

Abstract
Krabbe disease is an autosomal recessive demyelinating lysosomal storage disorder caused by a deficiency of galactocerebrosidase. The adult-onset variant is very rare. Hematopoietic stem cell transplantation (HSCT) is reported to be successful in treating infants with Krabbe disease prior to the onset of symptoms, but there are no reported cases of its use for adult-onset disease. We report the first follow-up data for a patient with adult-onset Krabbe disease who underwent HSCT at age 41, 16 years after the onset of symptoms. HSCT resulted in a sustained normalization of peripheral GALC enzyme activity, halted the progression of symptoms at 24 months post-allograft, and led to improvements in gait and balance. Serial imaging also confirmed that no significant progression of demyelination has occurred. Although long-term follow-up is needed to confirm the effects of HSCT, our 24-month results suggest that HSCT is a viable therapeutic option for symptomatic patients with adult-onset Krabbe disease.
Introduction
Krabbe disease, or globoid cell leukodystrophy, is an autosomal recessive demyelinating lysosomal storage disorder caused by a deficiency of galactocerebrosidase (GALC). The accumulation of psychosine results in death of oligodendrocytes and Schwann cells, both essential to myelin formation. Infantile- and juvenile-onset disease result in rapid neurological decline and death in most in the first few years of life. The adult-onset variant, which is the rarest, has a much milder and more protracted course typically presenting with slowly progressive spastic quadriplegia, bulbar signs, and demyelinating peripheral neuropathy (Kolodny et al. 1991; Suzuki 2003). Though the diagnosis is based on reduced GALC activity, MRI serves to support the diagnosis in late-onset cases by demonstrating regions of increased T2 signal in the pyramidal tracts, the posterior corpus callosum, and the parietooccipital white matter (Loes et al. 1999).
Therapeutic developments have focused on hematopoietic stem cell transplant (HSCT) strategies whereby the newly derived white blood cells (WBC) restore GALC levels thus halting accumulation of toxic metabolites (Sakai 2009). The evidence in support of HSCT comes from treatment of the infantile- and juvenile-onset disease. Outcomes have been most encouraging in patients treated prior to the development of neurological symptoms (Escolar et al. 2005; Krivit et al. 1998; Sakai 2009); however, there is some concern that most of these children do eventually develop symptoms (Duffner 2009). The largest study of symptomatic later-onset Krabbe disease describes only four patients. Following HCST, all patients had either improved or at least stabilized on neurological and MRI assessments (Krivit et al. 1998).
Even fewer data are available on the role of HSCT in the management of adult patients. Only one other adult patient has been reported to undergo HSCT for the management of Krabbe disease. Following HSCT at 24 years of age, she showed marked clinical improvement and had no progression of white matter abnormalities on MRI over a 7-year follow-up period (Lim et al. 2008). This patient had onset of symptoms at 3 years of age, while our patient developed symptoms in her 20s. Thus we present the first reported case of successful HSCT in a patient with adult-onset disease and who, to our knowledge, is also the oldest patient with Krabbe disease to be transplanted.
Report of a Case
The patient presented with spasticity, appendicular ataxia, dysarthria, and emotional lability that started in her 20s and progressed slowly over a 15-year period. White matter changes on MRI led to a possible diagnosis of primary progressive multiple sclerosis. She was seen at our center at 41 years of age at which point she had marked spasticity, dysmetria, bilateral foot drops, and ambulated with a walker. Krabbe disease was confirmed with a markedly reduced GALC activity of 0.2 nmol/h/mg protein in WBC (normal 2.1–10.44 nmol/h/mg protein using the substrate HMU-beta-Gal). Our patient was found to be a compound heterozygote for two mutations in trans: c.857G>A/p.G286D (previously described as p.G270D), a known pathogenic mutation (Furuya et al. 1997), and c.349A>G/p.M117V (previously p.M101V), a novel mutation found in several other patients with late-onset disease (De Gasperi et al. 1999). At the time of diagnosis, she had normal cerebrospinal fluid protein (261 mg/L, normal<450 mg/L), normal bilateral visual evoked potentials, and a normal EEG. Nerve conduction studies revealed a length-dependent axonal neuropathy that remained stable over the course of follow-up and was thought to be related to her diabetes. There was no demyelinating neuropathy (normal motor conduction velocities in the arm, 51–56 m/s, and in the legs, 41–45 m/s), which is the typical pattern of peripheral nerve involvement in Krabbe disease (Siddiqi et al. 2006). Lower extremity somatosensory evoked potentials revealed bilateral symmetrically delayed cortical responses (P40 latency 46.8–47.1 ms, normal<43.5 ms) with normal peripheral responses. Though the upper extremity responses were normal, this result was reproducible with repeated testing over 10 years and, though not definitive, suggests a delay in the dorsal columns of the spinal cord. Brain MRI revealed minimal progression over a 7-year period of bilateral symmetric T2 hyperintensities within the corticospinal tracts, cerebellar peduncles, and periventricular white matter (Fig. 1a). Findings on diffusion tensor imaging (DTI) and myelin water imaging were in support of a demyelinating process.
Fig. 1.
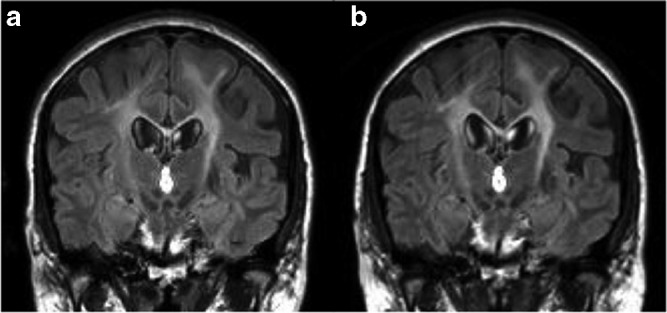
Serial MRI follow-up. Coronal FLAIR images obtained (a) prior to HSCT and (b) 24-month post-HSCT showing symmetrical confluent high-signal intensity changes along the corticospinal tracts with no progression of WM abnormality
The patient underwent 10/10 HLA-matched sibling allogenic blood stem cell transplant following a reduced-intensity conditioning regimen (fludarabine/melphalan/alemtuzumab), which did not cause neurological deterioration. The donor sibling was negative for both mutations. She received prophylaxis for graft versus host disease (GVHD) with cyclosporine delivered at 3 mg/kg/day and methotrexate 5 mg/m2 on days +1, +3, and +6. Her post-transplant course was relatively uncomplicated, and she was discharged on day +16 from hospital. She developed no significant GVHD. GALC activity was normalized at 1-year post-allograft (4.7 nmol/h/mg protein) and, though still within normal limits, has trended down to 2.7 nmol/h/mg protein at 24 months post-allograft. Though too early to determine if this trend is significant in our patient, it has been documented by others that the GALC enzyme activity peaked within 2 years of the transplantation and then decreased but remained within normal limits over the course of a 3- to 8-year follow-up period (Krivit et al. 1998). Full donor chimerism was confirmed on day +61 (myeloid 98% donor/lymphoid 95% donor) and maintained at 24 months (myeloid 97% donor/lymphoid 77% donor).
At 24 months post-allograft, on our assessments, her dysarthria is improved, her gait is less wide based, and she can tandem walk without support. Spasticity and hyperreflexia are unchanged. She can now walk unaided for 750 m and, with the help of a cane, she can even hike, an activity she could not enjoy pre-transplant. Her mood lability is also improved. Electrophysiological testing, including nerve conduction studies and somatosensory evoked potentials, is unchanged. Follow-up conventional and advanced MRI metrics at 6, 12, and 24 months post-allograft indicate the demyelinating process has not progressed (Fig. 1b), but given the slow rate of change on imaging pre-transplant, only ongoing follow-up will determine whether this trend is a significant change from her baseline.
In this case, HSCT resulted in improvements in gait and balance. Demyelination on serial imaging has not progressed. Although long-term follow-up is needed to confirm the effects of HSCT, our 24-month results suggest that HSCT is a viable therapeutic option for symptomatic patients with adult-onset Krabbe disease.
Disclosures and Funding
This study was not sponsored.
Dr. Sharp is the corresponding and principal author and reports no disclosures.
Dr. Laule was responsible for analysis and interpretation of the imaging data and reports no disclosures.
Dr. Nantel provided guidance in preparing and revising the manuscript and reports no disclosures.
Dr. Mädler was responsible for analysis and interpretation of the imaging data and reports no disclosures.
Dr. Aul provided guidance in preparing and revising the manuscript and reports no disclosures.
Dr. Yip provided guidance in preparing and revising the manuscript and reports no disclosures.
Dr. Sirrs is the senior author and has received speaking and consultancy fees or travel support from Genzyme, Shire Human Genetics Therapies, and Actelion Pharmaceuticals.
Footnotes
Competing interests: None declared
References
- De Gasperi R, Gama Sosa MA, Sartorato E, Battistini S, Raghavan S, Kolodny EH. Molecular basis of late-life globoid cell leukodystrophy. Human Mut. 1999;14:256–262. doi: 10.1002/(SICI)1098-1004(1999)14:3<256::AID-HUMU9>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Duffner PK. A model in response to newborn screening mandates. Pediatric Neurol. 2009;41:156. doi: 10.1016/j.pediatrneurol.2009.04.007. [DOI] [PubMed] [Google Scholar]
- Escolar ML, Poe MD, Provenzale JM, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N Engl J Med. 2005;352:2069–2081. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- Furuya H, Kukita Y, Nagano S, et al. Adult onset globoid cell leukodystrophy (Krabbe disease): analysis of galactosylceramidase cDNA from four Japanese patients. Human Genetics. 1997;100:450–456. doi: 10.1007/s004390050532. [DOI] [PubMed] [Google Scholar]
- Kolodny EH, Raghavan S, Krivit W. Late-onset Krabbe disease (globoid cell leukodystrophy): clinical and biochemical features of 15 cases. Dev Neurosci. 1991;13:232–239. doi: 10.1159/000112166. [DOI] [PubMed] [Google Scholar]
- Krivit W, Shapiro EG, Peters C, et al. Hematopoietic stem-cell transplantation in globoid-cell leukodystrophy. N Engl J Med. 1998;338:1119–1126. doi: 10.1056/NEJM199804163381605. [DOI] [PubMed] [Google Scholar]
- Lim ZY, Ho AYL, Abrahams S, et al. Sustained neurological improvement following reduced-intensity conditioning allogeneic haematopoietic stem cell transplantation for late-onset Krabbe disease. Bone Marrow Transplant. 2008;41:831–832. doi: 10.1038/sj.bmt.1705984. [DOI] [PubMed] [Google Scholar]
- Loes DJ, Peters C, Krivit W. Globoid cell leukodystrophy: distinguishing early-onset from late-onset disease using a brain MR imaging scoring method. AJNR Am J Neuroradiol. 1999;20:316–323. [PMC free article] [PubMed] [Google Scholar]
- Sakai N. Pathogenesis of leukodystrophy for Krabbe disease: molecular mechanism and clinical treatment. Brain Dev. 2009;31:485–487. doi: 10.1016/j.braindev.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Siddiqi ZA, Sanders DB, Massey JM. Peripheral neuropathy in Krabbe disease: electrodiagnostic findings. Neurology. 2006;67:263–267. doi: 10.1212/01.wnl.0000230153.34613.84. [DOI] [PubMed] [Google Scholar]
- Suzuki K. Globoid cell leukodystrophy (Krabbe's disease): update. J Child Neurol. 2003;18:595–603. doi: 10.1177/08830738030180090201. [DOI] [PubMed] [Google Scholar]