 12 years of age (Fig. 1a), his weight is on 25th percentile (35 kg) and his height on 75th percentile (154 cm). He showed mild proximal muscular hypotonia with difficulties in uprising from a squatting position. His exercise tolerance was reduced, and he climbed stairs by holding on to the hand rail. He also complained of pain after exercise. His symptoms worsen during febrile infections.
Clinical examination revealed mild ptosis and ophthalmoplegia with intermittent horizontal nystagmus and dysarthria, but normal deep tendon reflexes. He had mild cognitive impairment (Kaufman-Assessment Battery for Children: IQ 73); he attends a school for handicapped children. Cranial MRI at 8 years of age showed abnormal signal in the bilateral putamen (Fig. 1b). Echocardiography detected borderline left ventricular dysfunction (fractional shortening 25.6 %); electrocardiogram was normal, as well as his laboratory findings including CK and lactate.
Patient 3
The third child of the family is a girl, currently 9 years old. She reached her motor and mental milestones age appropriately. At 7 years of age she had mild generalized muscular hypotonia, scapular winging and mild knee contractures. Deep tendon reflexes were normal. She had difficulties in rising from a squatting position, but her walking distance was unlimited. She had myopia, intermittent nystagmus and dysmetria, but no gait ataxia. She had normal cognitive development until 9 years of age (Kaufman-Assessment Battery for Children: IQ 84), and she had been visiting a mainstream primary school from age 7. Because of mild symptoms, no laboratory findings or MRI were performed. Echocardiography and electrocardiogram were normal.
Results
Muscle biopsy of patient 1 at 5 years of age detected non-specific variation of fibre size, no ragged-fed fibres in the Gomori-Trichrome stain and enzyme histochemistry for NADH-CoQ-Oxidoreductase (NADH), cytochrome c oxidase (COX) and succinate dehydrogenase (SDH) revealed no abnormalities (data not shown).
Biochemical analysis of the respiratory chain enzymes in muscle was done by standard methods and showed a reduction of complex I activity, NADH-CoQ-Oxidoreductase (0.05U/U Citrate Synthase, normal range: 0.17–0.56). The activities of complex II, III and IV were normal.
Genetic analyses: mitochondrial DNA deletions, mtDNA depletion and point mutations were excluded by standard methods. Genetic analysis of POLG was negative.
Whole exome sequencing was performed as described previously in patient 1 and 2 (Horvath et al. 2012). We detected 62 novel homozygous changes shared in both patients, 30 were protein altering and 15 of these were predicted to be disease causing. Only one of these homozygous changes was in a mitochondrial complex I related gene, in NDUFS8 (Fig. 1c). The mutation c.160C>T, p.Arg54Trp (Fig. 1d) is predicted to be disease causing (Mutation Taster) (De Sury et al. 1998) and was absent in 222 control chromosomes. This mutation co-segregated within the family as determined by Sanger sequencing, it was found heterozygous in the healthy parents and homozygous in all three affected siblings.
Discussion
Although mutations in several nuclear and mitochondrial genes have been reported in association with complex I deficiencies, there are few reports describing clear genotype-phenotype correlations to direct diagnostic genetic testing (Tuppen et al. 2010). The patients reported here have – unlike deficiencies of structural nuclear complex I subunit genes – a subtle onset, slowly progressive neuromuscular condition, with ptosis, eye movement abnormalities, exercise intolerance, muscle weakness and pain, dysarthria and ataxia, resembling “progressive external ophthalmoplegia (PEO) plus”. Cognitive development was variable, although differences in the younger siblings could be explained by an earlier phase of the disease. Brain MRI showed Leigh syndrome and isolated complex I deficiency was detected in skeletal muscle of patient 1, pointing to a mitochondrial disease. The high number of nuclear genes involved in the structure and assembly of complex I and the lack of genotype/phenotype data prompted us to perform whole exome sequencing. We detected 15 possible disease causing homozygous changes in the two affected patients, but only c.160C>T, p.Arg54Trp altered a complex I related gene, NDUFS8. This mutation is most likely causative for the disease in our patients, since it co-segregates with the symptoms, and alters a highly conserved amino acid, but does not disrupt the [4Fe-4S] cluster, and thus may result in some residual protein function.
By searching the literature we identified eight patients carrying NDUFS8 mutations to date (Table 1); however, the published clinical information was very brief in five cases (Haack et al. 2012; Calvo et al. 2010; Tuppen et al. 2010; Procaccio et al. 2004; Loeffen et al. 1998). Some patients had a severe disease with feeding difficulties, respiratory problems, epilepsy and hypertrophic cardiomyopathy and died within the first months of life (Tuppen et al. 2010; Loeffen et al. 1998). Others, including our patients show normal or mildly impaired motor development in the first years of life and develop a slowly progressive neurological disease at the end of the first decade (Procaccio and Wallace 2004, this paper). All reported mutations are missense mutations (Table 1, Fig. 1d), and no patients are reported to date with nonsense mutations in NDUFS8, suggesting that a complete absence of this protein may have deleterious effect leading to intrauterine lethality. Relatively mild phenotypes associated with Leigh syndrome are not so uncommon in mitochondrial disorders due to mutations in some nuclear assembly genes, as reported in patients with mutations in SURF1 (Piekutowska-Abramczuk et al. 2009).
Table 1.
Summary of all previously reported patients compared to our patients with NDUFS8 mutations
| Reference | Onset | Age/ death |
Clinical presentation | Brain MRI | Muscle biopsy | RC in cell lines | Mutation |
|---|---|---|---|---|---|---|---|
| Previously described patients | |||||||
| Loeffen et al. 1998 | 5 weeks | Died at 11 weeks | Poor feeding, apnea, cyanosis, spasticity, seizures, hypertrophic cardiomyopathy | LS | Histology n.d. complex I↓ (39% of normal) |
Fibroblasts complex I↓ (69% of normal) | c.236C>T/p.Pro79Leu c.305G>A/p.Arg102His compound heterozygous |
| Procaccio and Wallace 2004 | 7 years | Alive at 9 years | Toe walking, nystagmus, dysarthria, dystonia, ataxic gait | LS | Histology norm complex I↓ (31% of normal) |
Lymphoblast complex I↓ (43% of normal) | c.254C>T/p.Pro85Leu c.413G>A/p.Arg138His compound heterozygous |
| Haack et al. 2012 | n.d. | n.d. | Mitochondrial encephalopathy, cardiomyopathy, muscular hypotonia, respiratory insufficiency | n.d. | Histology n.d. complex I↓ (38% of normal) |
Fibroblasts complex I↓ (52% of normal) |
c.279C>T/p.Arg79Trp c.476C>A/p.Ala159Asp compound heterozygous |
| n.d. | n.d. | Muscular hypotonia, dyskinesia, epilepsy, lactic acidosis | LS | Histology n.d. complex I↓ (8% of normal) |
Fibroblasts complex I↓ (54% of normal) |
c.187G>C/p.Glu63Gln homozygous | |
| n.d. | n.d. | Muscular hypotonia, hypertrophic cardiomyopathy, lactic acidosis | LS | n.d. | n.d. | ||
| Tuppen et al. 2010 | 4 weeks | Died at 12 weeks | Poor feeding, apnea, lactic acidosis, seizures, failure to thrive, respiratory insufficiency | n.d. | Histology n.d. complex I↓ (30% of normal) | n.d. | c.236C>T, p.Pro79Leu homozygous |
| Calvo et al. 2010 sibs |
n.d. | n.d. | Mitochondrial encephalomyopathy | Leuko-encepha-lopathy | Complex I↓ | n.d. | c.460G>A, p.Gly154Ser homozygous |
| Patients described in this paper | |||||||
| P1 | Birth | Alive at 13 years | Developmental delay, PEO, muscular hypotonia, ataxic gait, dysarthria, dystonia, respiratory problems | LS | Histology norm complex I↓ (25% of normal) |
n.d. | c.160C>T, p.Arg54Trp homozygous |
| P2 | 5 years | Alive at 11 years | Dysarthria, muscular hypotonia, myopia, ptosis, PEO, swallowing difficulties | LS | n.d. | n.d. | |
| P3 | 9 years | Alive at 9 years | Muscular hypotonia, scapular winging, dysmetria, nystagmus, myopia | n.d. | n.d. | n.d. | |
Abbreviations: + present, n.d. not determined; PEO progressive external ophthalmoparesis, LS Leigh syndrome, RC respiratory chain
Defining the molecular basis of human diseases is important for genetic counselling and for prenatal genetic testing. However, limited clinical data on new and existing disease genes makes it difficult to know which genes to target. More detailed clinical descriptions of patients carrying mutations in different genes would be highly valuable for clinicians, who need to diagnose and counsel patients with mitochondrial disease. This will be facilitated by next generation sequencing of either large panels of relevant genes (Calvo et al. 2010), whole exome (Haack et al. 2012) and ultimately whole genome analysis. Our chapter shows the success of this approach.
Acknowledgments
We thank Christine Dineiger and Ira Kaus for excellent technical assistance. The authors have no competing interest.
Synopsis
Here we report three children with a mild and unusual clinical presentation of Leigh syndrome with progressive external ophthalmoparesis due to a homozygous mutation in a nuclear complex I structural subunit gene detected through non-targeted whole exome sequencing.
Author Contribution
Adela Della Marina and Ulrike Schara participated in the clinical follow-up of the patients and in the drafting the manuscript.
Angela Pyle performed whole exome sequencing and participated in the drafting of the manuscript.
Claudia Möller-Hartmann analyzed and interpreted the MRI images.
Elke Holinski-Feder, Angela Abicht and Birgit Czermin did the diagnostic work-up.
Hanns Lochmüller was involved in the biochemical and histological analyses.
Helen Griffin and Mauro Santibanez-Koref did the bioinformatic analysis of the exome data.
Patrick F. Chinnery and Rita Horvath are responsible for the clinical and diagnostic work-up, the study designs and for drafting of the manuscript.
Funding
RH was supported by the Medical Research Council (UK) (G1000848). PFC is a Wellcome Trust Senior Fellow in Clinical Science and an NIHR Senior Investigator who also receives funding from the Medical Research Council (UK), the UK Parkinson’s Disease Society, and the UK NIHR Biomedical Research Centre for Ageing and Age-related disease award to the Newcastle upon Tyne Foundation Hospitals NHS Trust.
Ethical Issues
The study was approved by the County Durham & Tees Valley 1 Research Ethics Committee (08/H0905/106 “Nuclear genes in mitochondrial disease”).
The parents gave written consent for the study and for presenting the data and photographs of the patients.
Competing Interest Statement
The authors have no competing interests.
Footnotes
Competing interests: None declared
ADM, US and AP as first authors and RH and PFC as last authors contributed equally to this study
References
- Balsa E, Marco R, Perales-Clemente E, Szklarczyk R, Calvo E, Landázuri MO, Enríquez JA. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 2012;16:378–386. doi: 10.1016/j.cmet.2012.07.015. [DOI] [PubMed] [Google Scholar]
- Calvo SE, Tucker EJ, Compton AG, et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat Genet. 2010;42:851–858. doi: 10.1038/ng.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sury R, Martinez P, Procaccio V, Lunardi J, Issartel JP. Genomic structure of the human NDUFS8 gene coding for the iron-sulfur TYKY subunit of the mitochondrial NADH:ubiquinone oxidoreductase. Gene. 1998;215:1–10. doi: 10.1016/S0378-1119(98)00275-3. [DOI] [PubMed] [Google Scholar]
- Distelmaier F, Koopman WJ, van den Heuvel LP, Rodenburg RJ, Mayatepek E, Willems PH, Smeitink JA. Mitochondrial complex I deficiency: from organelle dysfunction to clinical disease. Brain. 2009;132:833–842. doi: 10.1093/brain/awp058. [DOI] [PubMed] [Google Scholar]
- Haack TB, Haberberger B, Frisch EM, et al. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J Med Genet. 2012;49:277–283. doi: 10.1136/jmedgenet-2012-100846. [DOI] [PubMed] [Google Scholar]
- Horvath R, Holinski-Feder E, Neeve VC, et al. A new phenotype of brain iron accumulation with dystonia, optic atrophy, and peripheral neuropathy. Mov Disord. 2012;27:789–793. doi: 10.1002/mds.24980. [DOI] [PubMed] [Google Scholar]
- Loeffen J, Smeitink J, Triepels R, et al. The first nuclear-encoded complex I mutation in a patient with Leigh syndrome. Am J Hum Genet. 1998;63:1598–1608. doi: 10.1086/302154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouws J, Nijtmans LG, Smeitink JA, Vogel RO. Assembly factors as a new class of disease genes for mitochondrial complex I deficiency: cause, pathology and treatment options. Brain. 2012;135:12–22. doi: 10.1093/brain/awr261. [DOI] [PubMed] [Google Scholar]
- Pagniez-Mammeri H, Loublier S, Legrand A, Bénit P, Rustin P, Slama A. Mitochondrial complex I deficiency of nuclear origin I. Structural genes. Mol Genet Metab. 2012;105:163–172. doi: 10.1016/j.ymgme.2011.11.188. [DOI] [PubMed] [Google Scholar]
- Piekutowska-Abramczuk D, Magner M, Popowska E, et al. SURF1 missense mutations promote a mild Leigh phenotype. Clin Genet. 2009;76:195–204. doi: 10.1111/j.1399-0004.2009.01195.x. [DOI] [PubMed] [Google Scholar]
- Procaccio V, Wallace DC. Late-onset Leigh syndrome in a patient with mitochondrial complex I NDUFS8 mutations. Neurology. 2004;62:1899–1901. doi: 10.1212/01.WNL.0000125251.56131.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuppen HA, Hogan VE, He L, et al. The p.M292T NDUFS2 mutation causes complex I-deficient Leigh syndrome in multiple families. Brain. 2010;133:2952–2963. doi: 10.1093/brain/awq232. [DOI] [PMC free article] [PubMed] [Google Scholar]