

Abstract
The direct synthesis of an acid-labile polyphosphoramidate by organobase-catalyzed ring-opening polymerization and an overall two-step preparation of polyphosphodiester ionomers (PPEI) by acid-assisted cleavage of the phosphoramidate bonds along the backbone of the polyphosphoramidate were developed in this study. The ultrafast organobase-catalyzed ring-opening polymerization of a cyclic phospholane methoxyethyl amidate monomer initiated by benzyl alcohol allowed for the preparation of well-defined polyphosphoramidates (PPA) with predictable molecular weights, narrow molecular weight distributions (PDI<1.10), and well-defined chain ends. Cleavage of the acid-labile phosphoramidate bonds on the polyphosphoramidate repeat units was evaluated under acidic conditions over a pH range of 1–5, and the complete hydrolysis produced polyphosphodiesters. The thermal properties of the resulting polyphosphoester ionomer acid and polyphosphoester ionomer sodium salt exhibited significant thermal stability. The parent PPA and both forms of the PPEIs showed low cytotoxicities toward HeLa cells and RAW 264.7 mouse macrophage cells. The synthetic methodology developed here has enriched the family of water-soluble polymers prepared by rapid and convenient organobase-catalyzed ring-opening polymerizations and straightforward chemical medication reactions, which are designed to be hydrolytically degradable and have promise for numerous biomedical and other applications.
Introduction
There has been considerable interest recently in the synthesis of synthetic functional bio-polymers that have compositions and architectures similar to those of polymers found in Nature and, thereby, possess ability for natural clearance mechanisms, and are explored for their biological and environmental applications, for instance tissue engineering, regenerative medicine, gene therapy, controlled drug delivery, oil recovery (viscosity control), paper manufacturing, agriculture (stimulation of plant growth) and packaging materials.1,2 Synthetic functional bio-polymers with various specific chemical structures and physical/biological properties are needed for biological and environmental applications because of the diversity and complexity of in vivo and natural environments. Inspired by our initial interest in polyphosphoesters,3–5 which were based upon phosphotriester repeat units that allowed for the introduction of various side chain functionalities, we expanded the synthetic methodology to the direct synthesis of acid-labile polyphosphoramidates (PPA) and their direct conversion to phosphodiester ionomeric repeat units. PPA and PPEI share the same polyphosphodiester backbone, PPA has side groups connecting to the backbone through an acid-labile phosphoramidate bond, while PPEI has a phosphate group on each repeat unit which is also represented on the natural nucleic and teichoic acids. This improved two-step preparation of polyphosphoester ionomers (PPEI) by organobase-catalyzed ring-opening polymerization (ROP) and acid-catalyzed side chain hydrolysis (Scheme 1) is a powerful alternative to the traditional metal catalyst-promoted ROP.6–11
Scheme 1.
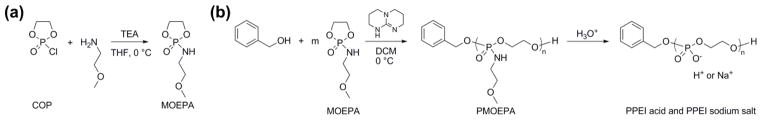
(a) Synthesis of cyclic phospholane amidate monomer from 2-chloro-2-oxo-1,3,2-dioxaphospholane (COP) and primary amine, methoxyethylamine (MOEA). (b) Polymerization of cyclic phospholane amidate monomer by using TBD as the catalyst and benzyl alcohol as the initiator and side chain hydrolytic cleavage of the PPA, PMOEPA, with the formation of PPEI (acid form and sodium salt form).
In this manuscript, we describe a direct synthesis of acid-labile PPA via organobase-catalyzed ROP of cyclic phospholane amidate monomer (Scheme 1). As reported previously, PPAs are conventionally prepared by the polymerization of 4-methyl-2-oxo-2-hydro-1,3,2-dioxaphospholane in the presence of metal catalyst triisobutylaluminum, followed by the modification of the resulting poly(1,2-propylene H-phosphonate) using amines via an Atherton–Todd reaction (Scheme 2).12 After the development of this method towards PPAs, a family of PPAs with different pendant amino groups was synthesized and applied for the delivery of nucleic acids.13,14 Due to the limited reactivity of the Atherton–Todd reaction, at least 25% of the P–H groups were converted into phosphates rather than phosphoramidate bonds,15 which resulted in a random copolymer of PPA and PPEI being isolated. The lack of a simple and reliable synthetic route to functional PPA constitutes a significant barrier to the widespread practical application of this degradable polymer platform. Recently, organobase-catalyzed ROP of cyclic phospholanes has greatly facilitated the preparation of polyphosphoesters, which share the similar polymer backbone with PPA but have side groups through the phosphoester linkage instead of the phosphoramidate linkage, with predictable molecular weight, narrow molecular weight distribution, well-defined chain ends and different polymer architectures.3–5,16–21 Herein, we took advantage of this state-of-the-art polymerization technique to synthesize PPA from a cyclic monomer in the presence of organocatalysts. The ultrafast organobase-catalyzed ROP offered control over the molecular weights, molecular weight distributions, compositions and structures, and also eliminates the usage of metal compounds, to fulfill the requirements of biomedical and environmental applications.
Scheme 2.
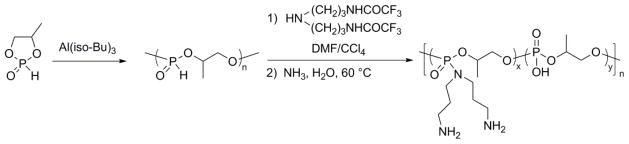
Reported synthetic approach for PPAs.12
After the complete side chain cleavage of the acid-labile phosphoramidate bond, the PPEI, a new type of degradable polymer, was prepared (Scheme 1). The PPEI has phosphodiester linkages along the backbone and a phosphate group on each repeat unit, and shares the same phosphate group with the nucleic and teichoic acids. Phosphate group-containing polymers, mostly based on non-degradable polymers, have served as dental materials and bone tissue engineering scaffolds, because of the formation of complexes of phosphonic acid or acidic phosphate groups with calcium in hydroxyapatite and improved adhesion on the tooth surface.22 Recently, increasing efforts have been devoted to degradable polymers with phosphate groups. Poly(propylene phosphate) was first synthesized by Penczek and colleagues in 1982 by the metal-catalyzed ROP of a cyclic phosphite monomer followed by oxidation (Scheme 3a).23 Later, the polymer was demonstrated as a thermo-sensitive injectable biomaterial whose gelation was induced by calcium ions.24 Another synthetic approach for PPEI-type materials was based on the demethylation of a methoxy side chain containing polyphosphoester, with an uncertain selectivity and a high risk of backbone degradation, by treating the polymer with sodium hydroxide (Scheme 3b). The free phosphate group on the PPEI provided the opportunity for the functionalization of the polymer backbone and ionical cross-linking by calcium ions.25 Poly(hydroxyoxyethylene phosphate) could be converted via Atherton-Todd reaction (Scheme 3c) from the poly(oxyethylene H-phosphonate), which was synthesized by condensation polymerization and had very broad molecular weight distribution and uncontrollable molecular weight.26 More recently, a PPEI-type copolymer was synthesized from the removal of benzyl protecting groups on a polyphosphoester precursor,27 which was synthesized by ROP from an unstable that was difficult to purify, 2-benzyl-2-oxo-1,3,2-dioxaphospholane (Scheme 3d).28 Even with some synthetic drawbacks, those advances on synthetic PPEI-type materials confirmed its capacity for aqueous solubility and for functionalization of the side chain phosphate group,25 and including by the simple complexation with calcium ions, for instance for use in tooth repair or as bone-specific drug delivery vehicles for the treatment of osteoporosis and osseous metastases.29,30 Here, the side chain cleavage of PPA provided a reliable strategy to synthesize PPEI homopolymer with controlled molecular weight, narrow molecular weight distribution and well-defined chain ends, which is expected to further advance the capabilities and development of these kinds of bio-mimicking, ionic, degradable polymer materials. The synthetic advances of PPA and PPEI, as well as their interesting chemical, physical and biological properties are discussed in this manuscript.
Scheme 3.

Reported synthetic approaches for PPEI-type materials.23,25–27
Experimental
Materials
N, N-Dimethylformamide (DMF), triethylamine (NEt3), acetone, diethyl ether, ethyl acetate, hexanes, glycine, hydrochloric acid, sodium acetate, acetic acid, trifluoroacetic acid (TFA), deuterium oxide (D2O), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), 2-methoxyethylamine and methanol were used as received from Sigma-Aldrich Company (St. Louis, MO). Amberlite™ IR120, H form, ion-exchange resin, Amberlite™ IR120, Na form, ion-exchange resin and 2-chloro-2-oxo-1,3,2-dioxaphospholane (95%) were used as received from Thermo Fisher Scientific Inc. (Pittsburgh, PA). Tetrahydrofuran (THF) and dichloromethane (DCM) were dried through columns (J. C. Meyer Solvent Systems, Inc. Laguna Beach, CA). Benzyl alcohol was purchased from Sigma-Aldrich and distilled from calcium hydride prior to use.
Instrumentation
1H NMR, 31P NMR and 13C NMR spectra were recorded on an Inova 300 or Mercury 300 spectrometer interfaced to a UNIX computer using VnmrJ software. Chemical shifts were referenced to the solvent resonance signals. The DMF gel permeation chromatography (GPC) was conducted on a Waters Chromatography, Inc. (Milford, MA) system equipped with an isocratic pump model 1515, a differential refractometer model 2414, and a four-column set of 5 μm Guard (50 × 7.5 mm), Styragel HR 4 5 μm DMF (300 × 7.5 mm), Styragel HR 4E 5 μm DMF (300 × 7.5 mm), and Styragel HR 2 5 μm DMF (300 × 7.5 mm). The system was equilibrated at 70 °C in pre-filtered DMF containing 0.05 M LiBr, which served as polymer solvent and eluent (flow rate set to 1.00 mL/min). Polymer solutions were prepared at a concentration of ca. 3 mg/mL and an injection volume of 200 μL was used. Data collection and analysis were performed with Empower 2 v. 6.10.01.00 software (Waters, Inc.). The system was calibrated with polystyrene standards (Polymer Laboratories, Amherst, MA) ranging from 615 to 442,800 Da. IR spectra were recorded on an IR Prestige 21 system (Shimadzu Corp.) and analyzed using IRsolution v. 1.40 software. Glass transition temperatures (Tg) were measured by differential scanning calorimetry on a Mettler-Toledo DSC822® (Mettler-Toledo, Inc., Columbus, OH), with a heating rate of 10 °C/min. Measurements were analyzed using Mettler-Toledo STARe v. 7.01 software. The Tg was taken as the midpoint of the transition recorded during the second heating scan. Thermogravimetric analysis was performed under N2 atmosphere using a Mettler-Toledo model TGA/SDTA851e, with a heating rate of 5 °C/min. Measurements were analyzed by using Mettler-Toledo STARe v. 7.01 software. Matrix assisted laser desorption ionization-time of flight (MALDI-TOF) experiments were performed on a Voyager DE-STR mass spectrometer (Applied Biosystems, Foster City, CA) under optimized conditions in positive linear mode. Ions were generated by a pulsed nitrogen laser at 337 nm and accelerated through 25 kV. 100 laser shots were used per spectrum. Trans-3-indoleacrylic acid (IAA) was used as the matrix and potassium trifluoroacetate was used as cationization agent. The sample and matrix were dissolved in dichloromethane at concentrations of 10 mg/mL. The sample solution was mixed with the matrix at a volume ratio of 1:1. About 0.5 μL of this mixture was deposited on a stainless steel sample holder. After allowing to air-dry, the sample was analyzed using MALDI-TOF MS.
Synthesis of 2-((2-methoxyethyl)amino)-1,3,2-dioxaphospholane 2-oxide, N-methoxyethyl phospholane amidate (MOEPA)
To a stirred solution of 2-methoxyethylamine (5.8 g, 77 mmol) and triethylamine (7.8 g, 77 mmol) in 200 mL of anhydrous THF in a cold room (4 – 6 °C) was added dropwise a solution of COP (10.0 g, 70 mmol) in 50 mL of anhydrous THF, and the reaction mixture was allowed to stir for 12 h. After complete conversion of COP, as confirmed by TLC, the reaction mixture was filtered and the filtrate was concentrated. The concentrated filtrate was purified by column chromatography on silica gel using ethyl acetate as eluent and gave a pale yellow liquid. The pale yellow liquid was further purified by recrystallization from ether/DCM 4:1 mixture and gave N-methoxyethyl phospholane amidate (MOEPA) as a white crystalline solid (8.0 g, yield: 63%). 1H NMR (CDCl3, ppm): δ 4.45-4.24 (m, 4H, POCH2CH2OP), 3.69–3.55 (b, 1H, PNHCH2), 3.42 (t, 3JH-H = 5.1 Hz, 2H, NHCH2CH2OCH3), 3.33 (s, 3H, NHCH2CH2OCH3), 3.07 (dt, 3JH-H = 5.1 Hz, 3JH-H = 6.2 Hz, 2H, NHCH2CH2OCH3). 13C NMR (CDCl3, ppm): δ 72.44, 65.72, 58.91, 41.28. 31P NMR (CDCl3, ppm): δ 25.82. +ESI MS: calculated [M+H]+ for C5H13NO4P: 182.0582, found: 182.0534. IR: 3300-3100, 3000-2800, 1446, 1241, 1108, 1087, 1033 cm−1.
General Procedure for Polymerization of MOEPA to afford PMOEPA
MOEPA was weighed inside a glove box and distributed into flame-dried 5 mL shell vials equipped with a rubber septum and a stir bar (about 0.200 g, 1.1 mmol for each) and stored in a vacuum desiccator under vacuum before being used for polymerizations. A solution of a given amount of benzyl alcohol (0.044 mmol to 0.011 mmol) in anhydrous dichloromethane (0.20 mL) was transferred via syringe into the shell vial. At 0 °C, a solution of a given amount of TBD (0.088 mmol to 0.011 mmol) in anhydrous dichloromethane (0.1 mL) was injected into the vial via syringe, while being maintained under a nitrogen gas atmosphere. After being stirred for a certain period of time (10 sec to 5 min), the reaction vial was unstoppered and 2 mL of dichloromethane was added via pipet into the reaction mixture to dilute the solution and quench the reaction. The poly(MOEPA) (PMOEPA, 2) was purified by precipitation from dichloromethane into diethyl ether (3×), and was then dried under vacuum, to give an average yield of 80%. 1H NMR (CDCl3, ppm): δ 7.40-7.28 (m, 5H, Ar-H), 5.00 (d, J = 7.5 Hz, 2H, OCH2Ar), 4.30-4.04 (b, 4nH, POCH2CH2OP), 4.02-3.83 (b, nH, PNHCH2), 3.40 (t, 3JH-H = 5.1 Hz, 2nH, NHCH2CH2OCH3), 3.31 (s, 3nH, NHCH2CH2OCH3), 3.07 (dt, 3JH-H = 5.1 Hz, 3JH-H = 6.2 Hz, 2nH, NHCH2CH2OCH3). 13C NMR (CDCl3, ppm): δ 128.63, 127.87, 72.77, 65.32, 58.70, 41.03. 31P NMR (CDCl3, ppm): δ 10.09. DSC: (Tg) = −23 °C. TGA in N2: 180–270 °C, 12% mass loss; 270–310 °C, 15% mass loss, 310–600 °C, 33% mass loss, 40 % mass remaining above 600 °C. IR: 3300-3100, 3000-2800, 1450, 1230, 1113, 1083, 1020, 953 cm−1.
Kinetic study of the side chain cleavage of PMOEPA
In a typical side chain cleavage experiment, PMOEPA (0.10 g) was dissolved into 1 mL of buffer solutions (100 mM hydrogen chloride solution as a pH 1.0 buffer, 100 mM glycine-HCl solution as a pH 3.0 buffer and 100 mM sodium acetate-acetic acid solution as a pH 5.0 buffer). 10 vol % of D2O (0.1 mL) was added to the buffer solutions. The mixture solution was stirred at room temperature allowing for the degradation. The 31P chemical shifts were measured by Inova 300 spectrometer during the degradation.
Preparation of polyphosphoester ionomer (PPEI)
PMOEPA (0.50 g) was dissolved into 10 mL of 100 mM HCl solution and stirred at room temperature for 11 h. The mixture solution was transferred to dialysis tubing (MWCO: 3.5 kDa) and dialyzed against nanopure water with the existence of Amberlite™ IR120, H form, ion-exchange resin or Amberlite™ IR120, Na form, ion-exchange resin in the cold room (4 – 6 °C) for 2 days, to remove small compounds. The clear solution was lyophilized to give white solids (for the H form PPI, 0.31 g, yield: 90 %; for the Na form PPI, 0.34 g, yield: 85 %). 1H NMR (D2O, ppm): δ 7.50-7.38 (m, 5H, Ar-H), 4.94 (d, J = 7.2 Hz, 2H, OCH2Ar), 4.01-3.92 (b, 4nH, POCH2CH2OP). 13C NMR (D2O, ppm): δ 128.54, 127.82, 67.58, 65.59, 60.67. 31P NMR (D2O, ppm): δ 1.01. Polyphosphoester ionomer sodium salt: DSC: (Tg) = 43 °C. TGA in N2: 260–320 °C, 30% mass loss, 70 % mass remaining above 600 °C. IR: 3650-3000, 3000-2800, 1650, 1455, 1224, 1077, 1033, 941 cm−1. Polyphosphoester ionomer acid: DSC: (Tg) = −14 °C. TGA in N2: 220–290 °C, 19% mass loss, 290–540 °C, 14% mass loss, 540–600 °C, 12% mass loss, 55 % mass remaining above 600 °C. IR: 3600-2400, 1650, 1450, 1214, 1077, 960 cm−1.
Cytotoxicity assays
HeLa cells and RAW 264.7 cells were seeded in a 96-well plate at a density of 1 × 104 cells/well and cultured in 100 μL Dulbecco’s Modified Eagle Medium (DMEM) containing 10% Fetal bovine serum (FBS) for 24 h. Then the medium was replaced with fresh medium, to which polyphosphoramidates and polyphosphoester ionomers (the PMOEA, PPEI acid and PPEI sodium salt) were added to make a final volume of 100 μL (final concentrations of polymers ranged from 5-to-2500 μg/mL). After 24 h, cytotoxicity was quantified by the CellTiter-Glo® Luminescent Cell Viability Assay (Promega) using 100 μL of CellTiter-Glo reagent. The contents were mixed and the plate was shaken for 5 min at 600 rpm, and then allowed to incubate at room temperature for 10 min to stabilize the luminescence signal. Luminescence intensities were recorded on a Luminoskan Ascent® luminometer (Thermo Scientific) with an integration time of 1 second/well. The relative cell viability was calculated by the following equation: Cell viability (%) = luminescence(sample)/luminescence(negative control)) × 100.
Results and Discussion
The synthetic efforts began from the one-step preparation and easy purification of cyclic phospholane amidate monomer, 2-((2-methoxyethyl)amino)-1,3,2-dioxaphospholane 2-oxide, N-methoxyethyl phospholane amidate (MOEPA). The polymerization behavior and kinetics of MOEPA by 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) were then carefully evaluated. The pH-dependent phosphoramidate bond cleavage and thermal properties of resulting PPEI acid and PPEI sodium salt were studied. All degradable polymers showed high biocompatibility toward HeLa cells and RAW 264.7 mouse macrophages.
Monomer design and synthesis
MOEPA was synthesized by coupling 2-chloro-2-oxo-1,3,2-dioxaphospholane (COP) to 2-methoxyethylamine in the presence of triethylamine (NEt3), according to the approach that has been widely used for the synthesis of cyclic phospholane ester monomers. As shown in Scheme 1a, the coupling reaction of COP to 2-methoxyethylamine was conducted at 4 °C in anhydrous THF with NEt3. MOEPA was purified by column chromatography on silica gel using ethyl acetate as eluent and followed by recrystallization from diethyl ether/DCM 1:1 mixture to give pure MOEPA as a white crystalline solid. Unlike most cyclic phospholane ester monomers, MOEPA was highly hygroscopic. To prevent absorption of moisture from the air, MOEPA was weighed and distributed into aliquots inside a glove box and stored in a vacuum desiccator under vacuum before being used for polymerizations or characterization. The 1H NMR spectrum of the monomer showed five groups of resonances at 4.45-4.24, 3.69–3.55, 3.42, 3.33 and 3.07 ppm, which were assigned to protons as shown in Figure 1 A–C. Resonances at 72.44, 65.72, 58.91 and 41.28 ppm in its 13C NMR spectrum also confirmed the chemical structure. In addition, the 31P NMR spectrum of the monomer exhibited a resonance at 25.82 ppm, consistent with the 31P chemical shift values of other reported cyclic phospholane amidate structures.31
Figure 1.

NMR spectra of MOEPA in CDCl3 (ppm): (A) 1H NMR (300 MHz), (B) 13C NMR (75 MHz), (C) 31P NMR (121 MHz). NMR spectra of PMOEPA in CDCl3 (ppm): (D) 1H NMR (300 MHz), (E) 13C NMR (75 MHz), (F) 31P NMR (121 MHz). NMR spectra of PPEI in D2O (ppm): (G) 1H NMR (300 MHz), (H) 13C NMR (75 MHz), (I) 31P NMR (121 MHz).
Polymerization of MOEPA by organobase catalysis
Two organocatalysts, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), were used to test the ROP of MOEPA (Table 1). Initially, DBU was employed to catalyze the polymerization of MOEPA initiated by benzyl alcohol at room temperature (entries 1–3 in Table 1). Although DBU has been reported to be efficient in promoting the polymerization of several cyclic phosphoester monomers at low catalyst-to-monomer molar ratios (in the range of 0.5 % to 5 % depending on the conditions),3,4,16–18 it was unable catalyze the ROP of MOEPA, even at a relatively high catalyst-to-monomer molar ratio of 10 mol%. Switching to the more basic catalyst TBD (Scheme 1b), which has dual activation effects: simultaneously serving as a hydrogen-bond donor to the monomer via the N–H site and also as a hydrogen-bond acceptor to the hydroxyl proton of the propagating alcohol,6,16,18 MOEPA polymerization proceeded rapidly at 0 °C (entries 4–12 in Table 1). For these polymerizations, the monomer, MOEPA, and initiator, benzyl alcohol, were dissolved in a certain amount of anhydrous dichloromethane (DCM) and allowed to stir at 0 °C. The conversion of MOEPA quickly reached over 55% within only 1 min with good control of the polymerization being retained (entry 5 in Table 1). After 1–5 min, the reaction was quenched by diluting the reaction mixture with ten times volume of DCM, instead of the typical method of quenching the polymerization by the addition of acid. Our control experiment showed by 31P NMR spectroscopy that the monomer conversion remained constant for several hours after the reaction mixture was diluted (data not shown). Although DBU was not effective, TBD was an efficient catalyst to promote the ROP of the cyclic phospholane amidate monomer, MOEPA.
Table 1.
Polymerization results of MOEPA with DBU and TBD under different conditions.
| entry | Catalyst | M : I : Catalyst (molar ratios) | Conc. (g/mL) | Time (min) | Conversion (31P NMR) | Mn, Da (GPC)a | Mw/Mn (GPC)b | Mn, Da (Theor)c | Mn, Da (1H NMR)d |
|---|---|---|---|---|---|---|---|---|---|
| 1 | DBU | 50 : 1 : 1.5 | 0.5 | 15 | 1 % | N.A. | N.A. | N.A. | N.A. |
| 2 | DBU | 50 : 1 : 2.5 | 0.5 | 30 | 2 % | N.A. | N.A. | N.A. | N.A. |
| 3 | DBU | 50 : 1 : 5.0 | 0.5 | 30 | 2 % | N.A. | N.A. | N.A. | N.A. |
| 4 | TBD | 50 : 1 : 1.5 | 0.5 | 1 | 77 % | 15000 | 1.34 | 7100 | 7300 |
| 5 | TBD | 50 : 1 : 1.0 | 0.5 | 1 | 58 % | 10000 | 1.08 | 5400 | 5500 |
| 6 | TBD | 50 : 1 : 1.0 | 0.5 | 2 | 75 % | 13000 | 1.17 | 6900 | 6800 |
| 7 | TBD | 50 : 1 : 1.0 | 0.25 | 1 | 41 % | 7200 | 1.07 | 3800 | 3900 |
| 8 | TBD | 50 : 1 : 1.0 | 0.25 | 3 | 66 % | 12000 | 1.07 | 6100 | 6300 |
| 9 | TBD | 50 : 1 : 0.5 | 0.5 | 1 | 34 % | 6200 | 1.08 | 3200 | 3300 |
| 10 | TBD | 50 : 1 : 0.5 | 0.5 | 4 | 62 % | 10000 | 1.07 | 5700 | 5400 |
| 11 | TBD | 100 : 1 : 1.0 | 0.5 | 5 | 51 % | 14000 | 1.05 | 9300 | 9000 |
| 12 | TBD | 25 : 1 : 0.25 | 0.5 | 1 | 68 % | 6200 | 1.08 | 3200 | 3400 |
Entries 1–3 were under room temperature; entries 4–12 were at 0 °C. Initiator (I) was benzyl alcohol for all entries. Solvent was anhydrous dichloromethane for all entries.
Mn (GPC) and Mw/Mn (GPC) were measured by DMF GPC calibrated using polystyrene standards.
Mn (Theor) was calculated from the monomer to initiator ratio and corrected for the conversion.
Mn (1H NMR) was calculated from the monomer to initiator ratio based on 1H NMR of final polymer product.
The catalytic behavior of TBD in the polymerization of MOEPA was studied further by tuning the polymerization conditions, including the catalyst-to-monomer molar ratio, the monomer concentration, the initiator-to-monomer molar ratio and the reaction time (Table 1). Polymerization with different catalyst-to-monomer molar ratios 1% (entries 9–10), 2% (entries 5–6) and 3% (entry 4) suggested that the amount of TBD didn’t alter the molecular weight of the PMOEPA polymer products when the conversions were similar, but only affected the rates of the polymerizations. The monomer concentration played a major role in the polymerization rate; the polymerization slowed significantly after diluting to twice the volume (entries 5–6 vs entries 7–8). The polydispersity indices (PDI) were all less than 1.10 and the gel permeation chromatography (GPC) traces were symmetrically mono-modal when the monomer conversion was limited to < 70% by quenching the reaction mixture at early stages (GPC traces see Figure 2.). After the conversion gradually reached over 70%, the GPC traces became increasingly asymmetrical, due to a growing high molecular weight shoulder, and eventually became bimodal (Figure 3). The increased breadths of the molecular weight distributions clearly indicated that adverse transesterification reactions of the polymer backbone were occurring and becoming dominant after the monomer conversion reached 70%. Therefore, quenching the polymerization reaction at conversions lower than 70% was found to be critical to obtain well-defined PMOEPA with low PDIs.
Figure 2.

GPC traces of PMOEPA52, PMOEPA29 and PMOEPA17.
Figure 3.

GPC traces of the polymerizations of MOEPA under conditions corresponding to those of entry 5 of Table 1: 0°C, I:M:catalyst = 1:50:1, concentration: 0.5 g/mL, however, allowed to proceed to longer time points and higher monomer conversions prior to quenching aliquots via dilution.
We further studied the kinetics of the ROP of the cyclic phospholane amidate by conducting parallel experiments, which allowed for monitoring of the extremely fast reaction. MOEPA and benzyl alcohol (at monomer-to-initiator molar ratios of 100 : 1 and 50 : 1) were premixed in anhydrous DCM and the solutions were divided equally into several portions, to each of which was added solutions of TBD (catalyst-to-monomer molar ratio 1%) in anhydrous DCM. After being stirred at 0 °C for preset periods of time, the reactions were quenched by diluting with DCM. The conversions were obtained from 31P NMR spectra, while the molecular weights and molecular weight distributions were determined by GPC. The linearity of Mn vs monomer conversion suggested that the numbers of macromolecules in the reactions were constant during polymerization (Figure 4 (A)). PDIs were all less than 1.10 when the monomer conversions were held to lower than 70%. Kinetic plots of ln[([M]0−[M]equil)/([M]−[M]equil)] vs time showed first order kinetics, characteristic of ROP of MOEPA (Figure 4 (B)), and suggesting that the rate constant of initiation was more than or equal to the rate constant of propagation and that the concentration of growing chains in the reaction was approximately constant during the polymerization. Those data, similar with that from ROP of cyclic phospholane monomers, confirmed the characteristics of a controlled/living polymerization of the ROP of cyclic phospholane amidate monomers into PMOEPA.
Figure 4.

(A) Plot of Mn and Mw/Mn (PDI) vs monomer conversion for the polymerizations of MOEPA by using TBD as the catalyst and benzyl alcohol as the initiator, obtained from a combination of GPC and 31P NMR analyses. Monomer : initiator ratios were 100 : 1 and 50 : 1. (B) Kinetic plots of ln[([M]0−[M]eq)/([M]−[M]eq)] vs time, obtained from 31P NMR spectroscopy data.
By controlling the monomer-to-initiator ratios as well as the reaction times, a series of poly(N-methoxyethyl phospholane amidates) (PMOEPAs) with different molecular weights were synthesized, and purified by precipitation from acetone into diethyl ether thrice. 31P NMR cleared showed only one phosphorus environment at a chemical shift of 10.09 ppm, which corresponds to the phosphorus on the PMOEPA (Figure 1 E).12,32 1H NMR and 13C NMR spectroscopic analyses also confirmed the structure of PMOEPA. Further analysis of the DP 7 polymer by MALDI-TOF MS revealed a single distribution with a spacing of 181 m/z, which is equal to that of a MOEPA unit (Figure 5). The main peak at m/z = 1415 corresponds to a potassium charged polymer chain of DP 7 with a benzyl alcohol end group. Their highly viscous liquid form at room temperature was attributed to the low glass transition temperatures (Tg) for all samples of PMOEPA, which were measured to be −23 ± 4 °C, independent of chain length over the range of degrees of polymerization measured, from 17 to 52. This ROP method allows the simple and efficient synthesis of well-defined PPAs with predictable molecular weights and chain ends from the cyclic phospholane amidate monomers. A variety of cyclic phospholane amidate monomers with different pendant functionalities, such as methyl, ethyl, isopropyl, alkenyl and alkynyl, for the syntheses of different functional PPAs are currently under further development.
Figure 5.

MALDI-TOF MS analysis of PMOEPA (DP = 7) initiated from benzyl alcohol.
Cleavage of acid-labile phosphoramidate bond
PPAs have side groups connecting to a phosphoester backbone through a phosphoramidate bond, which was reported to be labile in acidic environments, but relatively stable at neutral pH.33–35 Recently, acid-cleavable phosphoramidate linkages have been applied in biological nanomaterials to trigger release of conjugated drugs, macromolecules, coatings or other moieties.36–39 However, the acid cleavability of this phosphoramidate bond on the PPAs had not yet been studied. Therefore, we demonstrated the acid cleavability of the phosphoramidate bonds of PMOEPA and studied the side chain cleavage kinetics; furthermore, a novel PPEI homopolymer was synthesized by taking advantage of the acid-cleavable phosphoramidate linkage.
The side chain cleavage kinetics of the phosphoramidate bonds on the well-defined PMOEPAs were studied in three aqueous acidic buffer solutions, hydrogen chloride solution, glycine-HCl solution and sodium acetate-acetic acid solution having pH values of 1.0, 3.0 and 5.0, respectively (Figure 6). Cleavage of the phosphoramidate groups generated the PPEI, as shown in Scheme 1b. The distinct 31P chemical shift (1.01 ppm) of PPEI allowed for monitoring of the percentage conversion of side chain cleavage by 31P NMR spectroscopy. At pH 5.0, ca. 7% of the phosphoramidate bonds were converted into phosphate in 130 h. At the higher pHs, such as 7.4 and 9.0, the PMOEPA was found to be stable for more than 10 days with negligible changes (data not shown). In the more acidic environment, pH 3.0, greater than 23% of the phosphoramidate bonds were cleaved slowly in a controlled manner over 130 h. This slow side chain cleavage kinetics at pH 3.0 may enable PPAs to be responsive biomaterials for oral drug delivery, which requires sustained release in the acidic environment of the gastrointestinal tract. At pH 1.0, the side chain cleavage was greatly accelerated and complete hydrolysis was reached within about 10 h. These results indicate an interesting pH-dependent and gradual, tunable side cleavage rate for PMOEPA, which may be a general characteristic of PPA systems.
Figure 6.

Selective hydrolytic side chain cleavage kinetics of phosphoramidate bonds along the backbone of PMOEPA, monitored by 31P NMR spectroscopy.
Chemical and physical properties of PPEIs
After the complete side chain cleavage, the product PPEI was purified and ion exchanged by dialysis against nanopure water in the presence of Amberlite™ IR120, H form, ion-exchange resin or Amberlite™ IR120, Na form, ion-exchange resin to generate PPEI acid and PPEI sodium salt (Scheme 1b), respectively, and then confirmed by 1H, 13C and 31P NMR spectroscopies (Figure 1 (G)–(I)). The resulting PPEI acid and PPEI sodium salt had the identical 1H, 13C and 31P NMR spectra. Beyond the chain end group, there was only one peak in each of the three spectra, which confirmed the chemical structure of PPEI with only one chemical environment of the protons, carbons and phosphorus for the polymer repeat units, and no signals characteristic of the MOEA amine by-product. The PPEI acid and PPEI sodium salt had the same number of repeat units, calculated from the main chain units to chain end ratio based on 1H NMR, as the that of PMOEPA before the side chain cleavage. The fact that the phosphodiester backbone remained from the conditions employed for the side chain hydrolysis at pH 1.0 for 10 h, proved this method as a reliable synthetic methodology for the preparation of PPEI containing high contents of acidic phosphate groups, and points to relatively high hydrolytic stability for this polymer material. It is expected that DNAse and/or RNAse enzymes may be active against this polymer, and such studies are underway.
The PPEI acid and PPEI sodium salt had distinct thermal properties differences. The glass transition temperature (Tg) of the PPEI acid was −14 °C, while that of the PPEI sodium salt was 43 °C, measured by differential scanning calorimetry (DSC). Thermogravimetric analysis (TGA) of the PPEI acid exhibiteda multiple-stage mass loss profile: it was stable until 225 °C (Figure 7 (A)); from 225 °C to 273 °C (Figure 7 (B)), 21 % of mass was rapidly lost and the remaining mass agreed with the calculated value for H3PO4 on each repeat unit; from 273 °C to 520 °C (Figure 7 (C)), another 15 % of mass was gradually lost and the remaining mass was calculated as HPO3 for each repeat unit; to 600 °C (Figure 7 (D)), final 7 % of mass was lost and the remaining mass was calculated as PO2.5 for each repeat unit. In contrast, the PPEI sodium salt had only one mass loss temperature window: from 265 °C (Figure 7 (E)) to 310 °C (Figure 7 (F)), 30 % of mass was lost and the remaining mass corresponded to that calculated for NaPO3 from each repeat unit; and after 310 °C, no additional mass was lost. The higher decomposing temperature of PPEI sodium salt showed the higher stability when compared with PPEI acid. The ultra-high 70 wt% phosphorus and oxygen content, as much as that of ammonium polyphosphate, along with the high decomposition temperatures and decomposition products may enable PPEI sodium salt to perform as a promising fire-retardant material.40
Figure 7.
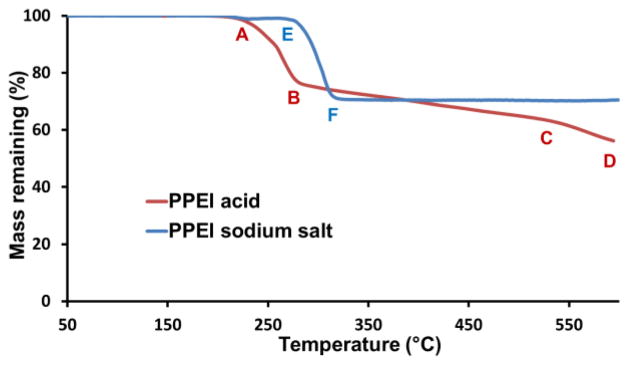
Thermogravimetric analyses of PPEI acid and PPEI sodium salt.
Cytotoxicity of degradable polymers
To demonstrate the biocompatibilities of PPAs and PPEIs, the PMOEPA, PPEI acid and PPEI sodium salt were tested for their cytotoxicity in both HeLa cells and RAW 264.7 mouse macrophage cells at different concentrations (Figure 8). No cytotoxicity was observed for all three polymers over the range of concentrations from 5-to-1250 μg/mL in both cell lines after 24 h-incubation. Only at the highest tested concentration (2500 μg/mL), the polymers exhibited dose-dependent toxicities against both cell lines. Therefore, PPAs and PPEIs could be used at a broad range of concentrations without causing cytotoxicity for biological applications.
Figure 8.

Cytotoxicity of PMOEPA, PPEI sodium salt and PPEI acidkin HeLa cells (A) and RAW 264.7 mouse macrophage cells (B) after treatment over a concentration range of 5-to-2500 μg/mL for 24 h.
Conclusions
In this study, we have demonstrated the direct polymerization of a cyclic phospholane amidate monomer by organobase catalysis in the presence of an alcohol initiator, and transformation of the resulting polyphosphoramidate to polyphosphoester ionomer acid and sodium salts by hydrolytically cleaving the acid-labile phosphoramidate bonds in acidic solutions. The prolonged chain cleavage profiles of the polyphosphoramidates even at pH 1.0 might be useful for polyphosphoramidates serving as responsive biomaterials for localized drug release in the gastrointestinal tract. The high thermal stability and ultrahigh 70 wt% phosphorus and oxygen content of the polyphosphoester ionomer sodium salt promises applicability as an alternative fire-retardant material. In addition, polyphosphoramidate, polyphosphoester ionomer acid and polyphosphoester ionomer sodium salt showed high biocompatibility toward HeLa cells and RAW 264.7 mouse macrophages. The phosphoramidate and phosphoester ionomer functionalities may also provide for bonding to inorganic substrates in vivo, for instance to achieve targeting to bone in the treatment of osteosarcomas, leukemia, etc. and bone and tooth repair. Overall, this work represents novel and reliable synthetic methodology to obtain two degradable polymer systems with diverse potential. A particularly interesting direction that we are taking for these materials is their incorporation as hydrophilic block segments together with saccharide-based polycarbonate chain segments41,42 to afford functional nanomaterials that are derived from renewable resources and produce biologically beneficial by-products upon degradation.
Acknowledgments
We gratefully acknowledge financial support from the National Heart Lung and Blood Institute of the National Institutes of Health as a Program of Excellence in Nanotechnology (HHSN268201000046C) and the National Science Foundation under grant numbers DMR-0906815 and DMR-1105304 (K.L.W). The Welch Foundation is gratefully acknowledged for support through the W. T. Doherty-Welch Chair in Chemistry, Grant No. A-0001 (K.L.W). A.P.D. also acknowledges financial assistance provided by the Royal Society of Chemistry’s Journals Grants for International Authors programme.
References
- 1.Nair LS, Laurencin CT. Prog Polym Sci. 2007;32:762. [Google Scholar]
- 2.Gross RA, Kalra B. Science. 2002;297:803. doi: 10.1126/science.297.5582.803. [DOI] [PubMed] [Google Scholar]
- 3.Zhang SY, Zou J, Zhang FW, Elsabahy M, Felder SE, Zhu JH, Pochan DJ, Wooley KL. J Am Chem Soc. 2012;134:18467. doi: 10.1021/ja309037m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang SY, Li A, Zou J, Lin LY, Wooley KL. ACS Macro Lett. 2012;1:328. doi: 10.1021/mz200226m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang S, Zou J, Elsabahy M, Karwa A, Li A, Moore DA, Dorshow RB, Wooley KL. Chem Sci. 2013;4:2122. doi: 10.1039/C3SC50252J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dove AP. ACS Macro Lett. 2012;1:1409. doi: 10.1021/mz3005956. [DOI] [PubMed] [Google Scholar]
- 7.Sanders DP, Fukushima K, Coady DJ, Nelson A, Fujiwara M, Yasumoto M, Hedrick JL. J Am Chem Soc. 2010;132:14724. doi: 10.1021/ja105332k. [DOI] [PubMed] [Google Scholar]
- 8.Jeong W, Hedrick JL, Waymouth RM. J Am Chem Soc. 2007;129:8414. doi: 10.1021/ja072037q. [DOI] [PubMed] [Google Scholar]
- 9.Dove AP, Pratt RC, Lohmeijer BGG, Waymouth RM, Hedrick JL. J Am Chem Soc. 2005;127:13798. doi: 10.1021/ja0543346. [DOI] [PubMed] [Google Scholar]
- 10.Coulembier O, Mespouille L, Hedrick JL, Waymouth RM, Dubois P. Macromolecules. 2006;39:4001. [Google Scholar]
- 11.Coulembier O, Moins S, Dubois P. Macromolecules. 2011;44:7493. [Google Scholar]
- 12.Wang J, Zhang PC, Lu HF, Ma N, Wang S, Mao HQ, Leong KW. J Control Release. 2002;83:157. doi: 10.1016/s0168-3659(02)00180-3. [DOI] [PubMed] [Google Scholar]
- 13.Zhang XQ, Wang XL, Huang SW, Zhuo RX, Liu ZL, Mao HQ, Leong KW. Biomacromolecules. 2005;6:341. doi: 10.1021/bm040060n. [DOI] [PubMed] [Google Scholar]
- 14.Jiang X, Qu W, Pan D, Ren Y, Williford JM, Cui H, Luijten E, Mao HQ. Adv Mater. 2013;25:227. doi: 10.1002/adma.201202932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren Y, Jiang XA, Pan D, Mao HQ. Biomacromolecules. 2010;11:3432. doi: 10.1021/bm1009574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iwasaki Y, Yamaguchi E. Macromolecules. 2010;43:2664. [Google Scholar]
- 17.Liu JY, Pang Y, Huang W, Zhai XA, Zhu XY, Zhou YF, Yan DY. Macromolecules. 2010;43:8416. [Google Scholar]
- 18.Clement B, Grignard B, Koole L, Jerome C, Lecomte P. Macromolecules. 2012;45:4476. [Google Scholar]
- 19.Du JZ, Du XJ, Mao CQ, Wang J. J Am Chem Soc. 2011;133:17560. doi: 10.1021/ja207150n. [DOI] [PubMed] [Google Scholar]
- 20.Liu J, Huang W, Zhou Y, Yan D. Macromolecules. 2009;42:4394. [Google Scholar]
- 21.Liu J, Huang W, Pang Y, Zhu X, Zhou Y, Yan D. Biomaterials. 2010;31:5643. doi: 10.1016/j.biomaterials.2010.03.068. [DOI] [PubMed] [Google Scholar]
- 22.Monge S, Canniccioni B, Graillot A, Robin JJ. Biomacromolecules. 2011;12:1973. doi: 10.1021/bm2004803. [DOI] [PubMed] [Google Scholar]
- 23.Biela T, Penczek S, Slomkowski S, Vogl O. Makromol Chem-Rapid. 1982;3:667. [Google Scholar]
- 24.Wang J, Sun DDN, Shin-ya Y, Leong KW. Macromolecules. 2003;37:670. [Google Scholar]
- 25.Wan ACA, Mao HQ, Wang S, Phua SH, Lee GP, Pan JS, Lu S, Wang J, Leong KW. J Biomed Mater Res. 2004;70B:91. doi: 10.1002/jbm.b.30022. [DOI] [PubMed] [Google Scholar]
- 26.Troev K, Tsatcheva I, Koseva N, Georgieva R, Gitsov I. J Polym Sci, Part A: Polym Chem. 2007;45:1349. [Google Scholar]
- 27.Iwasaki Y, Kawakita T, Yusa S. Chem Lett. 2009;38:1054. [Google Scholar]
- 28.Amigues EJ, Migaud ME. Tetrahedron Lett. 2004;45:1001. [Google Scholar]
- 29.Low SA, Kopecek J. Adv Drug Deliver Rev. 2012;64:1189. doi: 10.1016/j.addr.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ikeuchi R, Iwasaki Y. Journal of Biomed Mater Res Part A. 2013;101A:318. doi: 10.1002/jbm.a.34321. [DOI] [PubMed] [Google Scholar]
- 31.Modro AM, Modro TA, Bernatowicz P, Schilf W, Stefaniak L. Magn Reson Chem. 1997;35:774. [Google Scholar]
- 32.N’Guyen TTT, Oussadi K, Montembault V, Fontaine L. J Polym Sci, Part A: Polym Chem. 2013;51:415. [Google Scholar]
- 33.Rahil J, Haake P. J Am Chem Soc. 1981;103:1723. [Google Scholar]
- 34.Benkovic SJ, Sampson EJ. J Am Chem Soc. 1971;93:4009. [Google Scholar]
- 35.Garrison AW, Boozer CE. J Am Chem Soc. 1968;90:3486. [Google Scholar]
- 36.Leriche G, Chisholm L, Wagner A. Bioorgan Med Chem. 2012;20:571. doi: 10.1016/j.bmc.2011.07.048. [DOI] [PubMed] [Google Scholar]
- 37.Jeong JH, Kim SW, Park TG. Bioconjugate Chem. 2003;14:473. doi: 10.1021/bc025632k. [DOI] [PubMed] [Google Scholar]
- 38.Romberg B, Hennink WE, Storm G. Pharm Res. 2008;25:55. doi: 10.1007/s11095-007-9348-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoon S, Kim WJ, Yoo HS. Small. 2013;9:284. doi: 10.1002/smll.201200997. [DOI] [PubMed] [Google Scholar]
- 40.Li YC, Mannen S, Morgan AB, Chang SC, Yang YH, Condon B, Grunlan JC. Adv Mater. 2011;23:3926. doi: 10.1002/adma.201101871. [DOI] [PubMed] [Google Scholar]
- 41.Azechi M, Matsumoto K, Endo T. J Polym Sci, Part A: Polym Chem. 2013;51:1651. [Google Scholar]
- 42.Mikami K, Lonnecker AT, Gustafson TP, Zinnel NF, Pai PJ, Russell DH, Wooley KL. J Am Chem Soc. 2013;135:6826. doi: 10.1021/ja402319m. [DOI] [PubMed] [Google Scholar]